Introduction
A marked increase in the number of cases of thyroid
cancer has been consistently observed in several countries,
including China. It is estimated that approximately 90,000 new
thyroid cancer cases were diagnosed in China in 2015 (1). More than 90% of thyroid cancer cases
are differentiated thyroid cancers (DTC). Some DTC patients with
distant metastasis are resistant to 131iodine.
Traditional cancer treatments are usually not effective for
131iodine-resistant DTC patients, who have shorter
overall survival times than 131iodine-responsive DTC
patients (2,3).
The biological behaviors of tumors are driven by
oncogenes (4). The MAPK and
PI3K/AKT pathways are two important pathways driving thyroid tumor
development and progression (5).
The activities of BRAF and RAS, two key components of the MAPK
pathway, affect cancer cell proliferation and resistance to
anticancer treatment (6). Multiple
genes in the MAPK pathway are targeted by sorafenib, a kinase
inhibitor (7,8). Sorafenib was recently approved as the
first targeted drug for advanced metastatic DTC by the Chinese Food
and Drug Administration. However, sorafenib only improves
progression-free survival for ~5 months compared with patients on a
placebo (9), and the majority of
patients acquire resistance to sorafenib after 1 or 2 years of
administration, which is an obstacle in clinical practice (10).
Autophagy is an evolutionarily conserved process in
which cytoplasmic materials are delivered to lysosomes for
degradation (11). The relationship
between autophagy and cancer is complex due to the dual roles of
autophagy in tumor progression and cancer therapy (11,12).
Several studies have reported that sorafenib induced autophagy in
cancer cells (13), and it has been
reported that autophagy can be induced by sorafenib in a dose- and
time-dependent in several hepatocellular carcinoma (HCC) cell lines
(14,15). It has also been observed that
sorafenib modulated the expression of LC3, Beclin-1, Atg5 and
Atg12, and decreased the expression of p62 in HCC cells in
vitro (16,17). Several studies have reported that
sorafenib inhibited AKT phosphorylation in renal cancer cells and
HCC, as reviewed by Prieto-Dominguez et al (13). In addition, inhibition of AKT
enhanced sorafenib-induced autophagy, and switched protective
autophagy to autophagic cell death in HCC (17). Although the role of autophagy in
cancer has been extensively investigated, its role in the treatment
of thyroid cancer by sorafenib has not been clearly determined.
We previously observed that sorafenib exerted a
therapeutic effect by targeting the MAPK and AKT/mTOR pathways.
However, the AKT/mTOR pathway is a negative regulator of autophagy
and the role of autophagy in cancer treatment is complex (17,18).
In this study, we aimed to investigate whether autophagy was
induced by sorafenib treatment in thyroid cancer cells and to
elucidate whether autophagy inhibition increases or reduces
sorafenib-induced thyroid cancer cell death.
Materials and methods
Cell culture and materials
The human thyroid cancer cell lines 8505C and FTC133
were cultured at 37°C in Dulbecco's modified Eagle's medium (Gibco;
Thermo Fisher, Shanghai, China) supplemented with 10% fetal bovine
serum (Gibco; Thermo Fisher), 100 U/ml penicillin and 100 g/ml
streptomycin in a humidified incubator with 5% CO2.
Antibodies against Beclin-1 (mouse, 1:1,000, SC-10086), GAPDH
(mouse, 1:1,000, SC-51907), p-ERK (mouse, 1:200, SC-7383), ERK
(rabbit, 1:1,000, SC153) and AKT (mouse, 1:1,000, SC-5298) were
purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA),
and the anti-p-AKT (Ser473) antibody was purchased from Abcam
(rabbit, 1:200, Cambridge, Massachusetts, USA). Antibodies against
cleaved-caspase-3 (rabbit, 1:1,000, #9661), p62 (rabbit, 1:1,000,
#88588), LC3 (rabbit, 1:1,000, #12741), p70s6 (rabbit, 1:1,000,
#2708) and p-p70s6 (rabbit, 1:500, #2211) were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA). BEZ-234 was
purchased from Selleck (Shanghai, China), and sorafenib was
generously donated by the Bayer Company (Shanghai, China). Cells
were transfected with scrambled (negative control) small
interfering (si)RNA (5′-UUCUCCGAACGUGUCACGUTT-3′ and
5′-ACGUGACACGUUCGGAGAATT-3′), AKT siRNA
(5′-GACGGGCACAUUAAGAUCATT-3′ and 5′-UGAUCUUAAUGUGCCCGUCTT-3′), or
ATG5 siRNA (5′-GUCCAUCUAAGGAUGCAAUTT-3′ and
5′-AUUGCAUCCUUAGAUGGACTT-3′) and purchased from GenePharma
(Shanghai, China). Transfection was performed using Lipofectamine™
2000 (Invitrogen, Shanghai, China).
Mouse xenograft
Nude BALB/c mice (5–6 weeks old; male; Animal Core
Facility of Nanjing Medical University, Nanjing, China) were used
to generate xenograft tumor models. All animal studies complied
with the management rules of the National Health and Family
Planning Commission of China and were approved by the Ethics
Committee of Zhejiang Cancer Hospital. A suspension of
5×106 8505C or FTC133 cells in 100 µl PBS was inoculated
subcutaneously into the right flanks of the mice. When the tumor
sizes averaged approximately 5×5 mm, the mice were randomly divided
into four groups that were matched for tumor volume, and treatment
was initiated (6 mice per treatment group). Treatment groups were
as follows: the vehicle control group (DMSO, ≤0.1%), the
chloroquine group (CQ; Sigma-Aldrich, Shanghai, China), the
sorafenib group, and the sorafenib + CQ group. Sorafenib was
administered at doses of 30 mg/kg every day for 21 days. CQ was
administered at doses of 60 mg/kg every day for 21 days. In the
sorafenib + CQ group, sorafenib was administered 20 min after CQ
administration. All drugs were administered via intraperitoneal
injection. A caliper was used to assess the tumors every 4 days.
After 21 days of treatment, the mice were sacrificed and the tumor
tissues were harvested for study. The mice were sacrificed by
anesthetizing with an intraperitoneal injection of 0.8%
pentobarbital sodium (60 mg/kg), followed by cervical dislocation.
All efforts were made to reduce pain experienced by the mice.
Western blot analysis
Tumor tissues or cell pellets were lysed in RIPA
lysis buffer and a protease inhibitor cocktail (Sigma-Aldrich).
Total protein samples were separated by 10% or 12% sodium dodecyl
sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred onto nitrocellulose membranes. The membranes were
blocked with 5% non-fat dry milk and incubated overnight with the
various primary antibodies at 4°C. Goat anti-mouse (sc-2005) or
anti-rabbit (sc-2004) antibodies were purchased from Santa Cruz
Biotechnology, Inc. and diluted to 1:1,000 in 5% skim milk,
Tris-HCl (pH 7.5) and 0.1% Tween-20. The immunoblots were
subsequently washed and incubated with the goat anti-mouse or
anti-rabbit antibody for 1 h at room temperature. The results were
visualized using chemiluminescence reagent (sc-2048, Santa Cruz
Biotechnology, Inc.).
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was isolated using an RNA Prep Pure kit
(Tiangen, Beijing, China), according to the manufacturer's
instructions. The synthesis of cDNA was performed with HiScript II
Q RT Super Mix for qPCR (+gDNA wiper) (Vazyme Biotech Co., Nanjing,
China). qPCR was performed with an AceQ qPCR SYBR Green Master Mix
(without ROX) (Vazyme Biotech Co.). GAPDH was used as an internal
control. Primer sequences used in the qPCR analysis were purchased
from GenePharma (Shanghai, China), and the sequences were as
follows: AKT, 5′-CCAACACCTTCATCATCC-3′ and
5′-CTCCTCCTCCTGCTTCTT-3′; ATG5, 5′-GCAGATGGACAGTTGCACACA-3′ and
5′-TTTCCCCATCTTCAGGATCAA-3′; and GAPDH,
5′-CGGAGTCAACGGATTTGGTCGTAT-3′ and 5′-AGCCTTCTCCATGGTGGTGAAGAC-3′.
Melt curve was made to determine the optimal condition (95°C 15
sec, 60°C 30 sec, 95°C 15 sec). The PCR protocol is as follows:
denaturation 95 °C for 5 min, then 40 amplification cycles (95°C
for 5 sec and 60°C for 31 sec, at a ramp-rate of 1.6°C/sec).
Apoptosis assay
8505C and FTC133 cells (2×105/well) were
seeded into a 6-well plate. After treatment with ATG5Ri mimic,
sorafenib (10 µΜ), BEZ-235 (0.8 µM) or combined sorafenib +
BEZ-235, the cells were harvested and analyzed for apoptosis by
Annexin V and propidium iodide staining, using an FITC Annexin V
Apoptosis Detection kit (Life Technologies, Waltham, MA USA),
according to the manufacturer's instructions, and a flow cytometer
(FACS, Beckman Coulter, Miami, FL, USA).
Immunohistochemistry (IHC)
Paraffin-embedded mouse tumor samples were sliced
into 5-µm sections and mounted on microscope slides. Slides were
incubated at 37°C overnight for deparaffinization. The slides were
dipped in dimethylbenzene three times (10 min/dip). The slides were
then immediately dipped in 100, 95, 90, 80, 70 and 50% alcohol
continuously for 2 min at each percentage (%) of alcohol. Antigen
retrieval was performed by heating for 15 min in 0.01 M sodium
citrate buffer (pH 6.0). The slides were washed in TBS three times
(5 min/wash), treated with 3% H2O2 and
blocked with 5% BSA for 20 min, then incubated with 50 µl
cleaved-caspase-3 (1:200 dilution) at 4°C overnight. Subsequently,
the slides were washed, and incubated with a mouse anti-rabbit
antibody for 1 h at room temperature. A DAB horseradish peroxidase
color development kit (ZSGB-BIO, Beijing, China) was used to detect
positive staining. The slides were also counterstained with
hematoxylin for 25 sec, washed under running water for 3 min, then
washed with distilled water for 1 min followed by dipping in 50,
70, 80, 90 and 100% alcohol continuously for 1 min each. Finally,
the slides were dipped in dimethylbenzene for 3 min, air-dried and
mounted with Permount™ Mounting Medium (eBioscience, San Diego, CA,
USA).
Cell proliferation and viability
assays
8505C and FTC133 cells were plated at a density of
1×104 cells/well in 96-well microtiter plates, and each
plate was incubated for 24 h at 37°C in 5% CO2.
Following treatment, the absorbance of the contents of each well
was assessed at 470 nm following the use of a CCK-8 kit (Beyotime,
Guangzhou, China).
Statistical analysis
Data are presented as the mean ± standard error of
the mean. The results were analyzed using One-way ANOVA, and
multiple comparison using Student-Newman-Keuls (SNK) and least
significant difference (LSD) tests. P<0.05 was considered to
indicate a statistically significant difference.
Results
CQ increases the therapeutic effect of
sorafenib and suppresses tumor growth in xenograft models
To investigate whether autophagy influences the
therapeutic efficacy of sorafenib in vivo, 5–6-week-old male
BALB/c nude mice were used as an animal model. CQ is a potential
autophagy inhibitor that has been used to regulate autophagy in
vivo and in vitro (19,20).
Mice were randomly divided into control, sorafenib (30 mg/kg/day),
CQ (60 mg/kg/day), or sorafenib + CQ groups (n=6/group) at day 7
after subcutaneous injection of 5×106 cancer cells.
Tumor volumes were monitored every 4 days, and the mice were
sacrificed after 21 days of treatment for isolation of tumor
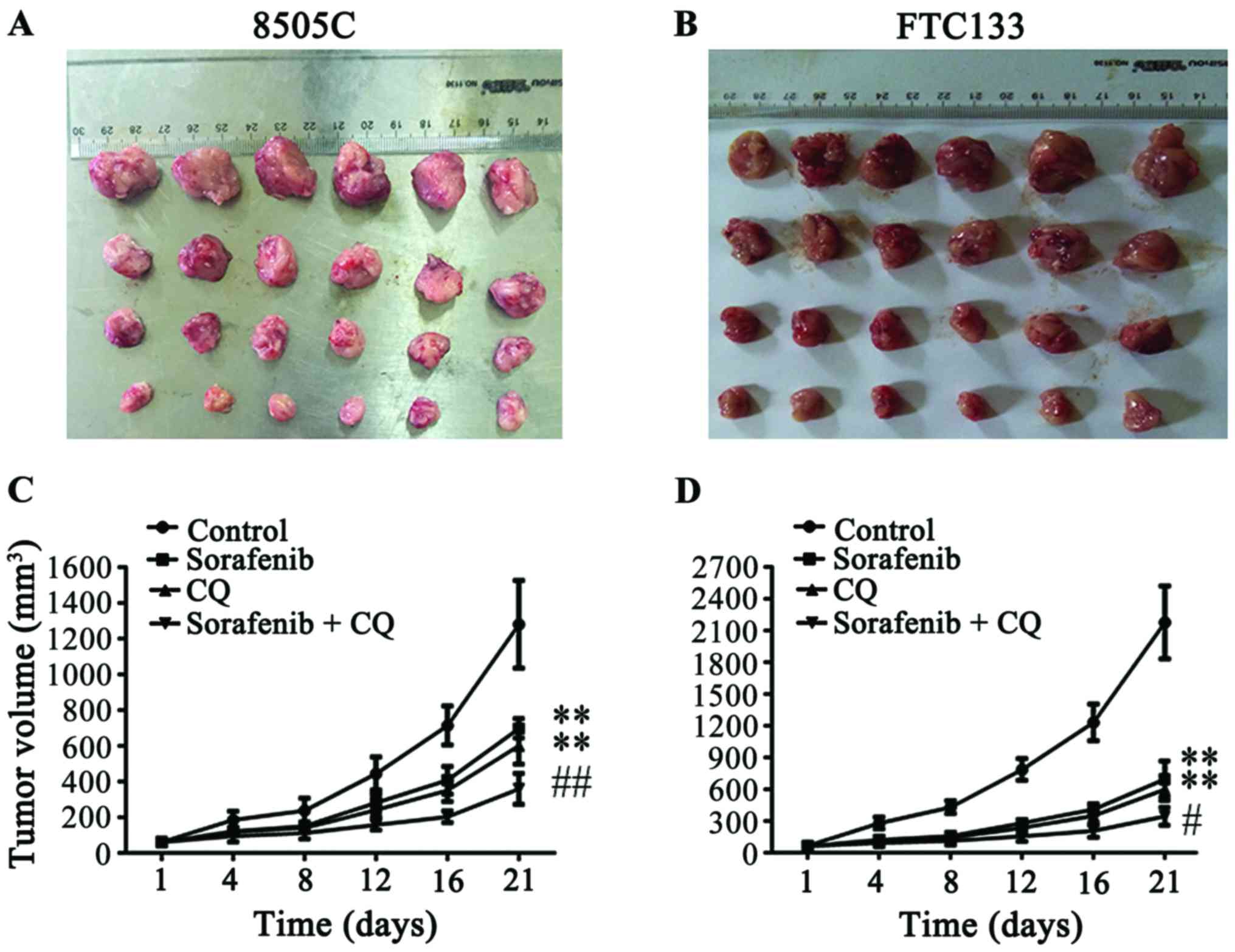
tissues (Fig. 1A and B). Compared
with the tumor sizes in the control group (1,280.93±245.27
mm3 in 8505C xenograft mice; 2,176.57±343.82
mm3 in FTC133 xenograft mice), the tumor volumes
significantly decreased after administration of CQ or sorafenib
alone (602.36±101.18 and 698.61±54.53 mm3 in 8505C
xenograft mice, respectively; 600.42±107.33 and 697.07±171.34
mm3 in FTC133 xenograft mice, respectively) (Fig. 1C and D), while the combined
treatment of CQ + sorafenib significantly suppressed tumor volumes
compared with CQ or sorafenib treatments alone in the 8505C and
FTC133 xenograft mice (358.99±86.54 and 344.36±84.36
mm3, respectively) (Fig. 1C
and D). There were no significant differences between CQ and
sorafenib treatment groups in 8505C or FTC133 xenograft mice.
Sorafenib induces autophagy and
apoptosis, and combined treatment with CQ increases autophagy and
apoptosis
8505C and FTC133 xenograft mice were sacrificed and
the tumor tissues were isolated for further studies after 21 days
of treatment. The proteins LC3 and p62 were used as autophagy
markers for monitoring autophagic flux and were examined by western
blotting of lysed tumor tissues. The expression level of
LC3II/GAPDH increased significantly in the sorafenib treatment
group compared with the control group, while the expression level
of p62 was reduced in the sorafenib treatment group compared with
the control group (Fig. 2A-C). To
confirm the effect of sorafenib on autophagy in thyroid cancer, we
treated 8505C and FTC133 cell lines with various concentrations of
sorafenib in vitro for 24 h. With an increase in sorafenib
concentration, LC3II/GAPDH levels increased significantly compared
with the control. When treated with 10 µΜ sorafenib, p62 protein
levels were obviously reduced compared with the control (Fig. 2D-F). These results indicated that
sorafenib increased autophagic flux in 8505C and FTC133 xenograft
mouse tumors and in vitro experiments. In CQ-treated tumor
tissues, the expression level of LC3II/GAPDH increased
significantly compared with the control. However, the expression
level of p62 in the CQ treatment group increased significantly
compared with the control group, which appeared to be contrary to
the sorafenib group (Fig. 2A-C).
These results indicated that autophagic flux was inhibited by
CQ.
Caspase-3 is activated in apoptotic cells by both
extrinsic and intrinsic pathways (21,22).
We used caspase-3 activation to assess apoptosis since caspase-3 is
an early marker for apoptosis (23). To assess the effect of sorafenib and
CQ on tumor apoptosis, we used IHC analysis of cleaved caspase-3 in
different treatment groups of mice. As expected, in the sorafenib-
and CQ-treated groups, the level of cleaved caspase-3 increased
compared with the control, suggesting CQ or sorafenib induced
apoptosis in 8505C and FTC133 xenograft mouse tumors. In the CQ +
sorafenib combined treatment group, a further increase of cleaved
caspase-3 was observed compared with CQ- or sorafenib-treated
groups in both 8505C and FTC133 xenograft mice (Fig. 2G). Our results demonstrated an
activation of caspase-3 in 8505C and FTC133 xenograft mouse tumors,
suggesting that caspase-mediated apoptosis occurs following
sorafenib and CQ treatment.
Sorafenib concurrently inhibits
activity of the MAPK and AKT/mTOR pathways in thyroid cancer
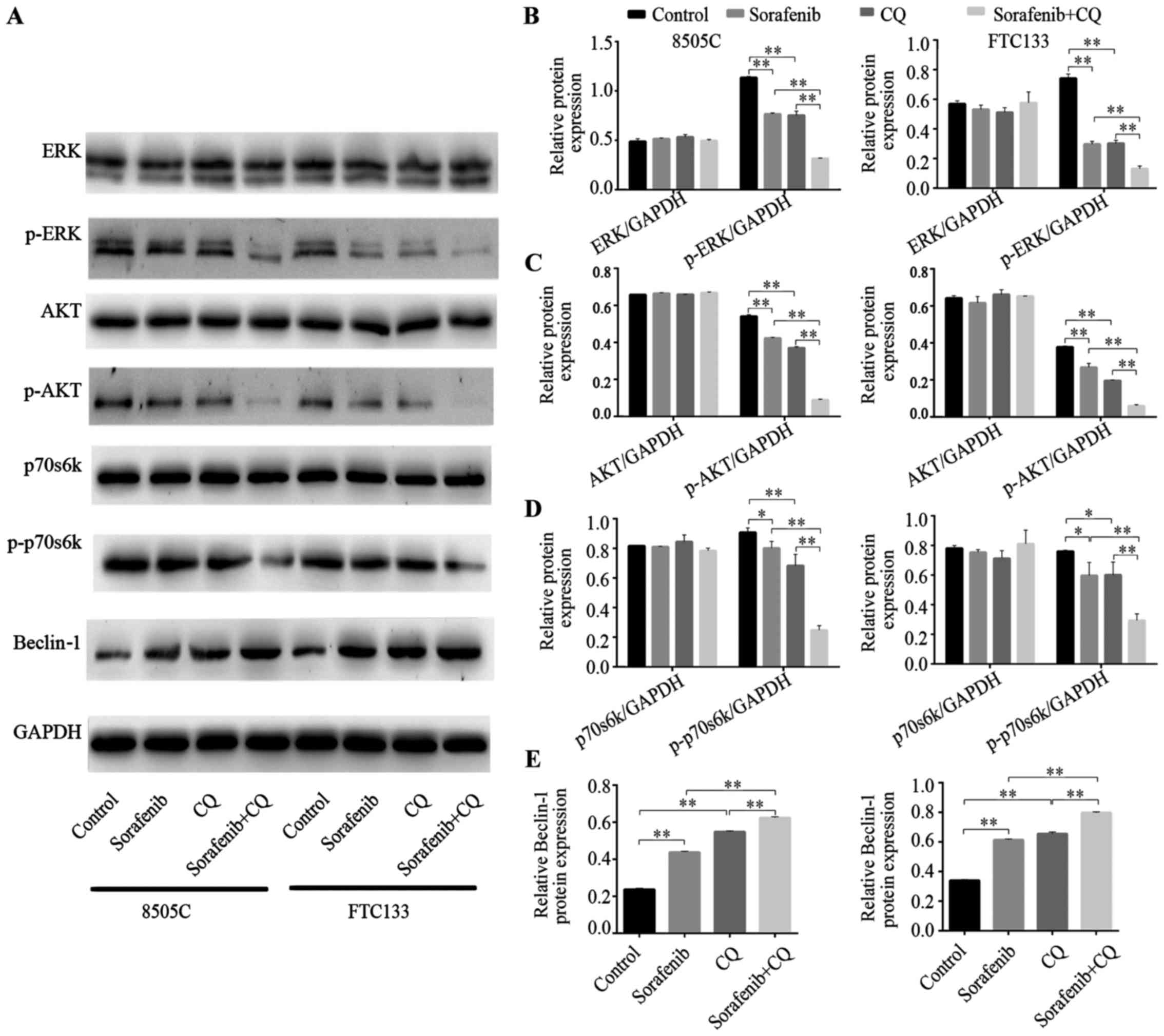
Tumor tissues were used to analyze the
target-suppressing effects of sorafenib. As shown in Fig. 3A-D, compared with the control
groups, the expression of the ERK protein was not significantly
different in the sorafenib treatment groups of 8505C and FTC133
xenograft mice. However, phosphorylation levels of ERK were
significantly inhibited in both 8505C and FTC133 xenograft mice
that were treated with sorafenib. Furthermore, we observed that the
phosphorylation levels of AKT and p70s6k were significantly reduced
in the sorafenib treatment groups compared with control groups. The
results indicated that CQ exerted similar effects to sorafenib. The
phosphorylation levels of ERK, AKT and p70s6k were significantly
reduced after CQ treatment. Furthermore, in 8505C and FTC133
xenograft mice treated with combined CQ + sorafenib, the
dephosphorylation effect was enhanced compared with that achieved
by either drug alone (Fig.
3A-D).
Beclin-1 is a key autophagy-related gene that takes
part in the initial process of autophagy (24). Beclin-1 protein expression was
increased in xenograft mice treated with CQ or sorafenib, while
combined CQ + sorafenib treatment increased the expression of
Beclin-1 protein more than CQ or sorafenib treatment alone
(Fig. 3A and E).
Inhibition of the AKT/mTOR pathway
activates autophagy and apoptosis, and enhances the effect of
sorafenib on autophagy and apoptosis induction
The MAPK pathway is a well-known target of
sorafenib. However, the AKT/mTOR pathway was inhibited by sorafenib
in the present study. The AKT/mTOR pathway is a major pathway that
regulates the process of autophagy (25,26).
Next, we demonstrated the role of the AKT/mTOR pathway in
sorafenib-treated thyroid cell lines. To construct an AKT
gene-silencing cell model, we transfected siRNAs into 8505C and
FTC133 cell lines. The silencing effect was examined by western
blotting. The protein levels of p-AKT were reduced significantly in
AKT siRNA-transfected cell line models (27). The LC3 and p62 proteins were
examined again for the evaluation of autophagy levels. Following
silencing of AKT by siRNA, LC3II increased and p62 decreased
compared with the control group (Fig.
4A-C). AKT silencing in combination with sorafenib treatment
significantly enhanced LC3II expression levels and reduced p62
compared with AKT silencing or sorafenib treatment alone (Fig. 4A-C). BEZ-235, a dual,
ATP-competitive PI3K and mTOR inhibitor, was selected to
demonstrate the role of the AKT/mTOR pathway in sorafenib-induced
autophagy. In a previous study, 0.8 µM BEZ-235 inhibited AKT/mTOR
pathway activity and inhibited 8505C and FTC133 cell line
proliferation (27). Thus, 8505C
and FTC133 cell lines were treated with 0.8 µM BEZ-235 for 24 h,
and western blotting was used to evaluate the expression levels of
LC3II and p62. LC3II expression increased and p62 expression
decreased compared with the control group in both 8505C and FTC133
cell lines (data not shown). Combined treatment with sorafenib and
BEZ-235 significantly enhanced LC3II expression and reduced p62
expression (data not shown). The AKT/mTOR pathway negatively
regulates autophagy and apoptosis (17). In the present study, 8505C and
FTC133 cell lines treated with AKT siRNA exhibited increased
cleaved capase-3 expression, and combined treatment of AKT siRNA
with sorafenib enhanced cleaved capase-3 expression compared with
either treatment alone (Fig. 4D and
E).
Effect of autophagy on sorafenib
treated thyroid cancer cell lines
In our results, sorafenib treatment or AKT/mTOR
pathway suppression induced autophagy in thyroid cancer cell lines.
As autophagy plays a dual role in cancer treatment, we examined the
role of autophagy in sorafenib treatment of thyroid cancer
(11,12). ATG5 is a key gene in the process of
autophagy that can be silenced to demonstrate the role of autophagy
in different treatments (6). Three
different siRNAs (ATG5Ri) were transfected into 8505C and FTC133
cell lines, and RT-qPCR was used to evaluate their silencing
effect. ATG5 siRNA2 appeared to have strong silencing effects and
was selected for use in further experiments (data not shown).
Treatment with sorafenib or ATG5Ri increased the
apoptosis of 8505C and FTC133 cell lines compared with the control
group (apoptosis rate, 4.9 and 5.6%); the apoptosis rates after
sorafenib or ATG5Ri treatment were 24.7 and 25.2%, respectively, in
8505C cells (Fig. 5A and B), and
53.2 and 27.3%, respectively, in FTC133 cells (Fig. 5C and D). Combined treatment with
sorafenib and ATG5Ri increased apoptosis rates more than either
treatment alone in both cell lines (42.8% in 8505C cells; 69.7% in
FTC133 cells; Fig. 5). Silencing of
ATG5 significantly increased the apoptosis rate after combined
treatment of sorafenib and BEZ235 in FTC133 cells (72.6 vs. 68.1%;
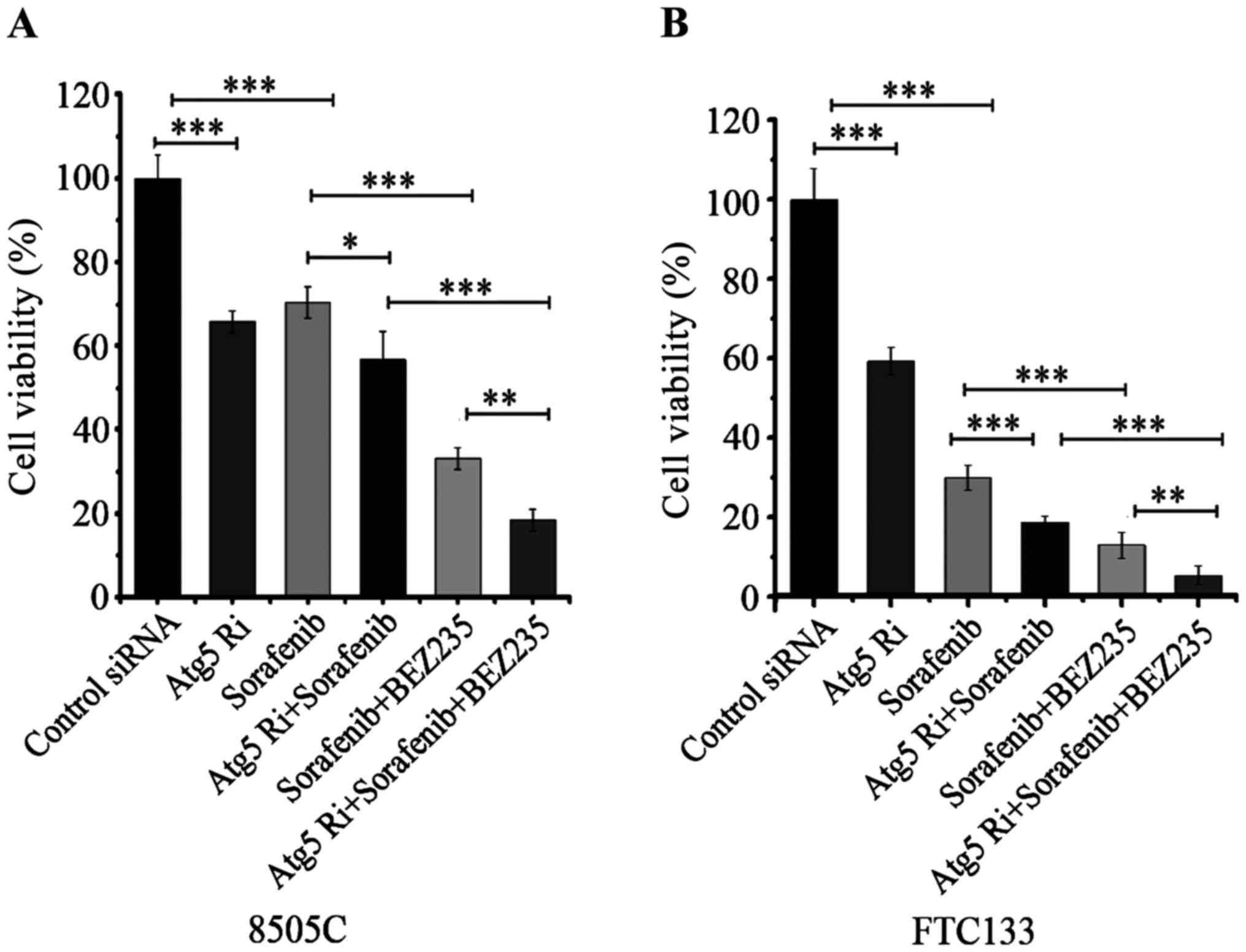
Fig. 5). Silencing of ATG5
increased the anti-proliferative effect of sorafenib and combined
treatment of sorafenib and BEZ-235 in both 8505C and FTC133 cell
lines (Fig. 6).
Discussion
Resistance to 131iodine therapy is an
important factor leading to poor prognosis for DTC patients
(2,3). Several targeted kinase inhibitors have
been used in clinical trials and administered to patients who have
advanced/progressive 131iodine-refractory DTC disease
(28). Sorafenib targets the RAF
serine/threonine kinases, and also potently inhibits tyrosine
kinase receptors, such as vascular endothelial growth factor
receptor and platelet-derived growth factor receptor-β, which
promote angiogenesis (7,8). It has been reported that sorafenib has
therapeutic effects in several types of cancer, such as HCC, acute
myeloid leukemia, advanced renal cell carcinoma or prostate cancer
(29–32). Sorafenib is the first tyrosine
kinase inhibitor (TKI) approved for advanced/progressive
131iodine-refractory DTC patients in China. After
sorafenib treatment, the majority of advanced/progressive
131iodine-refractory DTC patients exhibit a favorable
prognosis. However, most patients acquire resistance to sorafenib
after 1 or 2 years of administration, and the mechanism of this
acquired resistance remains unclear (10). In the treatment of various cancers,
including liver, kidney and prostate cancer, sorafenib can achieve
a few months of progression-free survival, but drug resistance
frequently emerges (31–33). Mechanisms of sorafenib resistance
may be related to EGFR activation, downstream signaling of EGFR
(particularly Ras/Raf/MEK/ERK), the PI3K/AKT pathway, autophagy or
epithelial-mesenchymal transition, as reviewed by Beardsley et
al (32), and Chen et al
(33).
The process of autophagy could be activated during
cancer treatment by radiation (34), chemotherapy (35) or TKI drugs (12,15).
The role of autophagy in cancer treatment is complex since it is a
‘double-edged sword’; autophagy may promote or inhibit cell death
(12,36,37).
The varying roles of autophagy in cancer treatment primarily depend
on different contexts and treatments (13,37,38).
In different HCC cell lines, the role of autophagy induced by
sorafenib was reversed, as detailed by Prieto-Dominguez et
al (13). It has been reported
that inhibition of AKT reverses acquired resistance to sorafenib by
switching protective autophagy to autophagic cell death (17).
It has been demonstrated that sorafenib triggers
autophagy in different cancer contexts (12,15).
In the present study, sorafenib induced autophagy in thyroid cancer
cell lines and mice xenograft models. Inhibition of autophagy by
either a pharmacological inhibitor (CQ) or siRNA knockdown of
essential autophagy genes increased the effect of sorafenib
treatment. CQ has been safely used for decades in patients for
malaria prophylaxis and for the treatment of rheumatoid arthritis
(39). The anticancer role of CQ
has primarily been attributed to its autophagy regulation role.
Autophagy is an evolutionarily conserved cellular degradation
process that engulfs cytoplasmic materials in double-membrane
structures (autophagosomes), which are subsequently transported to
lysosomes for degradation and recycling (11,40).
CQ is an autophagy inhibitor that prevents autophagosomes from
fusing with lysosomes, thereby disrupting the process of autophagy
(20). The primary role of CQ in
cancer treatment is to enhance the efficacy of a drug or other
treatment measure (41). However,
CQ does not markedly alter anticancer efficiency in certain types
of cancer; it may depend on the tumor type and context (42). To the best of our knowledge, no
previous studies have demonstrated the role of CQ in sorafenib
treatment of thyroid cancer. In the present study, thyroid cancer
xenograft mice were administered CQ. The results indicated that the
processes of autophagy were inhibited by CQ, and that the
anticancer efficiency of sorafenib was significantly increased.
Recent studies have indicated that the synergistic effects of CQ in
cancer treatment are independent of autophagy in several types of
cancer (43–45). The mechanism of the synergistic
effects of CQ is consistent with lysosomal cell death (43), vessel normalization (44) and other mechanisms (45). In order to confirm the role of
autophagy in sorafenib treatment of thyroid cancer, further
regulatory pathways and autophagy genes were evaluated.
Activation of the PI3K/AKT signaling pathway
mediates acquired resistance to sorafenib in HCC (46). Sorafenib has been reported to
inhibit both the MAPK and AKT/mTOR pathways in lymphoma xenografts
(47). Similarly, in the present
study, sorafenib inhibited both the MAPK and AKT/mTOR pathways.
mTOR is a negative regulator of autophagy and promotes the
proliferation of cancer cells (18). In the present study, the AKT/mTOR
pathway was selected to demonstrate the roles of autophagy in
thyroid cancer treatment by sorafenib. We silenced the AKT/mTOR
pathway by siRNA technology or with an inhibitor (BEZ-235 is a dual
inhibitor of PI3K and AKT). Subsequent results indicated that the
downstream gene activity of the AKT/mTOR pathway was suppressed,
cell proliferation was significantly reduced, and apoptosis and
autophagy were enhanced. These results indicated that sorafenib
induced autophagy, at least in part due to inhibition of the
AKT/mTOR pathway.
Beclin-1 is a key gene participating in the initial
process of autophagy (48). The
expression of Beclin-1 was promoted after sorafenib treatment in
the present study. This finding indicates that a complex mechanism
participates in sorafenib-induced autophagy. However, due to the
dual role of autophagy in cancer treatment, it is not clear whether
sorafenib-induced autophagy or AKT/mTOR pathway inhibition promote
or inhibit cell death (37).
ATG5 is an E3 ubiquitin ligase that is critical for
autophagy due to its role in autophagosome elongation (6). Silencing of ATG5 demonstrates the role
of autophagy in a cancer treatment process (6). Treatment with ATG5 siRNA in the
present study induced apoptosis in 8505C and FTC133 cell lines.
Combined treatment with an ATG5 siRNA increased the
tumor-suppressive effect of sorafenib in 8505C and FTC133 cell
lines. The apoptosis rate of the ATG5 silencing + sorafenib +
BEZ235 treatment group increased more than that of the sorafenib +
BEZ235 treatment group in FTC133 cells. We did not observe a
significantly different apoptosis rate between the ATG5 silencing +
sorafenib + BEZ-235 treatment group and the sorafenib + BEZ-235
treatment group in 8505C cells. In our data, silenced ATG5
suppressed the proliferation of 8505C and FTC133 cells, and ATG5
silencing combined with sorafenib treatment significantly
suppressed cell proliferation in 8505C and FTC133 cells. Our
previous study indicated that silencing the AKT/mTOR pathway
induced apoptosis, suppressed proliferation and induced autophagy
in both 8505C and FTC133 cells (27). In the present study, sorafenib
inhibited both the MAPK and AKT/mTOR pathways in 8505C and FTC133
cells. Thus, AKT/mTOR inhibition by sorafenib represents a positive
effect of sorafenib treatment in thyroid cancer. However, autophagy
induced by sorafenib involves a complex mechanism and can only be
partially attributed to AKT/mTOR suppression. Comprehensive
analysis of our results revealed that autophagy is a protective
mechanism in sorafenib treatment of thyroid cancer. Our study had
limitations. The additive or synergistic effect of sorafenib and CQ
in mouse xenograft models can not be easily established and can be
examined by future studies.
In summary, the present study demonstrated that
sorafenib induces autophagy in thyroid cancer, and that this effect
is, at least in part, due to AKT/mTOR pathway suppression. We
demonstrated that CQ increased the efficacy of sorafenib in
treating thyroid cancer in xenograft mice, resulting in decreased
tumor growth. Treatment with CQ or ATG5 silencers blocked the
processes of autophagy and increased apoptosis in sorafenib-treated
tumors. Autophagy has a cytoprotective role in sorafenib treatment
of thyroid cancer. Combined treatment of sorafenib with CQ or other
autophagy inhibitors is a novel and potentially useful clinical
strategy to improve the efficacy of sorafenib-targeted thyroid
cancer therapies. Our findings provide a basis for investigating
the use of sorafenib with autophagy inhibitors to treat
131iodine-resistant, advanced DTC.
Acknowledgements
The present study was supported by the Zhejiang
Provincial Natural Science Foundation of China (grant no.
LY15H180002), and the Medical and Health Research Program of
Zhejiang Province (grant no. 2015DTA003).
References
|
1
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Song HJ, Qiu ZL, Shen CT, Wei WJ and Luo
QY: Pulmonary metastases in differentiated thyroid cancer: Efficacy
of radioiodine therapy and prognostic factors. Eur J Endocrinol.
173:399–408. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Durante C, Haddy N, Baudin E, Leboulleux
S, Hartl D, Travagli JP, Caillou B, Ricard M, Lumbroso JD, De
Vathaire F, et al: Long-term outcome of 444 patients with distant
metastases from papillary and follicular thyroid carcinoma:
Benefits and limits of radioiodine therapy. J Clin Endocrinol
Metab. 91:2892–2899. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vogelstein B, Papadopoulos N, Velculescu
VE, Zhou S, Diaz LA Jr and Kinzler KW: Cancer genome landscapes.
Science. 339:1546–1558. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xing M, Haugen BR and Schlumberger M:
Progress in molecular-based management of differentiated thyroid
cancer. Lancet. 381:1058–1069. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang W, Kang H, Zhao Y, Min I, Wyrwas B,
Moore M, Teng L, Zarnegar R, Jiang X and Fahey TJ III: Targeting
autophagy sensitizes BRAF-mutant thyroid cancer to vemurafenib. J
Clin Endocrinol Metab. 102:634–643. 2017.PubMed/NCBI
|
|
7
|
Wilhelm SM, Adnane L, Newell P, Villanueva
A, Llovet JM and Lynch M: Preclinical overview of sorafenib, a
multikinase inhibitor that targets both Raf and VEGF and PDGF
receptor tyrosine kinase signaling. Mol Cancer Ther. 7:3129–3140.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Adnane L, Trail PA, Taylor I and Wilhelm
SM: Sorafenib (BAY 43-9006, Nexavar), a dual-action inhibitor that
targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases
VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 407:597–612.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brose MS, Nutting CM, Jarzab B, Elisei R,
Siena S, Bastholt L, de la Fouchardiere C, Pacini F, Paschke R,
Shong YK, et al DECISION investigators, : Sorafenib in radioactive
iodine-refractory, locally advanced or metastatic differentiated
thyroid cancer: A randomised, double-blind, phase 3 trial. Lancet.
384:319–328. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pitoia F and Jerkovich F: Selective use of
sorafenib in the treatment of thyroid cancer. Drug Des Devel Ther.
10:1119–1131. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yi H, Long B, Ye X, Zhang L, Liu X and
Zhang C: Autophagy: A potential target for thyroid cancer therapy
(Review). Mol Clin Oncol. 2:661–665. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Heqing Y, Bin L, Xuemei Y and Linfa L: The
role and mechanism of autophagy in sorafenib targeted cancer
therapy. Crit Rev Oncol Hematol. 100:137–140. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Prieto-Domínguez N, Ordóñez R, Fernández
A, García-Palomo A, Muntané J, González-Gallego J and Mauriz JL:
Modulation of autophagy by sorafenib: Effects on treatment
response. Front Pharmacol. 7:1512016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shi YH, Ding ZB, Zhou J, Hui B, Shi GM, Ke
AW, Wang XY, Dai Z, Peng YF, Gu CY, et al: Targeting autophagy
enhances sorafenib lethality for hepatocellular carcinoma via ER
stress-related apoptosis. Autophagy. 7:1159–1172. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shimizu S, Takehara T, Hikita H, Kodama T,
Tsunematsu H, Miyagi T, Hosui A, Ishida H, Tatsumi T, Kanto T, et
al: Inhibition of autophagy potentiates the antitumor effect of the
multikinase inhibitor sorafenib in hepatocellular carcinoma. Int J
Cancer. 131:548–557. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yuan H, Li AJ, Ma SL, Cui LJ, Wu B, Yin L
and Wu MC: Inhibition of autophagy significantly enhances
combination therapy with sorafenib and HDAC inhibitors for human
hepatoma cells. World J Gastroenterol. 20:4953–4962. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhai B, Hu F, Jiang X, Xu J, Zhao D, Liu
B, Pan S, Dong X, Tan G, Wei Z, et al: Inhibition of Akt reverses
the acquired resistance to sorafenib by switching protective
autophagy to autophagic cell death in hepatocellular carcinoma. Mol
Cancer Ther. 13:1589–1598. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yu L, McPhee CK, Zheng L, Mardones GA,
Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, et al: Termination of
autophagy and reformation of lysosomes regulated by mTOR. Nature.
465:942–946. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zou Y, Ling YH, Sironi J, Schwartz EL,
Perez-Soler R and Piperdi B: The autophagy inhibitor chloroquine
overcomes the innate resistance of wild-type EGFR non-small-cell
lung cancer cells to erlotinib. J Thorac Oncol. 8:693–702. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang C, Hu Q and Shen H-M: Pharmacological
inhibitors of autophagy as novel cancer therapeutic agents.
Pharmacol Res. 105:164–175. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Salvesen GS: Caspases: Opening the boxes
and interpreting the arrows. Cell Death Differ. 9:3–5. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ghavami S, Hashemi M, Ande SR, Yeganeh B,
Xiao W, Eshraghi M, Bus CJ, Kadkhoda K, Wiechec E, Halayko AJ, et
al: Apoptosis and cancer: Mutations within caspase genes. J Med
Genet. 46:497–510. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Günther A, Luczak V, Abel T and Baumann A:
Caspase-3 and GFAP as early markers for apoptosis and astrogliosis
in shRNA-induced hippocampal cytotoxicity. J Exp Biol.
220:1400–1404. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhong Y, Wang QJ, Li X, Yan Y, Backer JM,
Chait BT, Heintz N and Yue Z: Distinct regulation of autophagic
activity by Atg14L and Rubicon associated with Beclin
1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 11:468–476.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vlahakis A and Powers T: A role for TOR
complex 2 signaling in promoting autophagy. Autophagy.
10:2085–2086. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Díaz-Troya S, Pérez-Pérez ME, Florencio FJ
and Crespo JL: The role of TOR in autophagy regulation from yeast
to plants and mammals. Autophagy. 4:851–865. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yi H, Ye X, Long B, Ye T, Zhang L, Yan F,
Yang Y and Li L: Inhibition of the AKT/mTOR pathway augments the
anticancer effects of sorafenib in thyroid cancer. Cancer Biother
Radiopharm. 32:176–183. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Valerio L, Pieruzzi L, Giani C, Agate L,
Bottici V, Lorusso L, Cappagli V, Puleo L, Matrone A, Viola D, et
al: Targeted therapy in thyroid cancer: State of the art. Clin
Oncol (R Coll Radiol). 29:316–324. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bruix J, Takayama T, Mazzaferro V, Chau
GY, Yang J, Kudo M, Cai J, Poon RT, Han KH, Tak WY, et al STORM
investigators, : Adjuvant sorafenib for hepatocellular carcinoma
after resection or ablation (STORM): A phase 3, randomised,
double-blind, placebo-controlled trial. Lancet Oncol. 16:1344–1354.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tarlock K, Chang B, Cooper T, Gross T,
Gupta S, Neudorf S, Adlard K, Ho PA, McGoldrick S, Watt T, et al:
Sorafenib treatment following hematopoietic stem cell transplant in
pediatric FLT3/ITD acute myeloid leukemia. Pediatr Blood Cancer.
62:1048–1054. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hutson TE, Escudier B, Esteban E,
Bjarnason GA, Lim HY, Pittman KB, Senico P, Niethammer A, Lu DR,
Hariharan S, et al: Randomized phase III trial of temsirolimus
versus sorafenib as second-line therapy after sunitinib in patients
with metastatic renal cell carcinoma. J Clin Oncol. 32:760–767.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Beardsley EK, Hotte SJ, North S, Ellard
SL, Winquist E, Kollmannsberger C, Mukherjee SD and Chi KN: A phase
II study of sorafenib in combination with bicalutamide in patients
with chemotherapy-naive castration resistant prostate cancer.
Invest New Drugs. 30:1652–1659. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cheng AL, Kang YK, He AR, Lim HY, Ryoo BY,
Hung CH, Sheen IS, Izumi N, Austin T, Wang Q, et al Investigators'
Study Group, : Safety and efficacy of tigatuzumab plus sorafenib as
first-line therapy in subjects with advanced hepatocellular
carcinoma: A phase 2 randomized study. J Hepatol. 63:896–904. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hu L, Wang H, Huang L, Zhao Y and Wang J:
Crosstalk between autophagy and intracellular radiation response
(Review). Int J Oncol. 49:2217–2226. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liang B, Liu X, Liu Y, Kong D, Liu X,
Zhong R and Ma S: Inhibition of autophagy sensitizes MDR-phenotype
ovarian cancer SKVCR cells to chemotherapy. Biomed Pharmacother.
82:98–105. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Thorburn A, Thamm DH and Gustafson DL:
Autophagy and cancer therapy. Mol Pharmacol. 85:830–838. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
White E: The role for autophagy in cancer.
J Clin Invest. 125:42–46. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
O'Neill PM, Bray PG, Hawley SR, Ward SA
and Park BK: 4-Aminoquinolines-past, present, and future: A
chemical perspective. Pharmacol Ther. 77:29–58. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yin Z, Pascual C and Klionsky DJ:
Autophagy: Machinery and regulation. Microb Cell. 3:588–596. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Grimaldi A, Santini D, Zappavigna S,
Lombardi A, Misso G, Boccellino M, Desiderio V, Vitiello PP, Di
Lorenzo G, Zoccoli A, et al: Antagonistic effects of chloroquine on
autophagy occurrence potentiate the anticancer effects of
everolimus on renal cancer cells. Cancer Biol Ther. 16:567–579.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zinn RL, Gardner EE, Dobromilskaya I,
Murphy S, Marchionni L, Hann CL and Rudin CM: Combination treatment
with ABT-737 and chloroquine in preclinical models of small cell
lung cancer. Mol Cancer. 12:162013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
King MA, Ganley IG and Flemington V:
Inhibition of cholesterol metabolism underlies synergy between mTOR
pathway inhibition and chloroquine in bladder cancer cells.
Oncogene. 35:4518–4528. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Maes H, Kuchnio A, Peric A, Moens S, Nys
K, De Bock K, Quaegebeur A, Schoors S, Georgiadou M, Wouters J, et
al: Tumor vessel normalization by chloroquine independent of
autophagy. Cancer Cell. 26:190–206. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chen X, Clark J, Wunderlich M, Fan C,
Davis A, Chen S, Guan JL, Mulloy JC, Kumar A and Zheng Y: Autophagy
is dispensable for Kmt2a/Mll-Mllt3/Af9 AML maintenance and
anti-leukemic effect of chloroquine. Autophagy. 13:955–966. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen KF, Chen HL, Tai WT, Feng WC, Hsu CH,
Chen PJ and Cheng AL: Activation of phosphatidylinositol
3-kinase/Akt signaling pathway mediates acquired resistance to
sorafenib in hepatocellular carcinoma cells. J Pharmacol Exp Ther.
337:155–161. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Carlo-Stella C, Locatelli SL, Giacomini A,
Cleris L, Saba E, Righi M, Guidetti A and Gianni AM: Sorafenib
inhibits lymphoma xenografts by targeting MAPK/ERK and AKT pathways
in tumor and vascular cells. PLoS One. 8:e616032013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liu G, Yuan Y, Long M, Luo T, Bian J, Liu
X, Gu J, Zou H, Song R, Wang Y, et al: Beclin-1-mediated autophagy
protects against cadmium-activated apoptosis via the Fas/FasL
pathway in primary rat proximal tubular cell culture. Sci Rep.
7:9772017. View Article : Google Scholar : PubMed/NCBI
|