Introduction
Oral cancer incidence varies from 1.06 to
1.09/100,000 cases, and is steadily rising worldwide, with the age
at onset increasingly younger (1).
Tongue cancer is the most common oral malignancy (2), and more than 98% of cases are found at
the anterior lingual 2/3, mostly at the tongue base. In addition,
most cases diagnosed are squamous cell carcinoma and seldom
adenocarcinoma. Tongue cancer is usually accompanied with early
cervical lymph node metastasis, with a high incidence of 40–80%
(3). A 5-year survival rate of 80%
was documented for patients with no cervical lymph node metastasis;
while 30% was found in those with metastasis (4).
Invasion and metastasis of tongue squamous cell
carcinoma (TSCC) is a comprehensive, multistep and sequential
process, which encompasses inducible lymphogenesis and/or
angiogenesis, dissemination of tumor cells and their intravasation
into lymph or blood vessels, and random or specific residing at the
target microvessels. This is followed by extravasation out of
vessels, proliferation and invasion into distal tissues and organs,
and finally formation of metastatic foci (5). Chemokines are closely related to tumor
invasion and metastasis. Τhey bind to specific tumor cell surface
receptors and induce cytoskeleton rearrangement, promoting tight
attachment of tumor cells to lymphatic endothelial cells and their
directional migration (6).
Chemokines and their receptors extensively exist in
a wide range of cells, playing essential roles in embryogenesis,
hematopoiesis, HIV infection, tumor invasion, and metastasis
(7). Chemokines are secretory small
molecule proteins of the cytokine superfamily, with chemotactic
capabilities (8,9). Chemokine receptors belong to the
superfamily of G protein coupled seven transmembrane receptors
(10), whose N-terminal domains
bind to specific ligands, with cytosolic domains conjugating with G
protein; phosphorylation of their C-terminal serine/threonine
residues contributes to signal transduction. The chemokine receptor
CXCR3, including three spliced variants, termed CXCR3-A and CXCR3-B
and CXCR3-alt, which belongs to the CXC subfamily of chemokine
receptors, is highly expressed in colon cancer, melanoma, B
lymphoma and breast cancer cells. Its specific binding to its
ligand chemokine CXCL9 mediates the directional migration of cancer
cells, regulating cancer invasion and metastasis (11).
The chemokine CXCL9, also termed MIG, is a member of
the chemokine γ-subfamily (CXC family) of proteins. The human CXCL9
gene is located on chromosome 4q21, next to the gene encoding
CXCL10 (IFN2-γ inducible 10 kDa protein, IP210). CXCL9 is primarily
induced by IFN2-γ, and produced in lymphocytes,
monocytes/macrophages, and fibroblasts. CXCR3 belongs to the CXC
chemokine subfamily (12). The
human CXCR3 protein has 368 amino acids (AA) and a molecular weight
of 41 kDa (13). Its extracellular
N-terminal structural domain composed of AA 1–57 contributes to
ligand recognition and binding. Chemotaxis by CXCL9 is mainly
mediated by CXCR3, which, upon binding to CXCL9, initiates Src
phosphorylation and activates Src kinase, concomitantly enhancing
the activities of phosphatidylinositol 3 kinase (PI3K) and the
downstream Akt (14).
In the present study, high expression levels of the
chemokine CXCL9 and its receptor CXCR3 were detected in TSCC tissue
samples from patients with lymph node metastasis, indicating the
possible involvement of the CXCL9/CXCR3 axis in TSCC invasion and
metastasis. Furthermore, CXCL9/CXCR3 was demonstrated to activate
the Akt signaling pathway, which possibly mediates
epithelial-mesenchymal transition (EMT), thus promoting TSCC
invasion and metastasis.
Materials and methods
Reagents
The EnVision + HPR/DAB kit was purchased from
Shanghai Gene Technology Co., Ltd. (Shanghai, China), and the
cytokine CXCL9 from PeproTech Inc. (Rocky Hill, NJ, USA). CXCR3
neutralizing antibodies (2.0 µg/ml; mouse, monoclonal; cat. no.
49801) and IgG (2.0 µg/ml; mouse; cat. no. A7028) were purchased
from R&D Systems (Minneapolis, MN, USA) and Beyotime
Biotechnology Co., Ltd. (Shanghai, China), respectively.
Anti-E-cadherin (1:200; rabbit, polyclonal; cat. no. ab53226) and
anti-vimentin (1:100; rabbit, monoclonal; cat. no. ab16700) were
both purchased from Abcam (Cambridge, UK). The phalloidin-labeled
cytoskeleton staining kit was purchased from Sigma-Aldrich (St.
Louis, MO, USA), the CCK-8 kit from Dojindo Molecular Technologies,
Inc. (Kumamoto, Japan), the plasmid extraction kit from Qiagen GmbH
(Hilden, Germany), the high efficiency transfection (HET) kit from
Shenzhen Biowit Technologies Co., Ltd. (Shenzhen, China), and the
RNA reverse transcription kit from Thermo Fisher Scientific
(Waltham, MA, USA).
EnVision immunohistochemistry
Ten normal tongue mucosa specimens were used as
negative controls. TSCC specimens (provided by Professor Tiejun Li
from Peking University Stomatological Hospital) with pathologically
definitive diagnosis were collected from 51 cases without
preoperative treatment, averaging 22 years old, and including 30
males and 21 females. Among them, 28 cases had lymph node (LN)
metastasis while the remaining 23 had no LN metastasis. CXCL9 and
CXCR3 expression levels were detected by Envision
immunohistochemistry and intracellular brown particles reflected
positive signals. The semi-quantitative additive score method was
used to quantify positive staining by counting cells in 10 high
power fields randomly in every slide. Specific criteria were: i)
score based on the percentage of positive cells (0, ≤5%; 1, 6–25%;
2, 26–50%; 3, 51–75%; 4, >75%); ii) score based on signal
intensity in most cells (1, slight yellow; 2, yellow; 3, brown).
The product of the score based on the percentage of positive cells
and the signal intensity score were used as the final score:
negative, score of 0 (−); slightly positive, score of 1–4 (+);
positive, score of 5–8 (++); strongly positive, score of 9–12
(+++). Double blinded quantification of the slides by 2 people was
applied and a difference of 3 points between values required
re-examination.
Cell culture
The human TSCC cell line Cal-27 (ATCC; Manassas, VA,
USA) was cultured in high-glucose DMEM with 10% FBS, at 37°C and 5%
CO2. Cells at the logarithmic growth phase were used for
experiments.
Establishment of an hCXCR3 stable cell
line
The hCXCR3 expression vector
pLVX-hCXCR3-mCMV-ZsGreen-PGK-Puro was constructed by DNA synthesis
(1107 bp) with the following primers pLVX-hCXCR3-EcoRI-F,
5′-CCGGAATTCGCCACCATGGTCCTTGAGGTGAGTGACCACCAAG-3′;
pLVX-hCXCR3-BamHI-R,
5′-CGCGGATCCTCACAAGCCCGAGTAGGAGGCCTCTGAG-3′. Prior to large volume
plasmid extraction, the vector was characterized by enzymatic
digestion and sequencing. The lentivirus was packaged by infecting
293T cells and concentrated. Following purification, Cal-27 cells
were infected with the lentivirus under selection by puromycin.
Establishment of an hCXCR3 shRNA
stable cell line in the Cal-27 background
The shCXCR3 (pLVX-ShRNA-puro-hCXCR3) expression
vector was constructed by DNA synthesis with the following primers:
hCXCR3-F,
5′-GATCCGCCTACTGCTATGCCCACATCTTCAAGAGAGATGTGGGCATAGCAGTAGGCTTTTTTCTCGAGG-3′;
hCXCR3-R,
5′-AATTCCTCGAGAAAAAAGCCTACTGCTATGCCCACATCTCTCTTGAAGATGTGGGCATAGCAGTAGGCG-3′.
Cell proliferation assay
Cells at the log-phase were trypsinized, seeded in
96-well plates at a density of 1.2×104 cells/ml in 100
µl, and cultured overnight at 37°C. Then, the culture medium was
aspirated, and supplemented with 100 µl of CXCL9 protein, CXCR3
neutralizing antibodies (2.0 µg/ml), and IgG (2.0 µg/ml) for 24 h,
respectively, followed by the addition of 10 µl of CCK-8 reagent
for 3 h. The absorbance at 450 nm was assessed on a microplate
reader. Each experiment was repeated 3 times.
Scratch assay
Cells at the log-phase were seeded in 6-well plates
at a density of 5×105 cells/well. At a confluency of
95%, 0.5 ml of CXCL9, CXCR3 neutralizing antibodies (2.0 µg/ml) and
IgG (2.0 µg/ml) were added for 3 h. Then, the cell monolayer was
scratched with a 200-µl pipette tip. Detached or dead cells were
washed out with PBS, and serum-free culture media were added. A
total of 3 fields/group were photographed under a fluorescence
microscope at 12, 24, and 48 h, respectively. Cell migration rates
were calculated with ImageJ software 6.0 and each experiment was
repeated three times.
Cell invasion assay
Transwell chambers were used for cell invasion
assays. The upper chambers were seeded with 0.5 ml of Cal-27 cells
in high-glucose complete medium, with the lower chambers filled
with 0.7 ml of high-glucose complete culture medium. For each
group, three different interventions were applied: i) the lower
chamber was supplemented with 100 nM CXCL9; ii) the upper chamber
was supplemented with 2 µg/ml CXCR3 neutralizing antibodies in
combination with 100 nM CXCL9 protein in the lower chamber; and
iii) the upper chamber was supplemented with 2 µg/ml IgG combined
with 100 nM CXCL9 in the lower chamber. After 24 h, the membranes
were separated, stained, sealed, and assessed for invasive cells
under a microscope. Every experiment was repeated 3 times.
Cytoskeleton staining
Cells under routine culture were supplemented with 1
ml of the CXCL9 protein, CXCR3 neutralizing antibodies (2.0 µg/ml),
and IgG (2.0 µg/ml), followed by 4% paraformaldehyde fixation for
48 h. Then, 0.5 ml of 40 µg/ml phalloidin-rhodamine solution was
added for 60 min in the dark in a humid box at room temperature,
and sealed before analysis by fluorescence microscopy. Edge
aggregation of F-actin was assessed in Cal-27 cells.
Cell immunofluorescence staining
Cultured cells were fixed in paraformaldehyde at
4°C, permeabilized with 0.1% Triton X-100, blocked with 10% goat
serum, and incubated with anti-E-cadherin (1:200; rabbit,
polyclonal; cat. no. ab53226) or anti-vimentin (1:100; rabbit,
monoclonal; cat. no. ab16700, both from Abcam) primary antibodies
overnight at 4°C in the dark, in a humid box. This was followed by
incubation with fluorescence-labeled secondary antibody (2 µg/ml,
goat anti-rabbit IgG (H+L); antibody, cat. no. A11012; Thermo
Fisher Scientific) in a shaker incubator. Counterstaining was
performed with 100 µg/ml DAPI before analysis by fluorescence
microscopy.
Western blotting
Cal-27 cells were lysed on ice with lysis buffer.
Proteins in whole cell lysates were separated by SDS-PAGE,
electrically transferred onto Immobilon-P membranes, incubated with
primary antibodies anti-Akt2 (1:500; rabbit, polyclonal; cat. no.
ab8805), anti-p-Akt (1:500; rabbit, polyclonal; cat. no. ab38513),
anti-eIF4E (1:500; rabbit, monoclonal; cat. no. ab33766),
anti-p-eIF4E (1:1,000; rabbit, monoclonal; cat. no. ab76256; all
from Abcam) overnight at 4°C, followed by 1 h of incubation with a
horseradish peroxidase (HRP)-labeled secondary antibody (1:3,000;
goat anti-rabbit IgG-HRP; cat. no. sc2004; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) at room temperature
before chemiluminescence detection (Western Chemiluminescent HRP
substrate; Millipore, Billerica, MA, USA).
Statistical analysis
The Wilcoxon non-parametric test was applied to
assess intergroup differences for enumeration data. One-way ANOVA
was used for measurement data; intergroup differences were analyzed
using the LSD-t method. P<0.05 indicated a statistically
significant difference.
Results
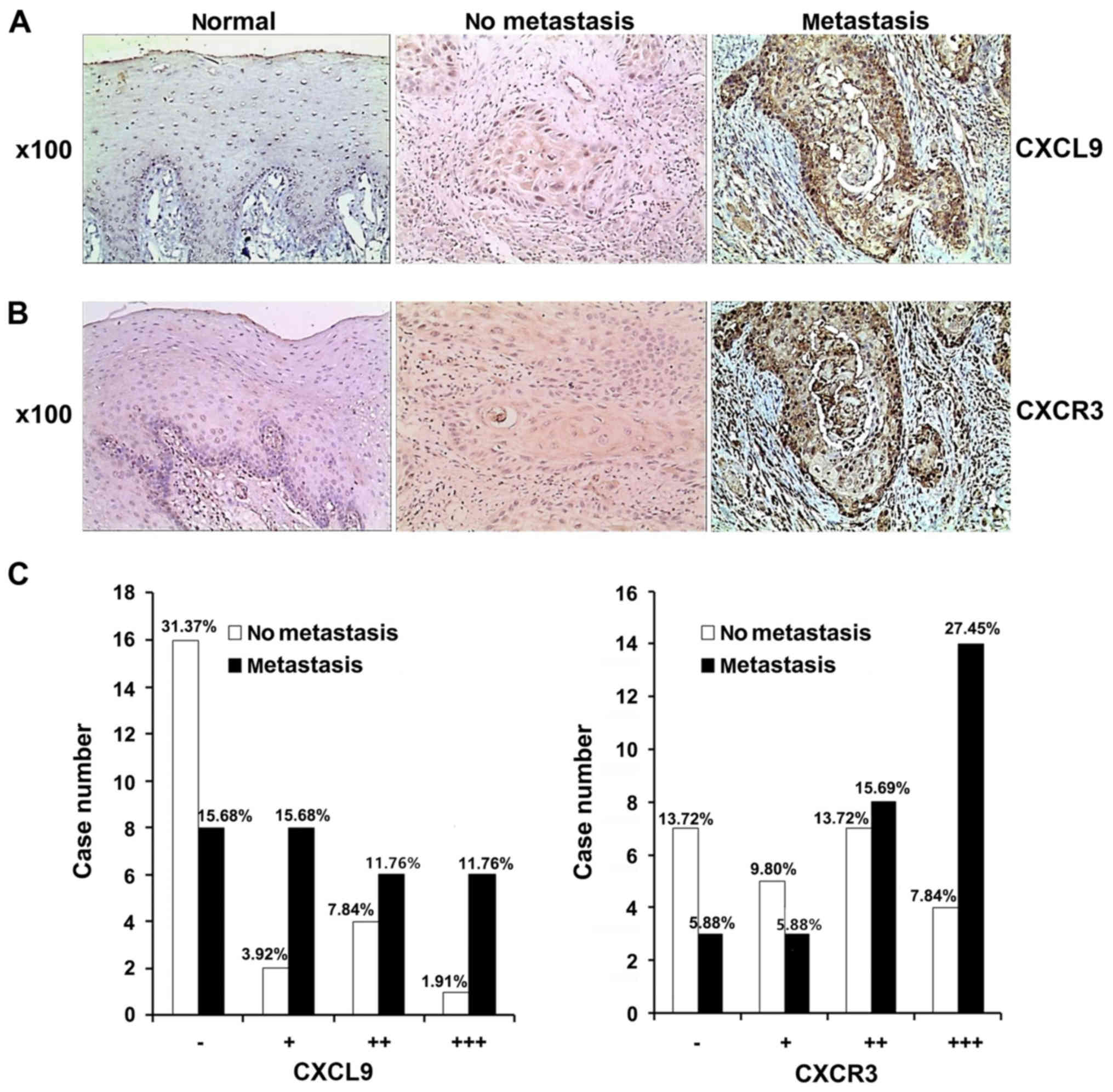
CXCL9 and CXCR3 levels are higher in
TSCC with LN metastasis than in normal tissues or TSCC without LN
metastasis
In tongue mucosa epithelial tissue specimens from
the 10 normal cases, no CXCL9-positive cells were detected, i.e.,
CXCL9 (−). In addition, CXCR3-positive cells were observed in
individual specimens, and positive cells were limited and primarily
located in the basal layer, i.e., CXCR3 (−) (Fig. 1A and B). Of the 51 TSCC specimens,
24 (47.06%) were CXCL9 (−), 10 (19.60%) CXCL9 (+), 10 (19.60%)
CXCL9 (++), and 7 (13.73%) CXCL9 (+++). The 23 TSCC specimens
without cervical LN metastasis were mostly CXCL9 (−)/(+), while the
20 out of 28 with cervical LN metastasis were CXCL9 (+) to (+++).
Statistical analysis revealed significantly higher CXCL9 expression
in TSCC with cervical LN metastasis compared with those without
metastasis (P<0.01) as assessed by the Wilcoxon non-parametric
test (Z= −2.68, p=0.0074) (Fig.
1C).
Of the 51 TSCC specimens, 10 (19.61%) were CXCR3
(−), 8 (15.69%) CXCR3 (+), 15 (29.41%) CXCR3 (++), and 18 (35.29%)
CXCR3 (+++). The 23 TSCC without cervical LN metastasis were mostly
CXCR3 (−) to (+), while the 28 with cervical LN metastasis were
mostly CXCR3 (++) to (+++); a statistically significant difference
(P<0.01) in CXCR3 expression in the LN metastasis group was
reflected in the Wilcoxon nonparametric test (Z= −2.66, P=0.0079)
(Fig. 1C).
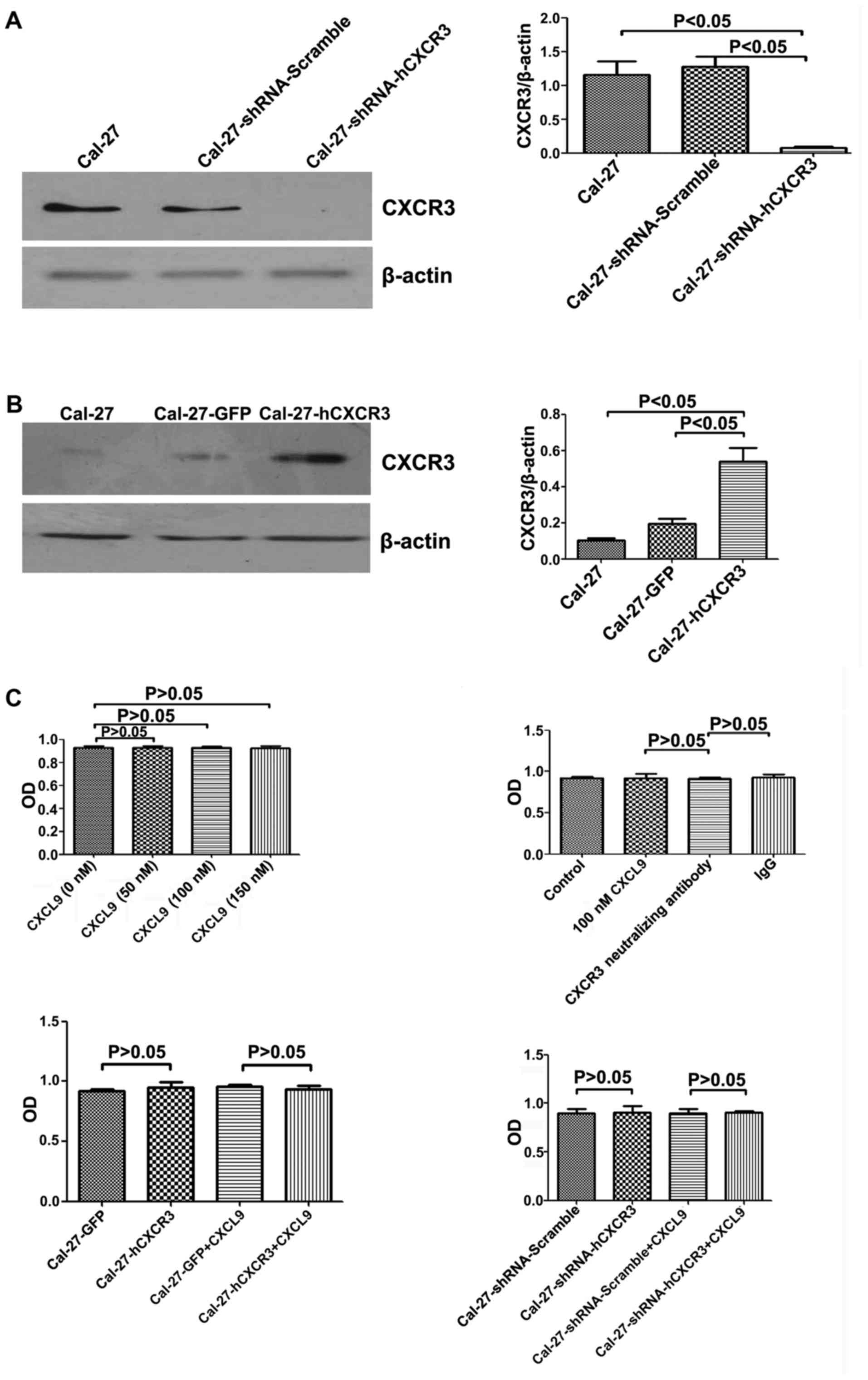
The CXCL9/CXCR3 axis does not promote
proliferation in Cal-27 cells
The hCXCR3 and shCXCR3 expression vectors were
constructed (Fig. 2A and B). Cal-27
cells were seeded in 96-well plates and incubated with CXCL9 at 0,
50, 100, and 150 nM, respectively, for 24 h, followed by 3 h of
incubation with CCK-8 reagent at room temperature before absorbance
assessment at 450 nm. Compared with the control group, no
statistical difference (P>0.05) was detected in terms of cell
proliferation (Fig. 2C).
CXCR3 neutralizing antibodies did not promote Cal-27
cell proliferation. Cells treated for 24 h with 100 nM CXCL9 and
CXCR3 neutralizing antibodies (2.0 µg/ml) or IgG (2.0 µg/ml)
exhibited no statistically significant differences (P>0.05) in
proliferation (Fig. 2C).
CXCR3 overexpression did not affect Cal-27 cell
proliferation. Cal-27-GFP and Cal-27-CXCR3 cells were incubated for
24 h with 100 nM CXCL9, followed by 3 h of incubation with CCK-8
reagent at room temperature before absorbance assessment at 450 nm.
Compared with the control group, cell proliferation exhibited no
statistically significant difference (P>0.05) (Fig. 2C).
Low CXCR3 expression was not related to Cal-27 cell
proliferation. Cal-27-shRNA-scramble and Cal-27-shRNA-hCXCR3
transfected cells were treated for 24 h with 100 nM CXCL9, followed
by 3 h of incubation with CCK-8 reagent at room temperature before
absorbance assessment at 450 nm. Compared with the control group,
cell proliferation exhibited no statistically significant
difference (P>0.05) (Fig. 2C).
The aforementioned data indicated that CXCL9 did not promote Cal-27
cell proliferation.
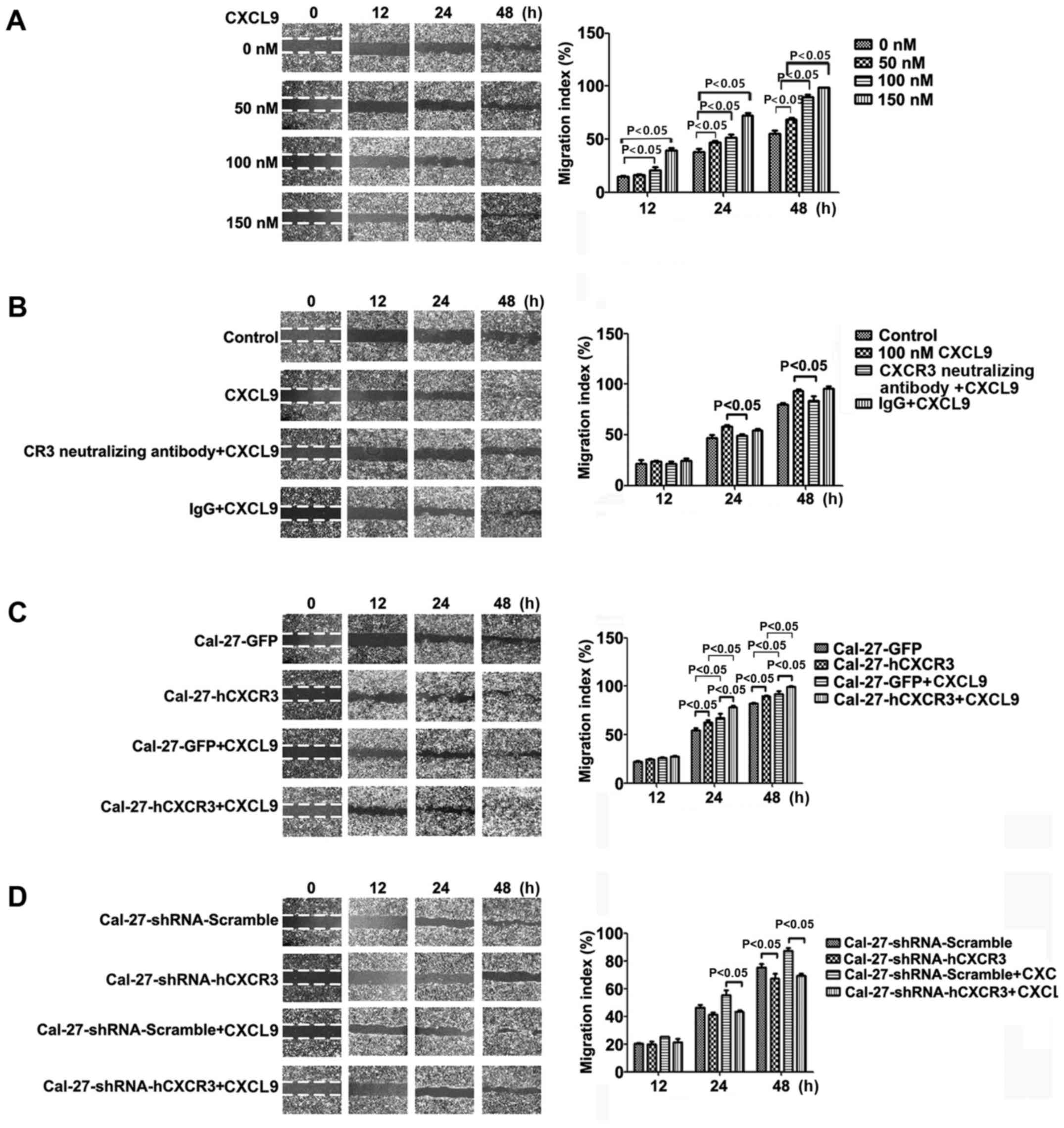
The CXCL9/CXCR3 axis promotes
migration in Cal-27 cells
Cal-27 cells seeded in 6-well plates were cultured
for 3 h with CXCL9 at 0, 50, 100 and 150 nM, respectively; then the
cell monolayer was scratched. After 0, 12, 24 and 48 h of
incubation, respectively, the samples were imaged, and migration
areas were determined. Compared with the control group, a higher
migration rate of Cal-27 cells was observed after treatment with
various concentrations of CXCL9 (except for 0 and 50 nM after 12
h), with statistical significance (P<0.05) (Fig. 3A). Compared with the CXCL9 group,
100 nM CXCL9 administered in combination with CXCR3 neutralizing
antibodies (2.0 µg/ml) and IgG (2.0 µg/ml) resulted in
significantly decreased migration at 48 h, from 93.7±6.13% to
83.8±3.40% (P<0.05) (Fig.
3B).
CXCR3 overexpression enhanced CXCL9-stimulating
effects on Cal-27 cell migration. Cal-27-GFP and Cal-27-CXCR3 cells
were treated for 3 h with 100 nM CXCL9, followed by scratching. The
samples were imaged at 0, 12, 24 and 48 h, respectively, to
evaluate the migration areas. Compared with the control group,
treatment resulted in significantly increased migration areas
(P<0.05) (Fig. 3C).
Low CXCR3 expression attenuated the effect of CXCL9
on Cal-27 cell migration. Cal-27-shRNA-Scramble and
Cal-27-shRNA-hCXCR3 cells were scratched and imaged at 0, 12, 24,
and 48 h of culture, respectively. Compared with the control group,
silencing of CXCR3 resulted in significantly reduced migration
areas (P<0.05) (Fig. 3D). These
results revealed that CXCL9 promoted Cal-27 migration, an effect
closely related to its receptor CXCR3.
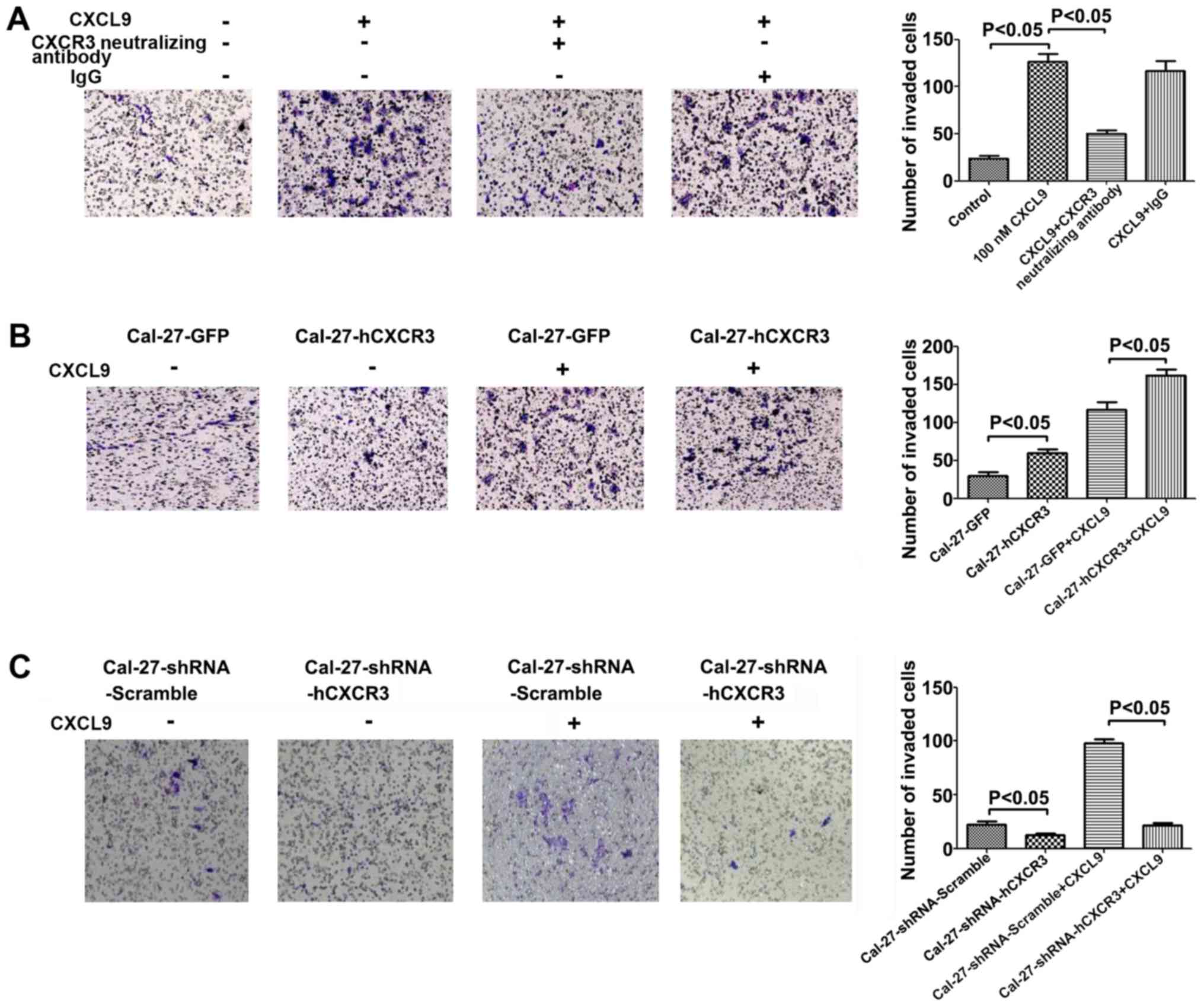
The CXCL9/CXCR3 axis promotes invasion
in Cal-27 cells
CXCR3 expressing Cal-27 cells seeded in invasion
chambers were cultured for 24 h with 100 nM CXCL9 in combination
with CXCR3 neutralizing antibodies (2.0 µg/ml) or IgG (2.0 µg/ml).
After staining with 0.1% crystal violet, the specimens were imaged.
Compared with the control group, a significant increase in the
number of invasive cells was observed (P<0.05). Compared with
the CXCL9 group, treatment with CXCL9 and CXCR3 neutralizing
antibodies resulted in significantly reduced cell invasion
(P<0.05) (Fig. 4A).
CXCR3 overexpression positively regulated the
inducive effect of CXCL9 on invasion in Cal-27 cells. Cal-27-GFP
and Cal-27-CXCR3 cells were seeded in invasion chambers and treated
for 24 h with 100 nM CXCL9, followed by staining with 0.1% crystal
violet. Compared with the control group, a significant increase in
the number of invasive cells was observed (P<0.05) (Fig. 4B).
Low CXCR3 expression inhibited the inducive effect
of CXCL9 on invasion in Cal-27 cells. Cal-27-shRNA-Scramble and
Cal-27-shRNA-hCXCR3 cells were seeded in invasion chambers, and
treated for 24 h with 100 nM CXCL9, followed by staining with 0.1%
crystal violet and imaging. Compared with the control group, a
significant decrease in the number of invasive cells was observed
(P<0.05) (Fig. 4C). In summary,
CXCL9 enhanced the invasive capability of Cal-27 cells, an effect
closely related to its receptor CXCR3.
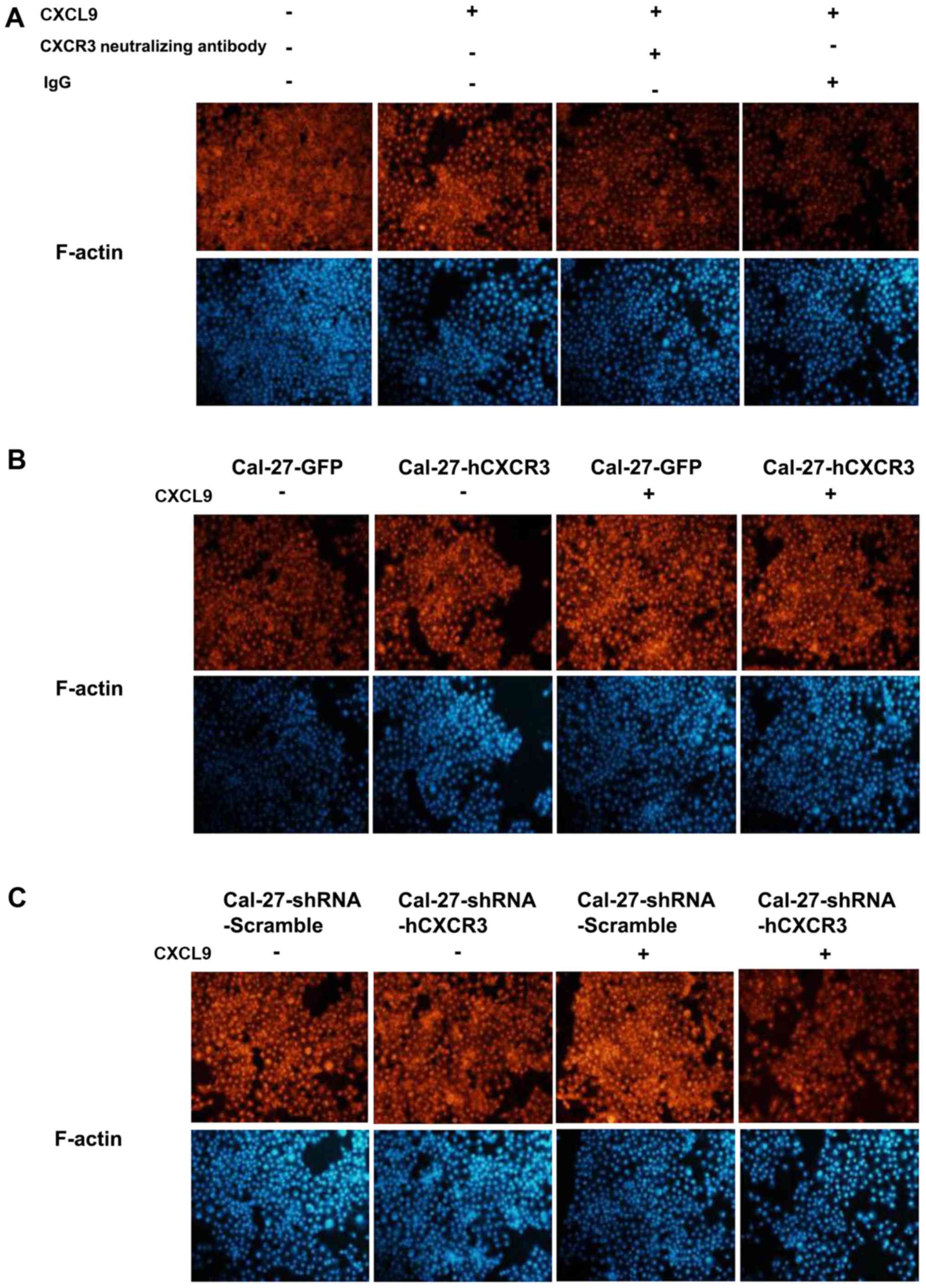
The mix/CXCR3 axis modifies the Cal-27
cytoskeleton
Routinely cultured Cal-27 cells were coated on
slides, and incubated for 48 h with 100 nM CXCL9, CXCR3
neutralizing antibodies (2.0 µg/ml) and IgG (2.0 µg/ml), and
incubated with phalloidin-rhodamine for 60 min before imaging.
Compared with the control group, edge aggregation of F-actin,
specifically binding to phalloidin-rhodamine, was evident. Compared
with the CXCL9 group, addition of CXCR3 neutralizing antibodies led
to significantly decreased edge aggregation of F-actin (Fig. 5A).
CXCR3 overexpression enhanced the inducive effect of
CXCL9 on edge aggregation of F-actin. Cal-27-GFP and Cal-27-CXCR3
cells were coated on slides and cultured routinely for 48 h with
CXCL9. After blocking with 1% BSA, incubation was performed with
rhodamine-labeled phalloidin for 60 min before imaging. Compared
with the Cal-27-GFP group, edge aggregation of F-actin was improved
in Cal-27-CXCR3 cells (Fig.
5B).
Reduced CXCR3 expression attenuated the inducive
effect of CXCL9 on edge aggregation of F-actin in Cal-27 cells.
Cal-27-shRNA-Scramble and Cal-27-shRNA-hCXCR3 cells were seeded on
slides and cultured routinely for 48 h with CXCL9. After blocking
with 1% BSA, incubation was performed with 40 µg/ml
rhodamine-labeled phalloidin for 60 min before imaging. Compared
with the shRNA-Scramble group, edge aggregation of F-actin was
reduced in Cal-27-shRNA-hCXCR3 cells (Fig. 5C). Overall, CXCL9 enhanced the edge
aggregation of F-actin in Cal-27 cells, an effect closely related
to its receptor.
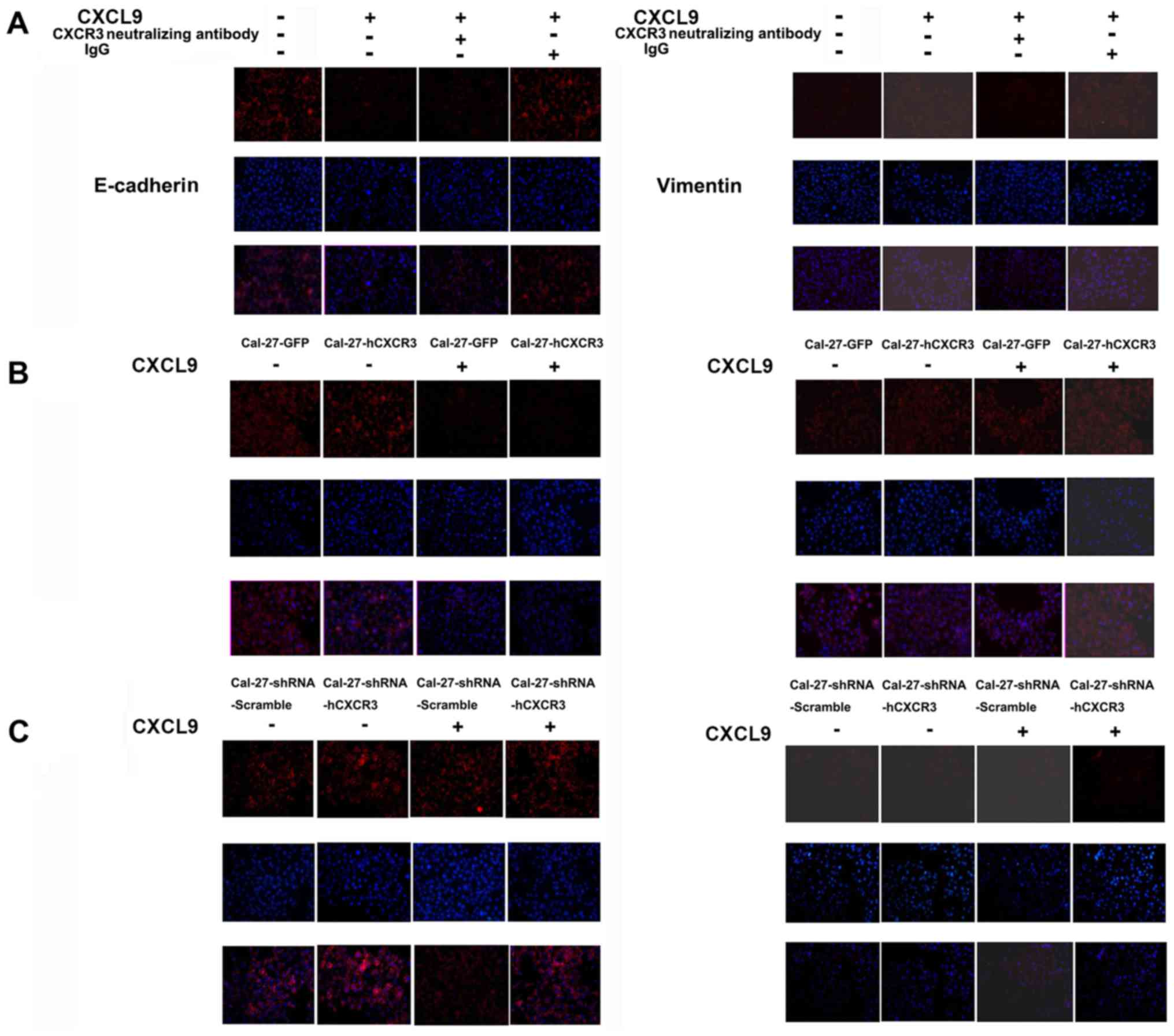
The CXCL9/CXCR3 axis induces EMT in
Cal-27 cells
Cal-27 cells from the test and control groups were
coated on slides and cultured routinely. Both groups underwent the
same process, except for 24-h treatment with 100 nM CXCL9 in the
test group. Cells were fixed with paraformaldehyde, permeabilized
for 20 min with 0.1% Triton X-100, blocked for 2 h in goat serum,
and incubated with anti-E-cadherin or anti-vimentin primary
antibodies, followed by rhodamine-labeled secondary antibodies and
DAPI staining before imaging. Compared with the control group,
E-cadherin expression in Cal-27 cells was significantly reduced in
the CXCL9 group while vimentin levels were increased. Compared with
the CXCL9 group, addition of CXCR3 neutralizing antibodies
attenuated the reduction of E-cadherin expression as well as
vimentin level increase. For western blotting, Cal-27 cells from
the CXCL9 and control groups were lysed, and proteins in the
resulting lysates were separated by SDS-PAGE and
electrophoretically transferred onto Immobilon-P membranes. The
membranes were then incubated with primary antibodies
(anti-E-cadherin and anti-vimentin) overnight at 4°C, followed by 1
h of incubation with HRP-labeled secondary antibodies at room
temperature before chemiluminescent detection. The results were
consistent with the aforementioned immunofluorescence findings
(Fig. 6A and D).
CXCR3 overexpression enhanced the inducive effect of
CXCL9 on expression increase of vimentin and E-cadherin level
decrease in Cal-27 cells. Cal-27-GFP and Cal-27-CXCR3 cells were
treated for 24 h with 100 nM CXCL9, fixed with paraformaldehyde and
permeabilized for 20 min with 0.1% Triton X-100. After blocking for
2 h in goat serum, the samples were sequentially incubated with
primary antibodies (anti-E-cadherin or anti-vimentin) and
rhodamine-labeled secondary antibodies, followed by DAPI
counterstaining before imaging. Compared with the control group,
E-cadherin expression in Cal-27 cells was reduced significantly
whereas vimentin levels were enhanced. For western blotting,
Cal-27-GFP and Cal-27-CXCR3 cells from the CXCL9 and control groups
were lysed. Proteins in whole cell lysates were separated by
SDS-PAGE, and electrophoretically transferred onto Immobilon-P
membranes. The latter were incubated with primary antibodies
(anti-E-cadherin and anti-vimentin) overnight at 4°C, followed by 1
h of incubation with HRP-labeled secondary antibodies at room
temperature before chemiluminescent detection. The results were
consistent with the immunofluorescence findings (Fig. 6B and D).
Silencing of CXCR3 attenuated the inducive effect of
CXCL9 on E-cadherin level reduction and vimentin expression
increase in Cal-27 cells. Cal-27-shRNA-Scramble and
Cal-27-shRNA-hCXCR3 cells were treated for 24 h with 100 nM CXCL9.
After sequential incubation with primary antibodies
(anti-E-cadherin or anti-vimentin) and rhodamine-labeled secondary
antibodies, the samples were treated with DAPI before imaging.
Compared with the Scramble group, CXCR3 silencing did not result in
E-cadherin reduction in Cal-27 cells but it did decrease the
expression level of vimentin. For western blotting,
Cal-27-shRNA-Scramble and Cal-27-shRNA-hCXCR3 cells of the CXCL9
and control groups were lysed. Proteins in whole cell lysates were
then separated by SDS-PAGE and electrophoretically transferred onto
Immobilon-P membranes. This was followed by sequential incubation
with primary antibodies (anti-E-cadherin or vimentin) overnight at
4°C, and HRP-labeled secondary antibodies at room temperature 1 h
before chemiluminescent detection. The results were consistent with
the immunofluorescence findings (Fig.
6C and D). Hence, in Cal-27 cells, CXCL9 downregulated
E-cadherin, an epithelial cell marker, while concomitantly
upregulating vimentin, a mesenchymal marker, an activity closely
related to its receptor CXCR3.
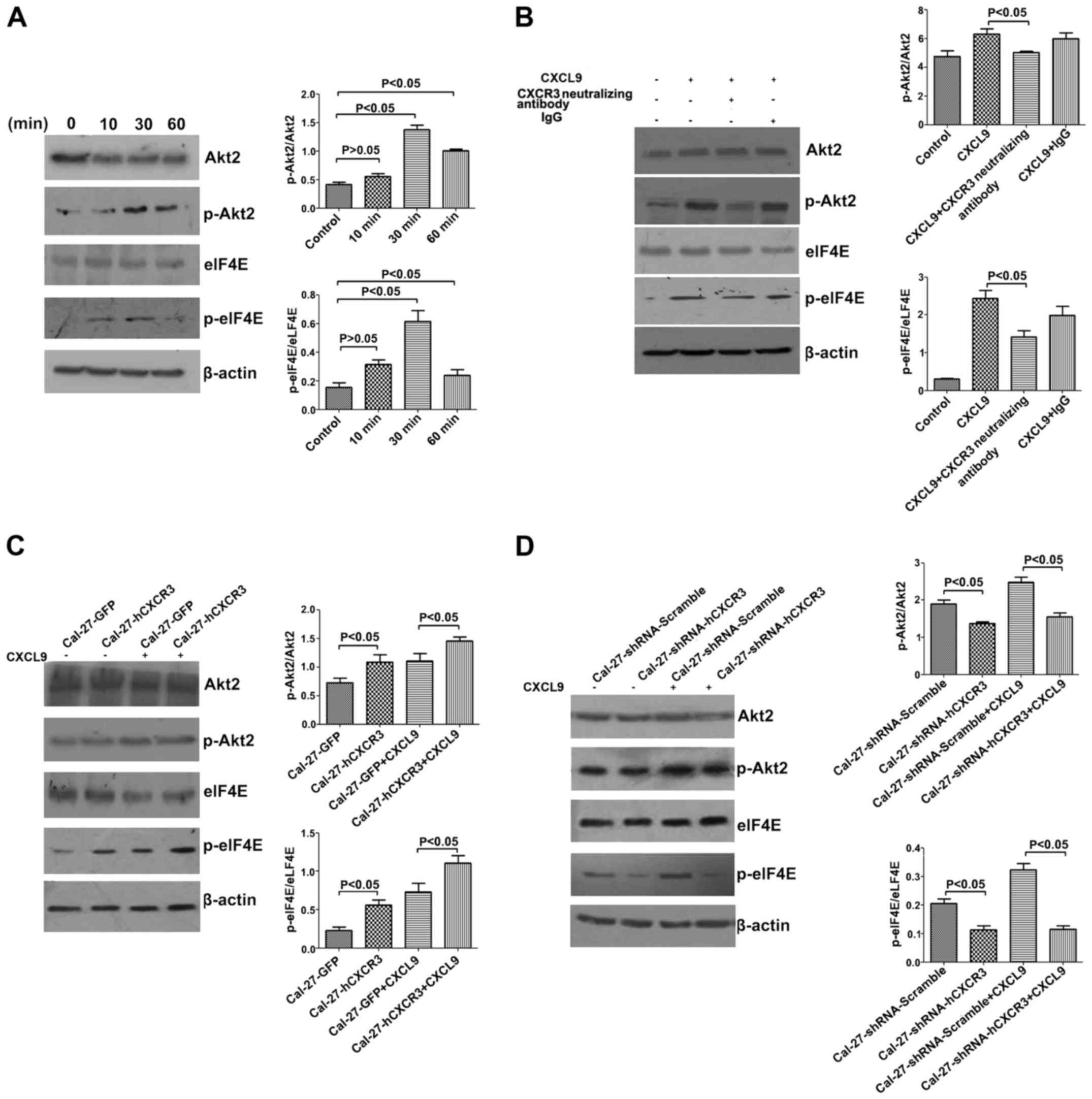
The CXCL9/CXCR3 axis activates Akt signaling in
Cal-27 cells. To further explore how the CXCL9/CXCR3 axis induces
EMT in Cal-27, we treated Cal-27 cells with 100 nM CXCL9, and whole
cell total protein was extracted at 10, 30, and 60 min,
respectively, for western blotting. The results indicated an
induction of phosphorylated Akt2 and eIF4E by CXCL9, which peaked
at 30 min, with statistically significant differences (P<0.05)
(Fig. 7A). According to the
aforementioned findings, the cells were incubated with 100 nM
CXCL9, CXCR3 neutralizing antibodies (2.0 µg/ml) and IgG (2.0
µg/ml). Compared with the CXCL9 group, addition of CXCR3
neutralizing antibodies resulted in decreased relative amounts of
p-Akt2 (from 6.28±0.39 to 5.01±0.07) and p-eIF4E (from 2.44±0.21 to
1.42±0.16), with statistically significant differences (P<0.05)
(Fig. 7B).
Moreover, CXCR3 overexpression enhanced the inducive
effect of CXCL9 on p-Akt2 and p-eIF4E in Cal-27 cells. Cal-27-GFP
and Cal-27-CXCR3 cells were stimulated for 30 min with 100 nM
CXCL9, and total cell protein was extracted for western blotting.
Compared with the control group, relative amounts of p-Akt2 in
Cal-27-CXCR3 cells were significantly increased, from 1.11±0.12 to
1.46±0.06, as well as p-eIF4E, from 0.73±0.11 to 1.11±0.09
(P<0.05) (Fig. 7C).
Silencing of CXCR3 attenuated the inducive effect of
CXCL9 on p-Akt2 and p-eIF4E in Cal-27 cells. Cal-27-shRNA-Scramble
and Cal-27-shRNA-hCXCR3 cells were stimulated for 30 min with 100
nM CXCL9, and total cell protein was extracted for western
blotting. Relative amounts of p-Akt2 were decreased from 2.48±0.14
in the control group to 1.55±0.10 in the interference group, as
well as relative p-eIF4E, from 0.32±0.03 to 0.12±0.02, with
statistically significant intergroup differences (P<0.05)
(Fig. 7D). In summary, the
CXCL9/CXCR3 axis upregulated the phosphorylated forms of Akt2 and
eIF4E in Cal-27 cells.
Discussion
TSCC invasion and metastasis is a comprehensive,
multifactorial, consecutive, and multistep biological process. The
underlying molecular mechanisms remain incompletely understood.
However, the critical roles of chemokines in tumor progression are
increasingly recognized along with the association of inflammation
with tumors. In the tumor microenvironment, paracrine or autocrine
chemokines bind to their cognate receptors on the tumor cell
surface, induce cytoskeleton rearrangement, and facilitate tight
attachment of tumor cells to lymphatic endothelial cells, promoting
directional migration (14). The
cytoskeleton is highly flexible and restructurable, mainly
involving the balance between aggregation and dispersion of
cellular F-actin, as the prerequisite for cell movement and
migration (15). EMT as
cytoskeleton reconstruction, is the primary factor promoting
invasion and metastasis in the tumor microenvironment (16). Occurrence of EMT involves
alterations of multiple biological behaviors, including the
downregulation of the epithelial marker E-cadherin, upregulation of
the mesenchymal marker vimentin, and cytoskeleton rearrangement
(17,18). Ploenes et al (19) revealed that the chemokine CCL18
promoted invasion and metastasis via EMT in the lung cancer A549
cell line. Another study reported that SDF-1, upon specific binding
to its receptor CXCR4, enhanced pancreatic cancer metastasis via
EMT (20). Protein levels of the
chemokine CXCL9 secreted by carcinoma-associated fibroblasts (CAFs)
was significantly higher in TSCC than those of fibroblasts in the
normal oral mucosa; meanwhile, the chemokine CXCL9 and its receptor
CXCR3 were highly expressed in TSCC tissues, and correlated with
cervical LN metastasis (21),
suggesting a critical role for CXCL9 and its receptor CXCR3 in
tumor invasion and metastasis. The underlying mechanisms remain
unknown.
CXCR3 is highly expressed in a wide range of poorly
differentiated neoplasms and malignant cells (22–26),
including prostate cancer, lung cancer, breast cancer and melanoma,
and mediates tumor cell migration and invasion through interaction
with its ligand CXCL9 (27). The
current study detected high CXCR3 and CXCL9 levels in TSCC tissues,
and significant expression differences due to cervical LN
metastasis. Moreover, CXCL9 promoted cell migration and invasion in
Cal-27 cells. To assess whether the effect of CXCL9 on the tumor
Cal-27 cell line was mediated by its receptor CXCR3,
CXCR3-overexpressing Cal-27 cells were incubated with CXCL9, and
improved migration and invasion was observed. To further confirm
the regulatory effect of CXCR3 on Cal-27 cell invasion and
metastasis, CXCR3-knockdown Cal-27 cells were treated with CXCL9,
and a reduction at 48 h of the migration rate was observed, from
87.2±5.23% in controls to 69.4±7.56% in the interference group,
with a reduction of invasive cells from 98.25±3.90 to 21.25±2.78,
indicating a close correlation between CXCL9 and its receptor CXCR3
in tumor cell migration and invasion, as well as the involvement of
the CXCL9/CXCR3 axis in the regulation of tumor cell migration and
invasion. Tumor cell migration and invasion is the initial stage of
distal metastasis. Ohshima et al (28) demonstrated high CXCR3 expression in
gastric cancer with LN metastasis, indicating the possibility that
CXCR3 promotes LN metastasis. In a metastatic lymph node melanoma
model, Kawada et al (29)
detected consecutive expression of CXCR3 in the melanoma B16F10
cell line, and the addition of the ligand CXCL9, CXCL10 or CXCL11
resulted in significantly enhanced cell invasion capability.
In order to explore the specific molecular
mechanisms of the CXCL9/CXCR3 axis in inducing TSCC cell migration
and invasion, Cal-27 cells were stimulated by CXCL9, followed by
staining with rhodamine-labeled phalloidin. As previously revealed,
CXCL9 stimulation resulted in edge aggregation of F-actin and
cytoskeleton rearrangement. The cytoskeleton is highly flexible and
restructurable, mainly involving the balance between the
aggregation and dispersion of cellular F-actin, which is the
prerequisite for cell movement and migration. Addition of CXCL9 to
CXCR3-overexpressing Cal-27 cells led to increased edge aggregation
of F-actin and cytoskeleton rearrangement. This effect was not
induced by CXCL9 in the CXCR3-knockdown Cal-27 cell line,
reflecting the close relationship between CXCL9 and its receptor
CXCR3 in tumor cytoskeleton rearrangement, as well as the
involvement of CXCL9/CXCR3 in the alteration of the TSCC
cytoskeleton. These data corroborated previous findings
demonstrating that CXCL12/CXCR4 mediates cytoskeleton rearrangement
in intestinal epithelial cells. Cadherin transition is believed to
play an important role in tumor cell EMT and cell movement. In
malignant tumors, especially those poorly differentiated or
metastatic, the epithelial marker E-cadherin is markedly
downregulated while the mesenchymal marker vimentin is upregulated.
The phenotype transition causes loss of intercellular junction in
tumor cells, and hence promotes invasion. In the current study,
immunofluorescence and western blotting indicated that CXCL9
promoted E-cadherin downregulation in Cal-27 cells as well as
vimentin upregulation. Concomitant addition of CXCL9 to
CXCR3-overexpressing Cal-27 cells resulted in significantly reduced
E-cadherin and increased vimentin, while no obvious expression
changes of the two markers were observed in CXCR3-knockdown Cal-27
cells following CXCL9 treatment, suggesting that the regulatory
effect of CXCL9 on EMT phenotype is closely related to its receptor
CXCR3, with the CXCL9/CXCR3 axis mediating EMT occurrence in
TSCC.
In agreement with Li et al (30) who demonstrated that the chemokine
CXCL12/CXCR3 axis modulated EMT in liver cancer, the present study
revealed the CXCL9/CXCR3 axis mediated EMT in TSCC. Binding of
CXCL9 with CXCR3 likely activated the tumor cytosolic Akt signaling
pathway, and induced EMT and cytoskeleton rearrangement, hence
mediating tumor invasion and metastasis. We stimulated Cal-27 cells
with 100 nM CXCL9, and extracted total protein for western blot at
10, 30 and 60 min, respectively. As previously revealed, Akt2 and
eIF4E phosphorylation was observed, peaking at 30 min of treatment,
corroborating Andersson et al and Zhang et al
(31,32). To explore the association of Akt
signaling with CXCR function, CXCR3 neutralizing antibodies were
applied to block CXCR3 activation. Notably, antibody addition
decreased phosphorylated Akt2 levels (P<0.05), indicating that
Akt2 may be downstream of CXCR3. Moreover, a CXCR3-overexpressing
cell line was established to explore whether CXCR3 modulated
biological behaviors in TSCC cells via the Akt signaling pathway.
Addition of CXCL9 to these cells resulted in increased p-Akt-2,
with higher relative amounts from 0.73±0.11 to 1.11±0.09. To
further assess the effect of CXCR3 on the Akt signaling pathway, a
CXCR3-knockdown tumor cell line was generated.
As previously revealed, silencing of CXCR3 resulted
in reduced phosphorylated levels of Akt2, indicating that
CXCL9/CXCR3 likely regulated Cal-27 cell migration, invasion and
EMT via Akt2 signaling. Prior studies revealed close associations
of aberrant Akt2 phosphorylation with tumor proliferation,
progression, invasion and metastasis (33), and considered Akt2 the main
connection between CXCR3 and its downstream molecules. Ma et
al (26) revealed close
associations of CXCR3 with the G protein subunits Gαi and Gαq, with
G protein being the key molecule in Akt activation. In glioma
cells, via the Gαq subunit, CXCR3 activated PI3K/Akt to increase
intracellular calcium ion levels, thus promoting cell proliferation
and chemotaxis (34). In addition,
the chemokine receptor CXCR3 modulated cell migration and
angiogenesis via Akt (35). A study
by Guan et al hinted that CXCR3 regulated
osteocarcinoma-related pain via the PI3K/Akt pathway (36).
eIF4E is highly expressed in most human malignancies
and positively correlated to tumor invasion and metastasis.
Sunavala-Dossabhoy et al (37) reported high expression of eIF4E in
head and neck squamous cell carcinoma (HNSCC), with no
overexpression in normal tissues and benign neoplasms, suggesting
an essential role for eIF4E in the entire process of tumor
development. As a downstream effector molecule of Akt signaling,
eIF4E phosphorylation is the main initiator of related biological
functions. In the current study, stimulation of TSCC cells with
CXCL9 induced eIF4E phosphorylation in Cal-27 cells, while addition
of CXCR3 neutralizing antibodies resulted in reduced eIF4E levels
by blocking CXCR3 activity. Moreover, stimulation of the
CXCR3-overexpressing cell line with CXCL9 enhanced eIF4E
phosphorylation. To further evaluate the regulatory effect of CXCR3
on eIF4E phosphorylation, a CXCR3-knockdown Cal-27 cell line was
established. As previously revealed, CXCR3 knockdown attenuated the
inducive effect of CXCL9 on eIF4E phosphorylation, indicating a
close relationship between CXCL9 and its receptor CXCR3 in the
regulation of eIF4E phosphorylation in tumor cells. As a survival
promoting signal, eIF4E alters the transcription levels of
malignant neoplasm-related mRNAs in various processes, including
cell mitosis, oncogene activation, autocrine improvement, cell
survival, apoptosis resistance, and communication with the
extracellular environment (38).
In summary, the CXCL9/CXCR3 axis is capable of
activating Akt signaling in TSCC cells, inducing EMT and
cytoskeleton rearrangement, and promoting cancer cell invasion and
metastasis.
Three spliced isoforms of CXCR3 receptors has been
described, including CXCR3-A (classic CXCR3), CXCR3-B and
CXCR3-alt. CXCR3-A is known as a pro-tumor receptor whereas CXCR3-B
exhibits antitumor properties (39). Meanwhile, CXCR3 splice variants and
its ligands (CXCL9, CXCL10, CXCL11) can activate different
signaling pathways in different diseases (40). In the present study, we focused on
the mechanisms of the CXCL9/CXCR3 axis promoting invasion and
metastasis in tongue squamous cell carcinoma. Notably, the
investigation of the functions of other CXCR3 variants and their
different ligands in the development and progression of TSCC are
warranted in a future study.
The chemokine CXCL9 and its receptor CXCR3 are
closely related to migration and invasion in TSCC cells, possibly
via ligand binding with its receptor, activating cytosolic Akt
signaling, and inducing EMT and skeleton rearrangement, thus
mediating tumor cell invasion and metastasis. The present study
provided new insights into the role of the CXCL9/CXCR3 axis in
inducing cell EMT as well as invasion and metastasis regulation in
TSCC. In addition, we elucidated the molecular mechanism of tumor
cell invasion and metastasis in relation to chemokines, providing a
new basis for clinical prevention and treatment in oral cancer.
Acknowledgements
We thank Professor Tiejun Li (Peking University
Stomatological Hospital) for providing clinical specimens. This
study was supported by the National Natural Science Foundation of
China (grant nos. 81660448 and 81360401), the Special Health
Technical Personnel Training program of Yunnan, China (grant no.
L-201612), and the Natural Science Foundation of Yunnan, China
(grant no. 2017FE468-006).
Glossary
Abbreviations
Abbreviations:
|
AA
|
amino acids
|
|
CAFs
|
carcinoma-associated fibroblasts
|
|
EMT
|
epithelial-mesenchymal transition
|
|
HNSCC
|
head and neck squamous cell
carcinoma
|
|
HRP
|
horseradish peroxidase
|
|
LN
|
lymph node
|
|
PI3K
|
phosphatidylinositol 3 kinase
|
|
TSCC
|
tongue squamous cell carcinoma
|
References
|
1
|
Kademani D, Bell RB, Schmidt BL,
Blanchaert R, Fernandes R, Lambert P and Tucker WM; American
Association of Oral and Maxillofacial Surgeons Task Force on Oral
Cancer, : Oral and maxillofacial surgeons treating oral cancer: A
preliminary report from the American Association of Oral and
Maxillofacial Surgeons Task Force on Oral Cancer. J Oral Maxillofac
Surg. 66:2151–2157. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kamangar F, Dores GM and Anderson WF:
Patterns of cancer incidence, mortality, and prevalence across five
continents: Defining priorities to reduce cancer disparities in
different geographic regions of the world. J Clin Oncol.
24:2137–2150. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang YF, Guo M and Zhang J: Nursing of
tissue defect after radical resection of tongue cancer with forearm
flap transplantation. Chin J Aesthetic Med. 21:514–515. 2012.(In
Chinese).
|
|
4
|
Landis SH, Murray T, Bolden S and Wingo
PA: Cancer statistics, 1999. CA Cancer J Clin. 49:8–31. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Steeg PS: Tumor metastasis: Mechanistic
insights and clinical challenges. Nat Med. 12:895–904. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Deryugina EI and Quigley JP: Matrix
metalloproteinases and tumor metastasis. Cancer Metastasis Rev.
25:9–34. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Balkwill F: Cancer and the chemokine
network. Nat Rev Cancer. 4:540–550. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zlotnik A and Yoshie O: Chemokines: A new
classification system and their role in immunity. Immunity.
12:121–127. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rollins BJ: Chemokines. Blood. 90:909–928.
1997.PubMed/NCBI
|
|
10
|
Strieter RM, Burdick MD, Mestas J,
Gomperts B, Keane MP and Belperio JA: Cancer CXC chemokine networks
and tumour angiogenesis. Eur J Cancer. 42:768–778. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vandercappellen J, Van Damme J and Struyf
S: The role of CXC chemokines and their receptors in cancer. Cancer
Lett. 267:226–244. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu L, Callahan MK, Huang D and Ransohoff
RM: Chemokine receptor CXCR3: An unexpected enigma. Curr Top Dev
Biol. 68:149–181. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bodnar RJ, Yates CC and Wells A: IP-10
blocks vascular endothelial growth factor-induced endothelial cell
motility and tube formation via inhibition of calpain. Circ Res.
98:617–625. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Friedl P and Alexander S: Cancer invasion
and the microenvironment: Plasticity and reciprocity. Cell.
147:992–1009. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Savagner P: Epithelial-mesenchymal
transitions: From cell plasticity to concept elasticity. Curr Top
Dev Biol. 112:273–300. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jung HY, Fattet L and Yang J: Molecular
pathways: Linking tumor microenvironment to epithelial-mesenchymal
transition in metastasis. Clin Cancer Res. 21:962–968. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nakaya Y and Sheng G: EMT in developmental
morphogenesis. Cancer Lett. 341:9–15. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tsai JH and Yang J: Epithelial-mesenchymal
plasticity in carcinoma metastasis. Genes Dev. 27:2192–2206. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ploenes T, Scholtes B, Krohn A, Burger M,
Passlick B, Müller-Quernheim J and Zissel G: CC-chemokine ligand 18
induces epithelial to mesenchymal transition in lung cancer A549
cells and elevates the invasive potential. PLoS One. 8:e530682013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li X, Ma Q, Xu Q, Liu H, Lei J, Duan W,
Bhat K, Wang F, Wu E and Wang Z: SDF-1/CXCR4 signaling induces
pancreatic cancer cell invasion and epithelial-mesenchymal
transition in vitro through non-canonical activation of Hedgehog
pathway. Cancer Lett. 322:169–176. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shao S: The role of chemokine Mig and
CXCR3 in invasion and metastasis of oral squamous cell carcinoma of
tongue. Kunming Medical University. 2013.(In Chinese).
|
|
22
|
Li L, Chen J, Lu ZH, Yu SN, Luo YF, Zhao
WG, Ma YH and Jia CW: Significance of chemokine receptor CXCR3
expression in breast cancer. Zhonghua Bing Li Xue Za Zhi. 40:85–88.
2011.(In Chinese). PubMed/NCBI
|
|
23
|
Wu Q, Dhir R and Wells A: Altered CXCR3
isoform expression regulates prostate cancer cell migration and
invasion. Mol Cancer. 11:32012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jenkins MH, Brinckerhoff CE and Mullins
DW: CXCR3 signaling in BRAFWT melanoma increases IL-8 expression
and tumorigenicity. PLoS One. 10:e01211402015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li Y, Reader JC, Ma X, Kundu N, Kochel T
and Fulton AM: Divergent roles of CXCR3 isoforms in promoting
cancer stem-like cell survival and metastasis. Breast Cancer Res
Treat. 149:403–415. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ma B, Khazali A and Wells A: CXCR3 in
carcinoma progression. Histol Histopathol. 30:781–792.
2015.PubMed/NCBI
|
|
27
|
Amatschek S, Lucas R, Eger A, Pflueger M,
Hundsberger H, Knoll C, Grosse-Kracht S, Schuett W, Koszik F,
Maurer D, et al: CXCL9 induces chemotaxis, chemorepulsion and
endothelial barrier disruption through CXCR3-mediated activation of
melanoma cells. Br J Cancer. 104:469–479. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ohshima K, Suefuji H, Karube K, Hamasaki
M, Hatano B, Tutiya T, Yamaguchi T, Suzuki K, Suzumiya J and
Kikuchi M: Expression of chemokine receptor CXCR3 and its ligand,
mig, in gastric and thyroid marginal zone lymphomas. Possible
migration and autocrine mechanism. Leuk Lymphoma. 44:329–336. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kawada K, Sonoshita M, Sakashita H,
Takabayashi A, Yamaoka Y, Manabe T, Inaba K, Minato N, Oshima M and
Taketo MM: Pivotal role of CXCR3 in melanoma cell metastasis to
lymph nodes. Cancer Res. 64:4010–4017. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li X, Li P, Chang Y, Xu Q, Wu Z, Ma Q and
Wang Z: The SDF-1/CXCR4 axis induces epithelial-mesenchymal
transition in hepatocellular carcinoma. Mol Cell Biochem.
392:77–84. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Andersson K and Sundler R:
Posttranscriptional regulation of TNFalpha expression via
eukaryotic initiation factor 4E (eIF4E) phosphorylation in mouse
macrophages. Cytokine. 33:52–57. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang B, Ma Y, Guo H, Sun B, Niu R, Ying G
and Zhang N: Akt2 is required for macrophage chemotaxis. Eur J
Immunol. 39:894–901. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pu R, Li WM, Liu D, Mo XM, Tang Y, Liu LX
and Chen BJ: The expression and clinical significance of AKT2,
phosphorylated AKT2 in non-small cell lung cancer. Sichuan Da Xue
Xue Bao Yi Xue Ban. 41:586–589. 2010.(In Chinese). PubMed/NCBI
|
|
34
|
Maru SV, Holloway KA, Flynn G, Lancashire
CL, Loughlin AJ, Male DK and Romero IA: Chemokine production and
chemokine receptor expression by human glioma cells: Role of CXCL10
in tumour cell proliferation. J Neuroimmunol. 199:35–45. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Willox I, Mirkina I, Westwick J and Ward
SG: Evidence for PI3K-dependent CXCR3 agonist-induced degranulation
of human cord blood-derived mast cells. Mol Immunol. 47:2367–2377.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Guan XH, Fu QC, Shi D, Bu HL, Song ZP,
Xiong BR, Shu B, Xiang HB, Xu B, Manyande A, et al: Activation of
spinal chemokine receptor CXCR3 mediates bone cancer pain through
an Akt-ERK crosstalk pathway in rats. Exp Neurol. 263:39–49. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sunavala-Dossabhoy G, Palaniyandi S, Clark
C, Nathan CO, Abreo FW and Caldito G: Analysis of eIF4E and 4EBP1
mRNAs in head and neck cancer. Laryngoscope. 121:2136–2141. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Siddiqui N and Sonenberg N: Signalling to
eIF4E in cancer. Biochem Soc Trans. 43:763–772. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Boyé K, Billottet C, Pujol N, Alves ID and
Bikfalvi A: Ligand activation induces different conformational
changes in CXCR3 receptor isoforms as evidenced by plasmon
waveguide resonance (PWR). Sci Rep. 7:107032017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Berchiche YA and Sakmar TP: CXC chemokine
receptor 3 alternative splice variants selectively activate
different signaling pathways. Mol Pharmacol. 90:483–495. 2016.
View Article : Google Scholar : PubMed/NCBI
|