Introduction
Lung cancer is the most common cancer and a leading
cause of cancer-associated mortality worldwide (1–3).
Non-small cell lung cancer (NSCLC) is the most common type of lung
cancer and is associated with high mortality and poor prognosis;
over 70% of the patients have advanced cancer at the time of
diagnosis and are not suitable for surgical treatment (4,5).
Therefore, chemotherapy is the main therapeutic option available
for these patients (6,7). Two platinum-containing chemotherapy
regimens are regularly used as the first-line treatment for
patients with lung cancer (8,9).
However, drug resistance is a major challenge with this line of
treatment (10,11). Intrinsic changes in tumor cells are
known to play a critical role in the emergence of chemoresistance
(12–14). Thus, the identification of the
associated mechanisms is essential in cancer therapeutics.
Cisplatin is the most active platinum agent for
patients with advanced lung cancer and early-stage patients who
require adjuvant therapy (15).
However, after treatment for a certain period of time, most
patients develop cisplatin resistance (16,17).
Inhibiting the apoptosis of cancer cells is one
mechanism of cisplatin resistance (18). Survivin is an apoptosis
protein-inhibitor that is upregulated in cancer cells and serves as
an attractive prognostic marker of malignancies (19).
Src homology 2 (SH2) domain containing protein
tyrosine phosphatase-2 (SHP2) promotes signaling for most growth
factors and cytokine receptors. SHP2 is required for growth
factor/cytokine-induced cell proliferation, migration and survival
in many cancers (20) and is
essentially mediated via activating the Ras/mitogen-activated
protein kinase (MAP) pathway (7,21–23).
However, the influence of SHP2 in inducing lung cancer cell
resistance to cisplatin is not well established.
The phosphoinositol 3-kinase (PI3K/Akt) pathway is
involved in cell survival, differentiation, proliferation,
apoptosis and metastasis and is implicated in the pathogenesis of
many tumors (24).
The Ras/MAPK pathway has been the preferential
pathway used in cancer research (25,26).
SHP2 is a direct activator of Ras and the downstream of MAPK
signaling, which are involved in tumor progression (27).
We previously discovered upregulated SHP2 in lung
cancer tissues. Inhibiting SHP2 downregulated the expression of Akt
(28). Furthermore, inhibiting the
PI3K/Akt pathway reversed resistance of the A549/CDDP cells to
cisplatin, while activation of the PI3K/Akt pathway enhanced
tolerance of the A549/CDDP cells to cisplatin. However, whether the
anti-apoptotic effect of the Ras-activated PI3K/Akt pathway, is
also involved in the SHP2-enhanced lung cancer resistance to
cisplatin is not known.
The aim of this study was to explore the role of
SHP2 in the development of cisplatin resistance in lung cancer as
well as the underlying mechanism.
Materials and methods
Statement of ethics
The experimental protocol was approved by the Human
Ethics Committee of the First Affiliated Hospital of the Third
Military Medical University.
Cell lines and reagents
The human small cell lung cancer (SCLC) H446 and
adenocarcinoma SPC-A-1 cell lines were obtained from the Shanghai
Institute of Biochemistry and Cell Biology (Shanghai, China). The
H446/CDDP and SPC-A-1/CDDP cells were sourced from Dr Linzhi Liu at
Xinqiao Hospital of the Third Military Medical University.
Cisplatin was obtained from North China Pharmaceutical Co., Ltd.
(Shijiazhuang, China). The Cell Counting Kit-8 (CCK-8) was obtained
from Dojindo Molecular Technologies, Inc. (Kumamoto, Japan) and the
H-Ras siRNA and antibodies from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA, USA). The bicinchoninic acid (BCA) protein assay
kit and the enhanced chemiluminescent detection kit were purchased
from Pierce Biotechnology, Inc. (Rockford, IL, USA). The RNAiso
reagent was obtained from Takara Bio, Inc. (Shiga, Japan). The cDNA
reverse transcription kit was purchased from Fermentas Inc.
(Burlington, ON, Canada). The FITC Annexin V and PI apoptosis
detection kit was obtained from BD Biosciences (San Diego, CA,
USA).
Cell culture and treatment
The cells were cultured in RPMI-1640 medium
containing 10% FBS at 37°C with 5% CO2 and 100% humidity
and treated with different concentrations of cisplatin for
predefined time-points.
Immunohistochemistry
The cells were seeded over cover slips, fixed with
4% paraformaldehyde, permeabilised with 1% Triton X-100, blocked
with 3% H2O2 and then incubated with primary
antibody after being blocked in 0.5% normal goat serum. The
immunoreactivity of the cells was assessed by staining intensity
and scored on a scale of 0 to 3 as follows: 0 (−), absence of
staining; 1 (+), weak staining; 2 (++), moderate staining; and 3
(+++), strongly immunoreacting. The cells incubated with primary
antibody diluents without the primary antibody served as a negative
control.
SHP2 enzyme activity assay
Protein (50 mg) was mixed with protein tyrosine
phosphatase (PTP) buffer (25 mM; pH 6.0 Tris-HCl, 50 mM NaCl, 2 mM
EDTA and 10 mM DTT) to a final volume of 99 µl. Subsequently, 1 µl
of 1 M para-nitrophenyl phosphate (pNPP) was added and incubated at
37°C for 10 min, and then terminated with 900 µl of termination
buffer (1 ml of 5 N NaOH plus 5 ml of 50% alcohol + 4 ml of Milli-Q
H2O). The optical density (OD) was determined at 405 nm.
Α PTP buffer (99 µl) without protein was used as a negative
control.
Construction of SHP2 lentiviral
vectors and cell transfection
To construct an SHP2 overexpression lentiviral
vector, we designed and synthesized the primers engineered with the
BamH1 restriction site in the forward direction and the
Xho1 restriction site in the reverse direction, according to
the full-length sequence of SHP2 in PubMed database (NM_002834) and
the restriction enzyme cutting sites of the lentiviral vector. cDNA
from the H446 cells was amplified and cloned into the PTA2 vector,
and then transfected into competent Escherichia coli (DH5α)
cells. The SHP2 overexpression lentiviral vector LV-SHP2-IRES-PURO
was constructed using conventional methods. An empty LV-IRES-PURO
vector was used as a negative control.
The knockdown of the SHP2 gene was also performed
using lentivirus-mediated RNA interference. Briefly,
oligonucleotides coding for the short hairpin RNA (shRNA) targeting
human SHP2 were annealed and inserted into the pFIV-GFP-PURO vector
to produce the lentiviral expression vector pFIV-GFP-PURO-shSHP2.
The integrity was confirmed on sequencing. An empty pFIV-GFP-PURO
vector was used as a negative control.
The SHP2 overexpression vector or SHP2 shRNA vector
and empty control vectors, together with the corresponding packaged
plasmids were transfected into the HEK293T cells to generate
lentiviral particles carrying target sequences. Virus supernatants
were collected 72 h post-transfection.
The H446 cells were infected with lentiviral
particles containing the SHP2 overexpression sequence to produce
stable H446-SHP2-OE and H446-control vector target cell lines after
puromycin selection (2 µg/ml). The H446/CDDP cells were infected
with lentiviral particles containing the SHP2 shRNA sequence to
produce stable H446/CDDP-SHP2-shRNA and H446/CDDP-mock target cell
lines after puromycin selection (2 µg/ml). The efficiency of
overexpression or knockdown of SHP2 in the H446-SHP2-OE and the
H446/CDDP-SHP2-shRNA cells respectively, was assessed by both
RT-PCR and western blot analysis.
Ras siRNA transfection
Three eppendorf tubes (A, B and C) were prepared
with 500 µl of RPMI-1640 medium in each. H-Ras siRNA (24 µl) was
added in tube A and 12 µl of Lipofectamine 2000 reagent was added
in tubes B and C, at room temperature (RT) for 5 min. Tubes A and B
were mixed and cultured at RT for 20 min. A 500 µl mixture of A and
B was added to cells in two culture bottles with 60% confluency and
1.5 ml fresh medium. A 500 µl of solution in C tube was added to
another bottle with cells and incubated for 8 h after which 5 ml of
complete growth medium without antibiotics was added. The
transfection mixture was removed and incubated for an additional 40
h. The efficiency of Ras RNA interference was assessed by both gel
based RT-PCR and western blot analysis.
Cell viability assay
Cell viability was detected using the Cell Counting
Kit-8 (CCK-8) according to the manufacturer's instructions. The
IC50 of cisplatin was calculated.
Flow cytometric analysis
An FITC Annexin V and PI apoptosis detection kit was
used to evaluate apoptosis, according to the manufacturer's
protocol.
Western blot analysis
Equal quantities of total protein were run on
SDS-PAGE and transferred onto PVDF membranes, blocked with 5%
nonfat milk, incubated with primary antibodies, incubated with
peroxidase-conjugated secondary antibodies and developed with
enhanced chemiluminescent detection reagent.
RNA extraction and gel based real-time
PCR analysis
Total RNA was isolated and cDNA was synthesized
routinely. SYBR Green I was used for RT-PCR analysis. The RT-PCR
products were run on the agarose gel and the quantitative gel
images were obtained.
Statistical analysis
Results are presented as the mean ± standard
deviation (SD). Statistical analysis was performed using SPSS 18.0
software (SPSS Inc., Chicago, IL, USA). Between-group differences
were assessed using the Student's t-test and one-way analysis of
variance (ANOVA) was used for multi-group comparisons. P<0.05
was considered to indicate a statistically significant
difference.
Results
Expression and enzyme activity of SHP2
in cisplatin-resistant human lung cancer cells
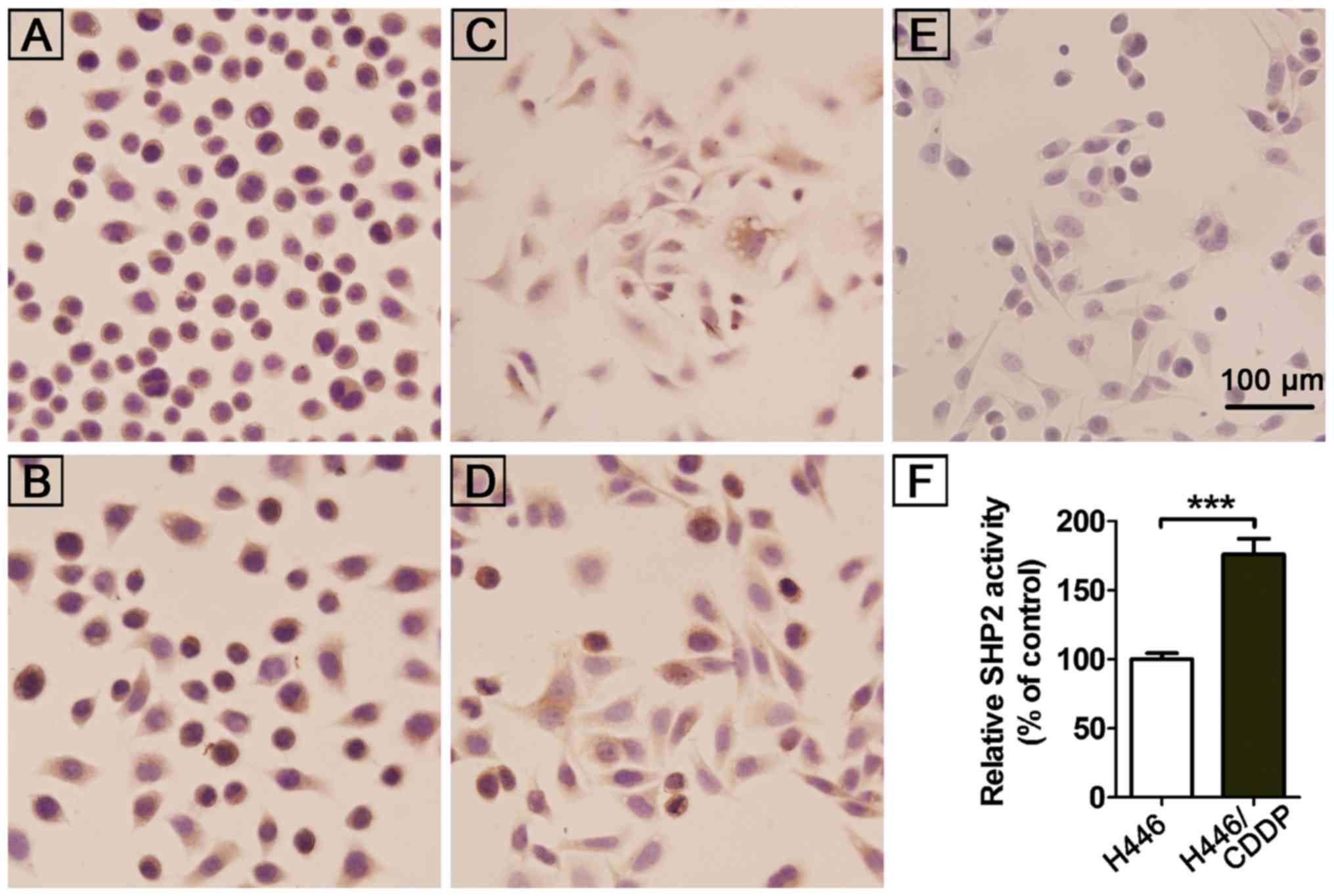
Our previous study found a significantly higher
expression of SHP2 in NSCLC by tissue microarray and also in NSCLC
patients with lymph node metastasis vs. patients without lymph node
metastasis (29). The results were
consistent with previous in vivo observations by
immunohistochemical detection of SHP2 expression in
cisplatin-induced drug resistant cells (H446/CDDP, SPC-A-1/CDDP)
and their parental cell lines (H446, SPC-A-1). A high expression of
SHP2 (+++) was observed in the cytoplasm of all tested cells
(Fig. 1A-E). There was no
significant difference in the SHP2 expression between the
cisplatin-resistant and the parental cells. However, further
investigation revealed a significantly higher activity of SHP2 in
cisplatin-resistant H446/CDDP cells vs. H446/CDDP cells (Fig. 1F). This indicates an involvement of
SHP2 in cisplatin resistance of lung cancer cells.
Validation of constructed recombinant
human lung cancer cells
To further investigate whether SHP2 is required for
the development of cisplatin resistance in lung cancer cells, we
separately induced overexpression and knockdown of the SHP2
expression in H446 and H446/CDDP cells. As illustrated in Fig. 2A-C, SHP2 expression in the
H446-SHP2-OE cells was significantly higher than that in the H446
and H446-control vector cells. In addition, SHP2 expression in the
H446/CDDP-SHP2-shRNA cells was significantly lower than that in the
H446/CDDP and H446/CDDP-mock cells. These results confirmed the
successful generation of transgenic cells with the overexpression
or knockdown of SHP2.
SHP2 protects lung cancer cells
exposed to cisplatin
To investigate the possible involvement of SHP2 in
cisplatin-induced drug resistance in lung cancer patients, cells
challenged with cisplatin were subjected to CCK-8 assay. The
results revealed no significant difference in the relative cell
viability of the H446, H446-control vector and the H446-SHP2-OE
cells without cisplatin (Fig. 3A),
which indicated that the inserted vectors had no influence on the
H446 cell viability. However, the relative cell viability of the
three cell lines was significantly inhibited with cisplatin (1
µg/ml) vs. the same type of cells without cisplatin (p<0.05). In
addition, the relative cell viability of the H446-SHP2-OE cells was
significantly increased than that of the other two cell lines
(p<0.05). This indicated that upregulation of SHP2 significantly
decreased the sensitivity of the H446 cells to cisplatin.
The influence of SHP2 knockdown on the sensitivity
of human lung cancer cells to cisplatin was further explored. The
H446/CDDP, H446/CDDP-mock and H446/CDDP-SHP2-shRNA cells exhibited
similar relative cell viability following treatment with cisplatin
(1 µg/ml) (Fig. 3B), which excluded
a possible influence of the inserted vector on cell viability.
However, the relative cell viability of the three cell lines was
significantly lower following treatment with cisplatin (1 µg/ml)
vs. that of the controls without cisplatin (p<0.05). The
relative cell viability of the H446/CDDP-SHP2-shRNA cells following
treatment with cisplatin was significantly lower vs. that of the
other two cell lines (p<0.05). This indicated that sensitivity
of the H446/CDDP cells to cisplatin was increased by downregulating
SHP2.
To quantitatively assess the influence of SHP2 on
lung cancer cell sensitivity to cisplatin, the IC50 for
each cell line was calculated. The H446-control vector and the
H446-SHP2-OE cells were treated with cisplatin (0.05, 0.25, 1.25, 5
and 10 µg/ml), and the H446/CDDP-mock and H446/CDDP-SHP2-shRNA
cells were treated with cisplatin (5, 10, 20, 40 and 80 µg/ml) for
24 h. The IC50 of the H446-control vector, the
H446-SHP2-OE, the H446/CDDP-mock and the H446/CDDP-SHP2-shRNA cells
to cisplatin was 1.01, 1.218, 11.92 and 4.382 µg/ml, respectively.
Collectively, these results confirmed the enhanced tolerance of
H446 cells to cisplatin after upregulating SHP2, while
downregulating SHP2 increased the sensitivity of H446 cells to
cisplatin.
SHP2 induces drug resistance via
inhibition of cisplatin-induced apoptosis in lung cancer cells
To investigate whether SHP2-induced chemoresistance
was mediated by anti-apoptosis, flow cytometric analysis was
performed to assess apoptosis of the H446-control vector, the
H446-SHP2-OE, H446/CDDP-mock and the H446/CDDP-SHP2-shRNA cells in
response to cisplatin (1.0 µg/ml).
The results revealed that SHP2 overexpression
significantly decreased lung cancer cell apoptosis and apoptosis
due to cisplatin was significantly lower in the H446-SHP2-OE vs.
the H446-control vector cells (average of 35.15 vs. 46.64%;
Fig. 4A and B). In contrast, SHP2
knockdown significantly increased apoptosis. The apoptosis rate due
to cisplatin was significantly higher in the H446/CDDP-SHP2-shRNA
vs. the H446/CDDP-mock cells (average of 17.35 vs. 6.73%; Fig. 4C and D). These observations
indicated that upregulation of SHP2 protected lung cancer cells
from apoptosis and enhanced their resistance to cisplatin.
SHP2 Ras/PI3K/Akt/survivin pathway is
critical to chemotherapeutic outcomes in lung cancer
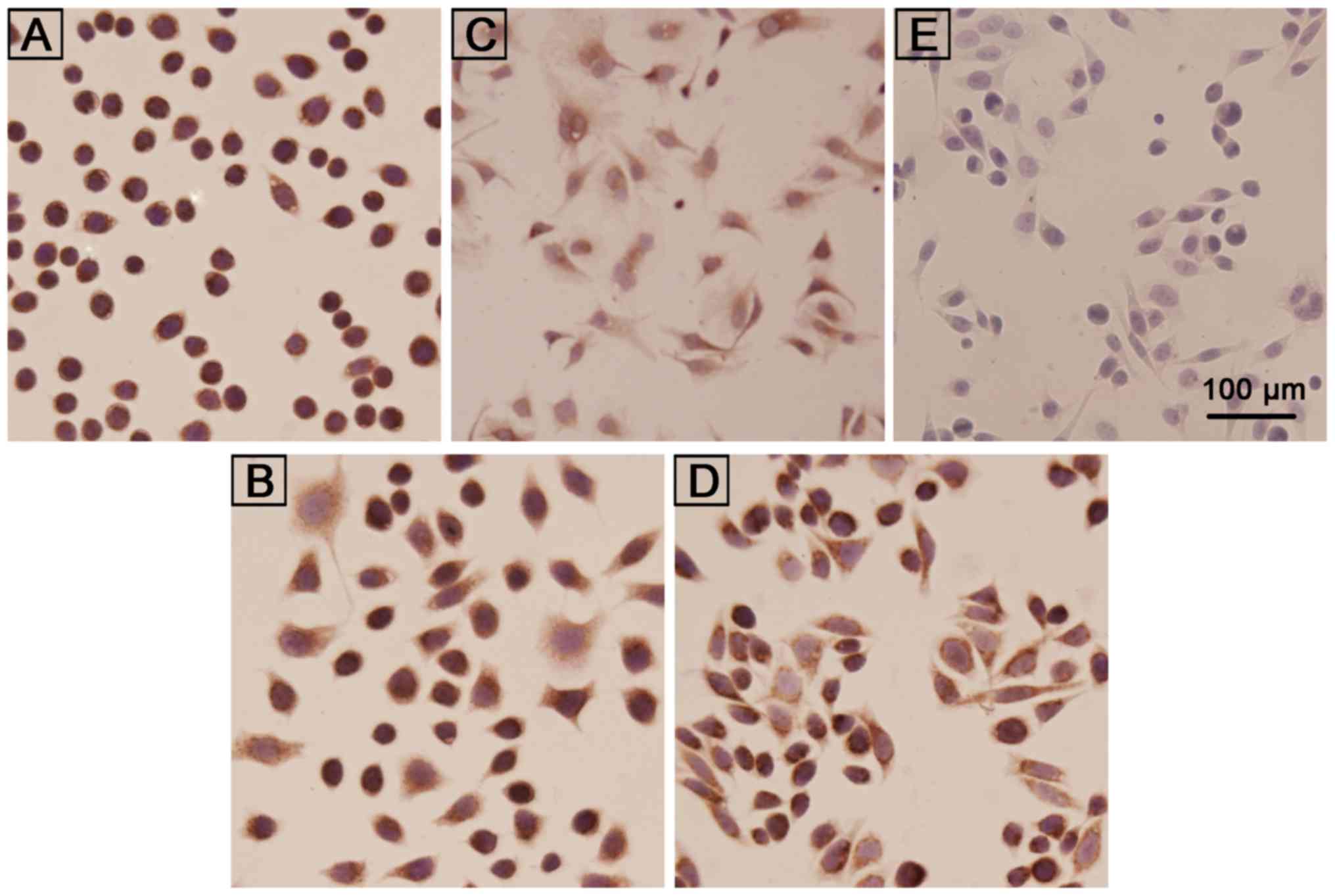
To investigate whether the Ras-activated PI3K/Akt
pathway, downstream of MAPK, plays a critical role in inhibiting
apoptosis and is involved in SHP2-induced lung cancer cell
resistance to cisplatin, the Ras expression in lung cancer cells
was assessed by immunohistochemistry. A high Ras expression (+++)
was observed in both SPC-A-1 and cisplatin-induced drug resistant
SPC-A-1/CDDP cells (Fig. 5A and B).
The expression of Ras in cisplatin-induced drug resistant H446/CDDP
cells (+++) was greater than that in the H446 cells (++) (Fig. 5C and D).
To gain an insight into the molecular mechanisms by
which Ras further promoted lung cancer cell resistance to
cisplatin, H446, H446/CDDP, H446/CDDP-mock and H446/CDDP-SHP2-shRNA
cells were treated with cisplatin (1.0 µg/ml) for 24 h. The changes
in expression of SHP2, Ras, pAkt1, Akt1 and survivin were assessed
by western blot analysis. The expression of Ras, pAkt1, Akt1 and
survivin in the H446/CDDP cells was significantly higher vs. that
in the H446 cells without cisplatin resistance, which indicated
that these proteins are closely associated with cisplatin
resistance (Fig. 6A and C).
Furthermore, the expression of Ras, Akt1, pAkt1 and survivin in
H446/CDDP-SHP2-shRNA cells was lower than that in the
H446/CDDP-mock cells, which is consistent with the SHP2 expression.
This indicated that SHP2 may be an upstream regulatory factor of
Ras, Akt1 and survivin, which may directly upregulate the PI3K/Akt1
pathway. The expression of pAkt1 and survivin was also increased
following exposure of these cells to cisplatin. The SHP2 expression
was not inhibited in the H446, H446/CDDP and H446/CDDP-mock cells,
which confirmed the involvement of these two factors and the
anti-apoptotic effect. However, no significant differences were
observed in the expression of these two proteins, before or after
cisplatin exposure, in the H446/CDDP-SHP2-shRNA cells after the
SHP2 knockdown. This indicates that cisplatin activated SHP2 which
in turn downregulated apoptosis via its effect on the
SHP2/Ras/PI3K/Akt1/survivin pathway. This further confirmed that
SHP2 participated in pAkt1- and survivin-induced cisplatin
resistance in lung cancer cells.
 | Figure 6.Ras-mediated PI3K/Akt pathway is
activated by SHP2 in cisplatin-resistant lung cancer cells. (A and
C) The expression of SHP2, Ras, Akt1, pAkt1 and survivin in the
H446/CDDP, H446/CDDP-mock and H446/CDDP-SHP2-shRNA cells treated
with cisplatin (1.0 µg/ml) for 24 h assessed by western blot
analysis. In (C) the bars in the histogram represent, from left to
right, the following cells: H446, H446 + CDDP, H446/CDDP, H446/CDDP
+ CDDP, H446/CDDP-mock, H446/CDDP-mock + CDDP, H446/CDDP-SHP2-shRNA
and H446/CDDP-SHP2-shRNA+ CDDP cells. (B and D) The expression of
SHP2, Ras, Akt1, pAkt1 and survivin in H446, H446-SHP2-OE and
H446/CDDP cells before and after Ras RNA interference assessed by
western blot analysis; In (B) the bars in the histogram represent,
from left to right, the following cells: H446, H446-Ras siRNA,
H446-SHP2-OE, H446-SHP2-OE-Ras siRNA, H446/CDDP and H446/CDDP-Ras
siRNA cells. (C) shows quantification of the relative expression
level of each test band in (A). (D) shows quantification of the
relative expression level of each test band in (B). *p<0.05,
**p<0.01, ***p<0.001. CDDP, cisplatin; SHP2, Src homology
phosphotyrosyl phosphatase 2; OE, overexpression. |
Upon Ras RNA silencing with cisplatin, the
expression of Ras was significantly decreased in the H446,
H446-SHP2-OE and the H446/CDDP cells. However, the SHP2 expression
was not significantly changed following Ras RNA interference
(Fig. 6B and D). The expression of
Akt1, pAkt1 and survivin was significantly increased in the
H446-SHP2-OE and H446/CDDP cells after the Ras RNA interference.
These results revealed that Ras was not the only mediator of
SHP2-induced apoptosis inhibition, via the PI3K/Akt1 pathway, in
inducing cisplatin resistance. There may be other compensatory ways
to increase the expression of Akt1 and pAkt1. Collectively, we
demonstrated that SHP2 was a direct activator of Ras. Inhibiting
the Ras expression regulated the PI3K/Akt pathway. Therefore, the
SHP2/Ras/PI3K/Akt/survivin pathway could be a potential therapeutic
target for cisplatin-related drug resistance in lung cancer
patients.
Discussion
Lung cancer is a highly malignant neoplasm. About
75% of all patients are diagnosed at an advanced stage and have
lost the opportunity for surgery, thus chemotherapy is often the
only viable option. Chemotherapy can considerably increase
survival, alleviate symptoms and improve the quality of life of
lung cancer patients. However, the development of chemoresistance
is a major cause of chemotherapy failure. Thus, combination therapy
directed to an appropriate target may improve therapeutic efficacy
(30).
We previously reported a positive expression of SHP2
in NSCLC tissues, but not in adjacent and normal lung tissues,
which indicated the importance of SHP2 in NSCLC (31). Indeed, SHP2 is considered as a
potential marker and therapeutic target for NSCLC and SHP2
inhibition may play a role in combination therapeutics. However,
the involvement of SHP2 in lung cancer resistance has not been
reported in the literature.
In the present study, cisplatin-induced
drug-resistant cells SPC-A-1/CDDP and H446/CDDP were generated from
SPC-A-1 and H446 cells, respectively (30). A high expression of SHP2 was
observed in these cells. We found significantly higher Ras
expression in the H446/CDDP cells as compared to that in the H446
cells, while no significant difference was observed between the
SPC-A-1/CDDP and the SPC-A-1 cells. This may be due to the fact
that lung adenocarcinoma typically exhibits primary resistance,
while SCLC exhibits acquired resistance to cisplatin treatment. Our
results support the hypothesis that SHP2 is associated with
cisplatin-induced drug resistance in lung cancer.
Drug sensitivity tests revealed that overexpression
of SHP2 reduced the sensitivity of the H446 cells to cisplatin
(IC50: 1.218 vs. 1.01 µg/ml) and inhibition of SHP2
significantly reversed the tolerance capacity of the H446/CDDP
cells to cisplatin (IC50: 4.382 vs. 11.92 µg/ml). Our
findings indicated that SHP2 is a cisplatin resistance-associated
protein in lung cancer. However, no significant differences were
observed in SHP2 mRNA and the protein expression between H446 and
H446/CDDP cells.
We further found significantly higher activity of
SHP2 in H446/CDDP cells vs. H446 cells (OD value: 0.488±0.015 vs.
0.249±0.011, P<0.05), which indicated that enzymatic activity of
SHP2 is involved in acquired resistance of H446 cells to
cisplatin.
Overexpression or knockdown of SHP2 had no
significant influence on the growth of H446 cells or H446/CDDP
cells without cisplatin. However, cell proliferation was
significantly inhibited with cisplatin. The lowest extent of
inhibition was in cells with SHP2 overexpression while the greatest
was in SHP2-knockdown cells. These results strongly indicated that
increased SHP2 expression enhanced the tolerance of lung cancer
cells to cisplatin. Inhibiting SHP2 reversed this tolerance and
increased the sensitivity of lung cancer cells to cisplatin.
Inhibition of apoptosis is a mechanism of cisplatin
resistance (14). After cisplatin
treatment, we found that overexpression of SHP2 reduced apoptosis
of the H446 cells (from 46.64 to 35.15%), while inhibition of SHP2
increased apoptosis of the H446/CDDP cells (from 6.73 to 17.35%).
The expression of Ras, pAkt1, Akt1 and survivin was also
significantly increased in the H446/CDDP cells vs. the H446 cells
and decreased in the H446/CDDP-SHP2-shRNA cells vs. the
H446/CDDP-mock cells, which was consistent with SHP2 expression.
These results confirmed the contribution of SHP2 in the acquisition
of cisplatin-induced drug resistance, which is mediated via the
regulation of cell apoptosis which may be mediated via the PI3K/Akt
pathway, an important anti-apoptotic pathway.
SHP2 is involved in cell growth, migration, invasion
and transformation via several signaling pathways. Among these, the
Ras/ERK and the PI3K/Akt pathways are considered to be closely
associated with oncogenesis and tumor development (16). The present study revealed that
inhibition of SHP2 was associated with reduced expression of Ras,
Akt1, pAkt1 and survivin. However, no significant difference in
SHP2 expression was found after silencing Ras in H446, H446-SHP2-OE
and H446/CDDP cells exposed to cisplatin. These findings indicated
that SHP2 is the upstream of Ras, Akt1 and survivin. SHP2 may be
involved in drug resistance induced by cisplatin in lung cancer
cells via the regulation of the Ras/PI3K/Akt1/survivin pathway.
Interestingly, the expression of Akt1 and pAkt1 was decreased in
the H446 cells, but increased in the H446-SHP2-OE and H446/CDDP
cells after the Ras inhibition. We assumed that the SHP2 activation
may increase the expression of both Akt1 and pAkt1 via other
compensatory pathways independent of Ras, such as the JAK/STAT
signaling pathway.
In conclusion, our findings revealed that SHP2 is
associated with cisplatin-induced drug resistance in lung cancer
and SHP2 directly activates Ras, which in turn regulates the
PI3K/Akt pathway. Only in vitro experiments were conducted
in the present study, whereas further in vivo studies are
currently ongoing by our research group.
Acknowledgements
This study was supported by grants from the National
Nature Science Foundation (no. 81071913).
References
|
1
|
Torre LA, Siegel RL and Jemal A: Lung
cancer statistics. Adv Exp Med Biol. 893:1–19. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rivera MP: Lung cancer in women:
Differences in epidemiology, biology, histology, and treatment
outcomes. Semin Respir Crit Care Med. 34:792–801. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mohan V, Agarwal R and Singh RP: A novel
alkaloid, evodiamine causes nuclear localization of cytochrome-c
and induces apoptosis independent of p53 in human lung cancer
cells. Biochem Biophys Res Commun. 477:1065–1071. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
D'Amico TA: Angiogenesis in non-small cell
lung cancer. Semin Thorac Cardiovasc Surg. 16:13–18. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Müller-Tidow C, Diederichs S, Thomas M and
Serve H: Genome-wide screening for prognosis-predicting genes in
early-stage non-small-cell lung cancer. Lung Cancer. 45:S145–S150.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rich JN and Bao S: Chemotherapy and cancer
stem cells. Cell Stem Cell. 1:353–355. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dittrich PS and Manz A: Lab-on-a-chip:
Microfluidics in drug discovery. Nat Rev Drug Discov. 5:210–218.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sampsonas F, Ryan D, McPhillips D and
Breen DP: Molecular testing and personalized treatment of lung
cancer. Curr Mol Pharmacol. 7:22–32. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dimou A and Papadimitrakopoulou V:
Non-small cell lung cancer beyond biomarkers: The evolving
landscape of clinical trial design. J Pers Med. 4:386–401. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pass HI, Lavilla C, Canino C, Goparaju C,
Preiss J, Noreen S, Blandino G and Cioce M: Inhibition of the
colony-stimulating-factor-1 receptor affects the resistance of lung
cancer cells to cisplatin. Oncotarget. 7:56408–56421. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gower A, Wang Y and Giaccone G: Oncogenic
drivers, targeted therapies, and acquired resistance in
non-small-cell lung cancer. J Mol Med (Berl). 92:697–707. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kachalaki S, Ebrahimi M, Mohamed
Khosroshahi L, Mohammadinejad S and Baradaran B: Cancer
chemoresistance; biochemical and molecular aspects: a brief
overview. Eur J Pharm Sci. 89:20–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen WX, Liu XM, Lv MM, Chen L, Zhao JH,
Zhong SL, Ji MH, Hu Q, Luo Z, Wu JZ, et al: Exosomes from
drug-resistant breast cancer cells transmit chemoresistance by a
horizontal transfer of microRNAs. PLoS One. 9:e952402014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Holohan C, Van Schaeybroeck S, Longley DB
and Johnston PG: Cancer drug resistance: An evolving paradigm. Nat
Rev Cancer. 13:714–726. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ardizzoni A, Boni L, Tiseo M, Fossella FV,
Schiller JH, Paesmans M, Radosavljevic D, Paccagnella A, Zatloukal
P, Mazzanti P, et al CISCA (Cisplatin vs. Carboplatin)
Meta-analysis Group, : Cisplatin- vs. carboplatin-based
chemotherapy in first-line treatment of advanced non-small-cell
lung cancer: An individual patient data meta-analysis. J Natl
Cancer Inst. 99:847–857. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Torigoe T, Izumi H, Ishiguchi H, Yoshida
Y, Tanabe M, Yoshida T, Igarashi T, Niina I, Wakasugi T, Imaizumi
T, et al: Cisplatin resistance and transcription factors. Curr Med
Chem Anticancer Agents. 5:15–27. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mo EP, Zhang RR, Xu J, Zhang H, Wang XX,
Tan QT, Liu FL, Jiang RW and Cai SH: Calotropin from Asclepias
curasavica induces cell cycle arrest and apoptosis in
cisplatin-resistant lung cancer cells. Biochem Biophys Res Commun.
478:710–715. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Florea AM and Büsselberg D: Cisplatin as
an anti-tumor drug: Cellular mechanisms of activity, drug
resistance and induced side effects. Cancers (Basel). 3:1351–1371.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xie Y, Ma X, Gu L, Li H, Chen L, Li X, Gao
Y, Fan Y, Zhang Y, Yao Y, et al: Prognostic and clinicopathological
significance of survivin expression in renal cell carcinoma: A
systematic review and meta-analysis. Sci Rep. 6:297942016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zeng LF, Zhang RY, Yu ZH, Li S, Wu L,
Gunawan AM, Lane BS, Mali RS, Li X, Chan RJ, et al: Therapeutic
potential of targeting the oncogenic SHP2 phosphatase. J Med Chem.
57:6594–6609. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ren Y, Chen Z, Chen L, Fang B, Win-Piazza
H, Haura E, Koomen JM and Wu J: Critical role of Shp2 in tumor
growth involving regulation of c-Myc. Genes Cancer. 1:994–1007.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chan G, Kalaitzidis D and Neel BG: The
tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis
Rev. 27:179–192. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang X, Dutta U and Shaw LM: SHP2 mediates
the localized activation of Fyn downstream of the α6β4 integrin to
promote carcinoma invasion. Mol Cell Biol. 30:5306–5317. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cheng SQ, Fan HY, Xu X, Gao WW, Lv SG, Ye
MH, Wu MJ, Shen XL, Cheng ZJ, Zhu XG and Zhang Y: Over-expression
of LRIG1 suppresses biological function of pituitary adenoma via
attenuation of PI3K/AKT and Ras/Raf/ERK pathways in vivo and in
vitro. JJ Huazhong Univ Sci Technolog Med Sci. 36:558–563. 2016.
View Article : Google Scholar
|
|
25
|
Malumbres M and Barbacid M: RAS oncogenes:
The first 30 years. Nat Rev Cancer. 3:459–465. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Giltnane JM and Balko JM: Rationale for
targeting the Ras/MAPK pathway in triple-negative breast cancer.
Discov Med. 17:275–283. 2014.PubMed/NCBI
|
|
27
|
Ksionda O, Melton AA, Bache J, Tenhagen M,
Bakker J, Harvey R, Winter SS, Rubio I and Roose JP: RasGRP1
overexpression in T-ALL increases basal nucleotide exchange on Ras
rendering the Ras/PI3K/Akt pathway responsive to protumorigenic
cytokines. Oncogene. 35:3658–3668. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhou XD and Agazie YM: Inhibition of SHP2
leads to mesenchymal to epithelial transition in breast cancer
cells. Cell Death Differ. 15:988–996. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tang C, Luo D, Yang H, Wang Q, Zhang R,
Liu G and Zhou X: Expression of SHP2 and related markers in
non-small cell lung cancer: a tissue microarray study of 80 cases.
Appl Immunohistochem Mol Morphol. 21:386–394. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li S, Shi H, Ji F, Wang B, Feng Q, Feng X,
Jia Z, Zhao Q and Qian G: The human lung cancer drug
resistance-related gene BC006151 regulates chemosensitivity in
H446/CDDP cells. Biol Pharm Bull. 33:1285–1290. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tang C, Zhou X, Yang H, Wang Q and Zhang
R: Expression and its clinical significance of SHP2 in non-small
cell lung cancer. Zhongguo Fei Ai Za Zhi. 13:98–101. 2010.(in
Chinese). PubMed/NCBI
|