Introduction
Hepatocarcinoma (HCC) is one of the most common
malignant tumors in China (1).
Surgical resection and liver transplantation are potential curative
therapies (2). Unfortunately, most
HCCs are too advanced at the time of diagnosis to benefit from
these surgical approaches. Currently, chemotherapy is ineffective
for HCC due to the inherent chemoresistance. However, the exact
mechanism of chemotherapy resistance in HCC is largely unknown.
Recently, accumulated evidence suggests that autophagy can promote
cancer resistance to chemotherapeutic agents (3–6).
Autophagy was first identified as a cellular defense response to
nutrient deprivation and is essential to maintaining cell survival
during nutrient starvation (7,8). It
has been reported that due to the insufficient blood supply
following poor vasculature, tumors are able to obtain a limited
supply of oxygen and nutrients during their progression (9). Moreover, certain treatments for HCC,
such as transarterial chemoembolization (TACE) (10) also result in a nutrient-deficient
microenvironment. Thus, it is crucial to explore whether nutrient
deprivation-induced autopahgy may contribute to the chemoresistance
of HCC.
Autophagy is an evolutionarily conserved
physiological process, and can be activated under various stimuli
such as starvation and hypoxia (11). The autophagic pathway begins with a
lipid bilayer structure called the isolation membrane. The
isolation membrane sequesters cytoplasmic materials, such as
organelles, to form autophagosomes. Subsequently, the autophagosome
fuses with the lysosome and matures into autolysosomes.
Sequestrated materials are digested to amino acids in the
autolysosomes by lysosomal enzymes (12–14).
Several pharmacologic autophagy inhibitors have been used to
evaluate the physiologic relevance of autophagy in culture cells.
For example, 3-methyladenine (3-MA) blocks autophagosome formation
to inhibit autophagy (15).
The role of autophagy in cancer is still
controversial. For example, prolonged autophagy has been suggested
to cause autophagic cell death, which is termed as type II
programmed cell death (16,17). In contrast, autophagy allows cancer
cells to survive in response to adverse conditions such as nutrient
deprivation by providing amino acid and other intermediates.
Inhibition of autophagy increases nutrient starvation-induced cell
death (18). Although, autophagy is
essential for helping cells against a shortage of nutrients, the
role of autophagy in chemotherapy during nutrient deprivation has
been rarely explored.
In the present study, we report that HCC cells
exhibited chemoresistance accompanied by activation of autophagy
during nutrient deprivation. Inhibition of autophagy increased
nutrient deprivation-induced apoptosis. Decreased mitochondrial
mass was detected when cells underwent autophagy. Furthermore,
inhibition of autophagy enhanced the chemosensitivity of HCC cells
to chemotherapeutic agents.
Materials and methods
Cell culture
Human hepatocarcinoma cell lines SMMC-7721, Hep3B
and HepG2 [obtained from the Tumor Immunology and Gene Therapy
Center of the Eastern Hepatobiliary Surgery Hospital (Shanghai,
China)] were maintained in Dulbecco's modified Eagle's medium (high
glucose) (Gibco, Invitrogen, Carlsbad, CA, USA) and supplemented
with 10% fetal bovine serum, 100 U/ml penicillin and 100 mg/ml
streptomycin in a humidified incubator under 5% CO2 at
37°C. Nutrient deprivation was carried out in Earles balanced salt
solution (EBSS) medium (Sigma-Aldrich, St. Louis, MO, USA).
Regents
Cisplatin and 5-fluorouracil (5-FU) were purchased
from Qilu Pharmaceutical Co., Ltd. (Jinan, Shandong, China). 3-MA
was obtained from Sigma-Aldrich (Shanghai, China) and dissolved in
sterile double distilled water.
Cell viability assay
The measurement of viable cell mass was performed
with a Cell Counting Kit (Cell Counting Kit-8, Dojin Laboratories,
Kumamoto, Japan) to count living cells using WST-8. Cells were
seeded (0.6×104 cells/well) on a 96-well plate and 24 h
later, the cells were treated with nutrient-containing and
nutrient-deprived medium with or without the indicated
chemotherapeutic agents; in some experiment, 3-MA (10 mM) was
added. For quantitative analysis of cell viability, 10 µl of cell
counting kit solution was added to each well. After incubation at
37°C for 2 h in a humidified CO2 incubator, absorbance
at 450 nm was monitored with a microplate reader (Synergy HT;
BioTek Instruments, Inc., Winooski, VT, USA). The values obtained
were normalized to those of the control cells incubated with
vehicle only.
4,6-Diamidino-2-phenylindole (DAPI)
staining
Cells were seeded into a 96-well plate and 24 h
later cells were subjected with nutrient-containing and
nutrient-deprived medium with or without the indicated
chemotherapeutic agents; in some experiment, 3-MA was added. For
DAPI staining, the cells were fixed with 4% paraformaldehyde for 10
min at room temperature. After being washed with 1X PBS for 2
times, the cells were then treated with 0.1% Triton X-100 for 5 min
prior to staining with 1 µg/ml DAPI. Cells were visualized by
fluorescence microscopy (Olympus IX71; Olympus, Tokyo, Japan).
Cells in which the nuclei contained clearly condensed chromatin or
cells exhibiting fragmented nuclei were scored as apoptotic.
Apoptotic data are reported as percentage of apoptosis, obtained by
determining the numbers of apoptotic cells vs. the total number of
cells. For each sample, a minimum of 5 counts involving a minimum
of 100–200 cells/count were scored.
Annexin V/PI staining
SMMC-7721, Hep3B and HepG2 cells were allowed to
reach 70–80% confluency, and then were subjected to
nutrient-containing and nutrient-deprived medium with or without
the indicated chemotherapeutic agents; in some experiment, 3-MA was
added. Cells (1×106) were collected by trypsinization at
the times indicated. All cells were washed with ice-cold
phosphate-buffered saline (PBS) twice and resuspended in 300 µl 1X
binding buffer containing 5 µl Annexin V and 5 µl PI for 30 min at
room temperature in the dark (Annexin V-FITC apoptosis detection
kit; Nanjing Keygen Biotech, China). Cell survival was measured by
flow cytometric analysis using a FACSAria flow cytometer
(Becton-Dickinson, Franklin Lakes, NJ, USA). Annexin V and PI
single-positive, and double-positive populations were collectively
counted as ‘dead’, whereas double-negative cells were considered
viable.
Transient transfection
SMMC-7721, Hep3B and HepG2 cells were seeded
(1×104 cells/well) into 96-well plates overnight, and
then GFP-LC3-expressing plasmids were transiently transfected into
the cells using FuGENE HD transfection reagent (Roche, Mannheim,
Germany) according to the manufacturer's instructions. Cultured for
24 h to ensure the expression of GFP-LC3, the cells were subjected
to EBSS medium in the absence or presence of 3-MA. At the end of
the treatment, autophagy was detected by counting the percentage of
cells with GFP-LC3-positive dots under a fluorescence microscope
(Olympus IX71). A minimum of 200 cells/sample were counted in
triplicate for each experiment.
Transmission electron microscopy
Cells were fixed with 2.5% glutaraldehyde in
phosphate buffer, and stored at 4°C until embedding. Cells were
post-fixed with 1% osmium tetroxide followed by an increasing
gradient dehydration step using ethanol and acetone. Cells were
then embedded in araldite, and ultrathin section were obtained
(50–60 nm), placed on uncoated copper grids, and stained with 3%
lead citrate-uranyl acetate. Images were examined with a CM-120
electron microscope (Philips, Andover, MA, USA).
siRNA
The Stealth™ RNAi negative control duplex (cat.
12935-200) and Stealth™ RNAi siRNA duplex oligoribonucleotides
targeting human Beclin 1 (cat. 1299003) were obtained from
Invitrogen. The siRNA was transfected into SMMC-7721 cells using
siRNA transfection reagent (cat. sc-29528; Santa Cruz
Biotechnology, Santa Cruz, CA, USA) according to the manufacturer's
protocol.
Western blot analysis
At the end of the designated treatments, cells were
lysed in RIPA lysis buffer (Beyotime Biotechnology, Shanghai,
China) with 1 mM phenylmethanesulfonyl fluoride (PMSF). Equal
amounts of protein were separated by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred onto an NC membrane. After blocking with 5% non-fat
milk, the membrane was probed with anti-Beclin 1 (Novus
Biologicals, Inc., Cat. NB500-249H, Host: Rabbit, dilution
1:1,000), anti-LC3 (Novus Biologicals, Inc. Cat. NB100-2220H, Host:
Rabbit, dilution 1:1,000) and anti-PARP (Cell Signaling Technology,
Beverly, MA, USA, Cat. 9532, Isotype: Rabbit, dilution 1:1,000),
developed with the BeyoECL Plus substrate system (Beyotime, China,
Cat. P0018). Blots were stripped and re-probed with β-actin
antibody (Santa Cruz, Cat. sc-47778, Host: mouse, dilution 1:2,000)
to confirm equal protein loading.
Semi-quantitative real-time PCR
RNA was extracted from cells using TRIzol
(Invitrogen, Carlsbad, CA, USA). cDNA was synthesized using MMLV
reverse transcriptase (Promega, Madison, WI, USA), and 2 µg total
RNA and oligo(dT)18 primers. Two-microliter aliquots of
cDNA were used for PCR amplification. Real-time RT-PCR was
performed in triplicate using the SYBR PrimeScript RT-PCR kit
(Takara, Dalian, China), and primers used were as follows: sense,
5-CCATCTTT GCAGGCACACTCATC-3′, and antisense
5′-ATCCACCTCAACTGCCTGCTATG-3′ for Nd2 (19); sense, 5′-ACAGCTCGTGTAATCTACCA-3′ and
antisense, 5′-GACCGTCCATTCTTTGC-3′ for Nme1. PCR used 40 cycles of
5 sec at 95°C, 20 sec at 60°C for Nd2 and Nme1. The expression of
Nme1 served as internal control. PCR products were separated by 2%
agarose gel electrophoresis, and bands were visualized under
ultraviolet (UV) radiation after staining with ethidium bromide.
Gels were photographed and bands were analyzed by computerized
densitometry.
Mitochondrial membrane potential
detection
The Mitochondrial Membrane Potential Assay kit with
JC-1 was obtained from Beyotime Biotechnology and the procedure was
carried out according to the manufacturer's instructions,
respectively.
Statistical analysis
All of the experiments were repeated at least 3
times. The data are expressed as means ± SD. Statistical analysis
was performed using the Student's t-test (two-tailed). The
criterion for statistical significance was taken as P<0.05.
Results
Chemotherapeutic agent-induced cell
death is reduced in HCC cells during nutrient deprivation
To investigate the functional significance of
nutrient shortage in chemotherapy, we incubated 3 HCC cell lines
(HepG2, Hep3B and SMMC-7721) in EBSS medium (nutrient-deprived
medium) and assessed the chemosensitivity of these cells. HCC cells
were treated with 5-FU (100 µg/ml) or cisplatin (8 µg/ml) during
nutrient deprivation for 12 h. As shown in Fig. 1A, WTS-8 assay results revealed that
HCC cells incubated in nutrient-deprived medium exhibited
significantly reduced susceptibility to the chemotherapeutic
agents, when compared to their counterpart cells which were
cultured in nutrient-containing medium. These results were further
confirmed by flow cytometric analysis with Annexin V/PI staining.
Nutrient-deficient HCC cells were more insensitive to
chemotherapeutic agents (Fig. 1B).
Thus, these data suggest that HCC cells acquire a protective effect
against chemotherapy during nutrient deprivation.
Autophagy is activated in HCC cells
during nutrient deprivation
Nutrient deprivation is a strong inducer of
autophagy. Thus, we then investigated whether autophagy was
activated in HCC cells during nutrient deprivation. LC3, one of the
mammalian homologues of yeast ATG8, is activated and relocalizes to
intracellular vesicles during the formation of autophagosomes
(20). The LC3 pro-form is cleaved
into soluble LC3-I, then LC3-I is modified to be membrane-bound,
which is known as LC3-II, and recruited onto autophagosomes. LC3-I
to LC3-II protein processing is considered a hallmark of autophagy.
As shown in Fig. 2A, levels of
endogenous LC3-II were markedly increased in the SMMC-7721 cells
incubated in nutrient-deprived medium and were attenuated by 3-MA
treatment.
To confirm the involvement of autophagy using
additional independent assays, 3 HCC cell lines, transfected with
the GFP-LC3 plasmid, were cultured for 12 h in nutrient-deprived
medium with or without autophagy inhibitor 3-MA. The distribution
of GFP-LC3 was determined by fluorescence microscopy. As shown in
Fig. 2B and C, cells cultured in
nutrient-deprived medium exhibited a significantly higher
percentage of punctuate GFP (green dots), while nutrient-containing
cells showed primarily diffusion. Furthermore, 3-MA, which inhibits
autophagosome formation, significantly reduced the visible green
dots in the nutrient-deprived cells and redistributed GFP-LC3 to
the cytoplasm. The morphological hallmark of autophagy is the
formation of a double-layered membrane structure, termed
autophagosome. To date, transmission electron microscopy is the
gold standard to monitor the formation of autophagosomes. As shown
in Fig. 2D, increased
autophagosomes were observed in the nutrient-deprived HCC cells,
and 3-MA treatment largely reduced the amount of autophagosomes. In
conclusion, these findings suggest that autophagy was activated in
these HCC cells under nutrient-deprived condition.
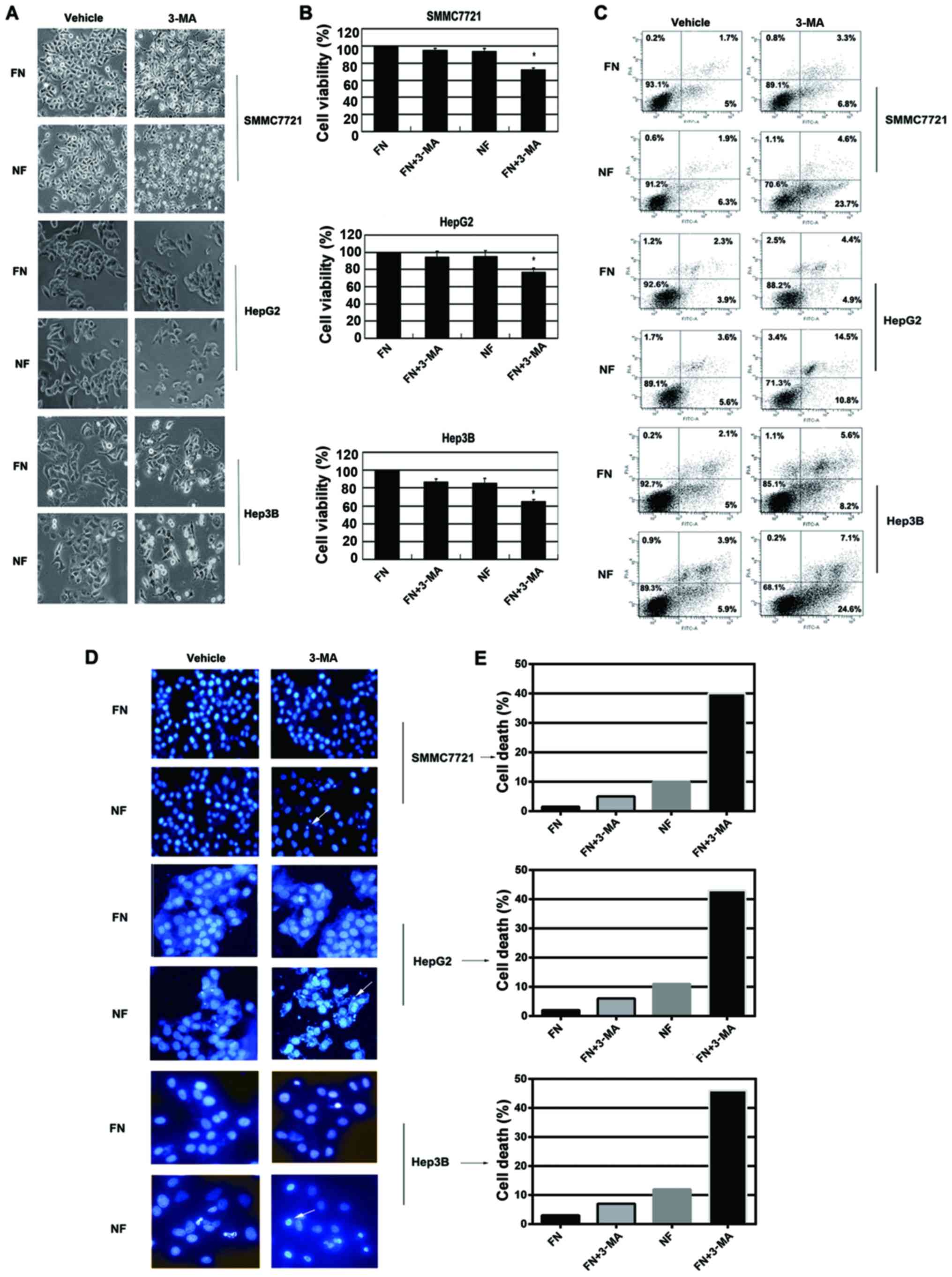
Inhibition of autophagy by 3-MA
increases chemosensitivity of HCC cells during nutrient
deprivation
Autophagy can be triggered by nutrient deprivation
and plays a role in protecting cells from nutrient shortage.
However, the role of autophagy in chemotherapy during nutrient
starvation has rarely been explored. Therefore, we first tested
whether autophagy contributes to cell escape from nutrient
shortage-induced cell death. HCC cells were cultured for 12 h in
nutrient-deprived medium and the autophagy inhibitor (3-MA) was
applied to examine the viability of the cells. Cell morphology was
detected by inverted phase contrast microscope. Typical apoptotic
changes were observed in 3-MA-treated cells during nutrient
deprivation, including marked rounding, shrinkage and detachment
from the culture dish (Fig. 3A).
These results were confirmed by WTS-8 assay. 3-MA treatment
resulted in significantly increased cell death (Fig. 3B). Similar results were further
obtained by Annexin V/PI staining analysis; 3-MA treatment in
nutrient-deprived medium induced significantly increased the
apoptosis of cells (Fig. 3C). DAPI
staining also demonstrated that 3-MA treatment markedly induced
chromatin condensation in HCC cells during nutrient deprivation
(Fig. 3D and E). The concentration
of these inhibitors did not affect the cell viability in
nutrient-containing medium. These data suggested that inhibition of
autophagy by 3-MA resulted in increased apoptosis in HCC cells
during nutrient deprivation.
To determine whether inhibition of autophagy by 3-MA
enhanced the chemotherapeutic sensitivity of the HCC cells, Hep3B
and SMMC-7721 cells were treated with 3-MA in the presence of the
chemotherapeutic agents, 5-FU or cisplatin during nutrient
deprivation. As shown in Fig. 4A, a
significantly increased amount of cell death was observed following
the combination treatment of 3-MA and 5-FU or cisplatin by
observation of cell morphology. WTS-8 assay also showed that the
combination treatment (3-MA and 5-FU or cisplatin) exhibited a much
greater extent of cell death (Fig.
4B). Similar effects were further confirmed by Annexin V/PI
staining analysis; co-treatment of 3-MA and 5-FU or cisplatin
induced significantly more robust cell death under the same
conditions (Fig. 4C). DAPI staining
also demonstrated that 3-MA treatment markedly induced chromatin
condensation in HCC cells following treatment with the
chemotherapeutic agents during nutrient deprivation (data not
shown). These results were further determined by immunoblotting,
where the combination treatment caused an increased level of
cleaved PARP in the SMMC-7721 cells (data not shown). JC-1 staining
also confirmed that the combination treatment (3-MA and 5-FU or
cisplatin) caused a significant loss of mitochondrial membrane
potential (data not shown). These results suggested that autophagy
contributes to chemotherapy insensitivity in HCC cells during
nutrient deprivation.
Knockdown of Beclin 1 by specific
siRNA enhances chemosensitivity of HCC cells during nutrient
deprivation
To confirm the role of the autophagic machinery in
nutrient deprivation-induced cell apoptosis, we used siRNA to
inhibit the Beclin 1 gene, essential to autophagosome generation
(21,22). Silencing of Beclin 1 (Fig. 5A) significantly increased nutrient
deprivation-induced cell death (Fig. 5B
and C). DAPI staining revealed that silencing of Beclin 1
induced cell apoptosis in the HCC cells (Fig. 5D and E). Together, those data
strongly suggest that HCC cells respond to nutrient shortage by
activating autophagy to resist apoptosis.
To further confirm that inhibition of autophagy
enhances chemotherapy-induced apoptosis, HCC cells were treated
with chemotherapeutic agents for 12 h after exposure to Beclin 1
siRNA. Knockdown of Beclin 1 significantly augmented
chemotherapeutic agent-induced apoptosis (Fig. 6), suggesting that this pathway
promotes drug resistance. Together with the findings in Figs. 1 and 4, these results suggest that autophagy
contributes to chemotherapy resistance in HCC cells during nutrient
deprivation. Thus, nutrient deprivation-induced autophagy in HCC
cells is a survival mechanism which protects cells from nutrient
shortage and chemotherapy.
Effect of nutrient deprivation-induced
autophagy on mitochondria in HCC cells
Autophagy is known as the only pathway for degrading
organelles, such as mitochondria (8). Previous studies have shown that
decreased mitochondrial mass by autophagy may be accompanied by
reduced levels of apoptosis induced by pro-apoptotic agents
(23–25). To characterize the mechanism by
which autophagy promotes the survival of HCC cells during nutrient
deprivation, we tested whether enhanced autophagy during nutrient
deprivation can reduce mitochondrial mass in HCC cells. SMMC-7721
cells were cultured for 12 h in nutrient-deprived medium with or
without 3-MA. The expression level of mitochondrial gene Nd2
was used to indirectly measure mitochondria mass. Mne1 gene
was used as the internal control. As shown in Fig. 7A and B, Nd2 mRNA expression was
significantly reduced in the SMMC-7721 cells during nutrient
deprivation, and significantly restored when 3-MA was used.
Moreover, ultrastructural analyses by transmission electron
microscopy (TEM) showed that the mitochondrion is enclosed by
double membrane-bound vacuolar structures in HepG2 cells (Fig. 2D). Thus, the changes of mitochondria
may be a critical step for autophagy inhibition-induced cell
apoptosis.
Discussion
Autophagy plays an important role in maintaining
cellular homeostasis and facilitates cell survival against adverse
conditions, such as nutrient deprivation. However, the role of
nutrient deprivation-induced autophagy in chemotherapy is poorly
understood. In the present study, we showed that autophagy performs
a vital role in hepatocarcinoma (HCC) cell survival during nutrient
deprivation, not only since it protects cells from nutrient
deprivation-induced apoptosis, but also provides cellular
protection from exposure to chemotherapeutic agents. In the present
study, we found that HCC cells cultured in nutrient-deficient
medium showed more resistance to chemotherapeutic agents than their
counterparts in nutrient-containing medium. Meanwhile,
nutrient-deficient HCC cells exhibited the increased autophagy
activity, as is shown by elevated levels of endogenous LC3-II,
increased amount of characteristic green dots in the cytoplasm and
massive accumulation of autophagosomes; inhibition of autophagy by
3-MA apparently decreased formation of the green dots and the
accumulation of LC3II and autophagosomes. In summary, these data
indicated that HCC cells exhibited chemoresistance accompanied by
autophagy activation under nutrient deprivation.
Recently, several studies have suggested that
autophagy plays an important role in maintaining cell survival
during nutrient deficiency (26).
Consistent with those findings, we observed that autophagy directly
contributes to the survival of HCC cells in response to the
shortage of nutrients. Nutrient-starved HCC cells treated with an
inhibitor of autophagy (3-MA or si-Beclin 1) underwent mass cell
death. The results of cell viability assays indicated that the
increased cell death by autophagy inhibition was associated by
increased apoptosis. Thus, the survival mechanism in HCC during
nutrient deprivation is likely due to the reduced apoptotic
potential of the cells.
Our previous data showed that HCC exhibited
properties of chemoresistance accompanied by autophagy activation
under nutrient deprivation. Therefore, we tested the effect of
autophagy inhibition on chemosensitivity. We observed that
inhibition of autophagy by 3-MA significantly enhanced sensitivity
of the HCC cells to cisplatin and 5-FU. Identical data were
obtained by inhibiting autophagy with siRNA against Beclin 1. These
data implied that autophagy mediates the chemoresistance of HCC
cells under nutrient deprivation. These results can be supported by
several studies suggesting that autophagy-delayed apoptosis may
contribute to drug resistance (3,27). The
relationship between autophagy and apoptosis is quite complicated.
Under some stress condition, autophagy restrains stress-induced
apoptosis to facilitate tumor cell survival (28). Moreover, several studies have
demonstrated that autophagy may promote apoptosis (29). Meanwhile, in a certain context,
autophagy and apoptosis could be simultaneously activated by the
same stimulus without any connection (30). Taken together, our current data
suggest that induction of autophagy confers two advantages for
survival of HCC cells during nutrient deprivation. One is rescuing
cells from nutrient deficiency-induced cell apoptosis, and the
other is keeping cells from chemotherapy-induced cell death.
What is the functional relevance of nutrient
deprivation-induced autophagy activation in HCC? Recent studies
have revealed that tumors always suffer the adversity of nutrient
deficiency, even after the construction of tumor vessels (31,32).
This phenomenon suggests that tumor cells including HCC may
physiologically face a shortage of nutrients during development.
Moreover, treatment of HCC, such as TACE and TAE (10) also leads to nutrient deficiency.
Thus, nutrient shortage-induced autophagy is commonly activated in
HCC. Although the role of autophagy in tumors is controversial, the
present study clearly showed that autophagy is the primary cell
survival mechanism following nutrient deficiency in HCC cells. It
has been well demonstrated that failure to induce cell death by
anticancer treatment contributes to chemotherapeutic failure and
tumor progression. Thus, the present study indicates that in a
nutrient deficient microenvironment, activation of autophagy may be
an adaptive and a protective mechanism for HCC development, and the
combination of the inhibition of autophagy and conventional
chemotherapeutic agents could be an effective therapy for HCC.
Nutrient deprivation is a type of metabolic stress that causes ROS
accumulation. The relationship between ROS and autophagy is quite
complicated. ROS, particularly mitochondrial ROS, could induce
autophagy activation (33,34). However, removal of ROS by induction
of autophagy could promote cell survival (35). In the present study, the role of ROS
in autophagy activation and HCC cell survival needs further
studies.
Although autophagy was suggested by numerous studies
that it can promote cell survival in response to adverse conditions
such as nutrient deprivation, the mechanism underlying the effect
of autophagy on promoting cell survival remains poorly defined. It
has been suggested that clearance of mitochondria and then
prevention of the diffusion of pro-apoptotic factors, such as
cytochrome c, may help cells to escape apoptosis in response
to cell death stimuli (23,24). In the present study, we observed
that the mitochondria mass was significant decreased under
nutrient-deficient condition, and inhibition of autophagy by 3-MA
distinctly restored the number of mitochondria. These results
suggest that enhanced autophagy may reduce mitochondrial mass.
Although it is possible that there exist other factors that
contribute to the survival of HCC cells during nutrient deprivation
(36), we provide a persuasive data
to suggest that reduced mitochondrial mass by activation of
autophagy may play a beneficial role in promoting HCC cell survival
under nutrient deprivation.
Taken together, the present study suggests that
enhanced autophagy protects HCC cells from nutrient deprivation and
chemotherapy-induced cell death. This is likely consistent with
decreased mitochondrial mass. The physiologic roles of autophagy in
tumor progression may closely connect with their microenvironments.
These results further expand our understanding of the relevant role
of autophagy in cancer formation and progression and may provide
new strategies for HCC treatment. It may be reasonable to predict
that drugs impairing the nutrient deprivation-induced autophagy
pathway may be beneficial for chemotherapy of HCC during TACE. We
also would like to extend the present study to other cancers.
Further studies on the molecular mechanism by which autophagy
promotes chemoresistance are warranted, and may possibly facilitate
cancer therapy.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (no. 81472623).
Glossary
Abbreviations
Abbreviations:
|
HCC
|
hepatocarcinoma
|
|
5-FU
|
5-fluorouracil
|
|
LC3
|
microtubule-associated protein 1 light
chain 3
|
|
DAPI
|
4′,6′-diamidino-2-phenylindole
dihydrochloride
|
|
3-MA
|
3-methyladenine
|
|
TACE
|
transarterial chemoembolization
|
|
GFP-LC3
|
green fluorescent protein-tagged
LC3
|
|
PI
|
propidium iodide
|
|
FITC
|
fluorescein isothiocyannate
|
|
GFP
|
green fluorescent protein
|
|
siRNA
|
small interfering RNA
|
|
TEM
|
transmission electron microscopy
|
References
|
1
|
Tang ZY: Hepatocellular carcinoma - cause,
treatment and metastasis. World J Gastroenterol. 7:445–454. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bartlett A and Heaton N: Hepatocellular
carcinoma: Defining the place of surgery in an era of organ
shortage. World J Gastroenterol. 14:4445–4453. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Abedin MJ, Wang D, McDonnell MA, Lehmann U
and Kelekar A: Autophagy delays apoptotic death in breast cancer
cells following DNA damage. Cell Death Differ. 14:500–510. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Katayama M, Kawaguchi T, Berger MS and
Pieper RO: DNA damaging agent-induced autophagy produces a
cytoprotective adenosine triphosphate surge in malignant glioma
cells. Cell Death Differ. 14:548–558. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kaushal GP, Kaushal V, Herzog C and Yang
C: Autophagy delays apoptosis in renal tubular epithelial cells in
cisplatin cytotoxicity. Autophagy. 4:710–712. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Amaravadi RK, Yu D, Lum JJ, Bui T,
Christophorou MA, Evan GI, Thomas-Tikhonenko A and Thompson CB:
Autophagy inhibition enhances therapy-induced apoptosis in a
Myc-induced model of lymphoma. J Clin Invest. 117:326–336. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Levine B and Yuan J: Autophagy in cell
death: An innocent convict? J Clin Invest. 115:2679–2688. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ogier-Denis E and Codogno P: Autophagy: A
barrier or an adaptive response to cancer. Biochim Biophys Acta.
1603:113–128. 2003.PubMed/NCBI
|
|
10
|
Qian J, Feng GS and Vogl T: Combined
interventional therapies of hepatocellular carcinoma. World J
Gastroenterol. 9:1885–1891. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Levine B and Klionsky DJ: Development by
self-digestion: Molecular mechanisms and biological functions of
autophagy. Dev Cell. 6:463–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kroemer G and Jäättelä M: Lysosomes and
autophagy in cell death control. Nat Rev Cancer. 5:886–897. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Klionsky DJ, Abeliovich H, Agostinis P,
Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA,
Ballabio A, et al: Guidelines for the use and interpretation of
assays for monitoring autophagy in higher eukaryotes. Autophagy.
4:151–175. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu SW, Baek SH, Brennan RT, Bradley CJ,
Park SK, Lee YS, Jun EJ, Lookingland KJ, Kim EK, Lee H, et al:
Autophagic death of adult hippocampal neural stem cells following
insulin withdrawal. Stem Cells. 26:2602–2610. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gozuacik D and Kimchi A: Autophagy as a
cell death and tumor suppressor mechanism. Oncogene. 23:2891–2906.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sato K, Tsuchihara K, Fujii S, Sugiyama M,
Goya T, Atomi Y, Ueno T, Ochiai A and Esumi H: Autophagy is
activated in colorectal cancer cells and contributes to the
tolerance to nutrient deprivation. Cancer Res. 67:9677–9684. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
De Flora S, Scarfì S, Izzotti A,
D'Agostini F, Chang CC, Bagnasco M, De Flora A and Trosko JE:
Induction by 7,12-dimethylbenz(a)anthracene of molecular and
biochemical alterations in transformed human mammary epithelial
stem cells, and protection by N-acetylcysteine. Int J Oncol.
29:521–529. 2006.PubMed/NCBI
|
|
20
|
Tanida I, Minematsu-Ikeguchi N, Ueno T and
Kominami E: Lysosomal turnover, but not a cellular level, of
endogenous LC3 is a marker for autophagy. Autophagy. 1:84–91. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pattingre S, Tassa A, Qu X, Garuti R,
Liang XH, Mizushima N, Packer M, Schneider MD and Levine B: Bcl-2
antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell.
122:927–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cao Y and Klionsky DJ: Physiological
functions of Atg6/Beclin 1: A unique autophagy-related protein.
Cell Res. 17:839–849. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Colell A, Ricci JE, Tait S, Milasta S,
Maurer U, Bouchier-Hayes L, Fitzgerald P, Guio-Carrion A,
Waterhouse NJ, Li CW, et al: GAPDH and autophagy preserve survival
after apoptotic cytochrome c release in the absence of caspase
activation. Cell. 129:983–997. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ravikumar B, Berger Z, Vacher C, O'Kane CJ
and Rubinsztein DC: Rapamycin pre-treatment protects against
apoptosis. Hum Mol Genet. 15:1209–1216. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang H, Bosch-Marce M, Shimoda LA, Tan
YS, Baek JH, Wesley JB, Gonzalez FJ and Semenza GL: Mitochondrial
autophagy is an HIF-1-dependent adaptive metabolic response to
hypoxia. J Biol Chem. 283:10892–10903. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Amaravadi RK and Thompson CB: The roles of
therapy-induced autophagy and necrosis in cancer treatment. Clin
Cancer Res. 13:7271–7279. 2009. View Article : Google Scholar
|
|
27
|
Bauvy C, Gane P, Arico S, Codogno P and
Ogier-Denis E: Autophagy delays sulindac sulfide-induced apoptosis
in the human intestinal colon cancer cell line HT-29. Exp Cell Res.
268:139–149. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Viola G, Bortolozzi R, Hamel E, Moro S,
Brun P, Castagliuolo I, Ferlin MG and Basso G: MG-2477, a new
tubulin inhibitor, induces autophagy through inhibition of the
Akt/mTOR pathway and delayed apoptosis in A549 cells. Biochem
Pharmacol. 83:16–26. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liao A, Hu R, Zhao Q, Li J, Li Y, Yao K,
Zhang R, Wang H, Yang W and Liu Z: Autophagy induced by FTY720
promotes apoptosis in U266 cells. Eur J Pharm Sci. 45:600–605.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang YH, Wu YL, Tashiro S, Onodera S and
Ikejima T: Reactive oxygen species contribute to oridonin-induced
apoptosis and autophagy in human cervical carcinoma HeLa cells.
Acta Pharmacol Sin. 32:1266–1275. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fujii S, Mitsunaga S, Yamazaki M, Hasebe
T, Ishii G, Kojima M, Kinoshita T, Ueno T, Esumi H and Ochiai A:
Autophagy is activated in pancreatic cancer cells and correlates
with poor patient outcome. Cancer Sci. 99:1813–1819.
2008.PubMed/NCBI
|
|
32
|
Ogata A, Yanagie H, Ishikawa E, Morishita
Y, Mitsui S, Yamashita A, Hasumi K, Takamoto S, Yamase T and
Eriguchi M: Antitumour effect of polyoxomolybdates: Induction of
apoptotic cell death and autophagy in in vitro and in vivo models.
Br J Cancer. 98:399–409. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Scherz-Shouval R, Shvets E, Fass E, Shorer
H, Gil L and Elazar Z: Reactive oxygen species are essential for
autophagy and specifically regulate the activity of Atg4. EMBO J.
26:1749–1760. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen Y, Azad MB and Gibson SB: Superoxide
is the major reactive oxygen species regulating autophagy. Cell
Death Differ. 16:1040–1052. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bensaad K, Cheung EC and Vousden KH:
Modulation of intracellular ROS levels by TIGAR controls autophagy.
EMBO J. 28:3015–3026. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bruno P, Calastretti A, Priulla M, Asnaghi
L, Scarlatti F, Nicolin A and Canti G: Cell survival under nutrient
stress is dependent on metabolic conditions regulated by Akt and
not by autophagic vacuoles. Cell Signal. 19:2118–2126. 2007.
View Article : Google Scholar : PubMed/NCBI
|