Introduction
Quinolizidine alkaloids have previously been
demonstrated to exhibit antitumour activities in vitro and
in vivo (1,2). Cytisine, a quinolizidine alkaloid from
Sophora Alopecuraides L., has been widely used for the
treatment of central nervous system diseases. It has previously
been demonstrated that cytisine may significantly inhibit neuronal
apoptosis induced by NMDA exposure by reversing intracellular
Ca2+ overload and balancing the expression levels of
Bcl-2 and Bax (3). Furthermore,
several in vitro studies have also confirmed the antitumour
activities of cytisine. Cytisine was found to inhibit human cancer
cell lines, including HepG2, A549, K562, Ec109, HL-60 and U937
cells (4). Our team confirmed the
molecular mechanisms underlying cytisine-induced apoptosis of HepG2
via a mitochondrial pathway; however, the specific mechanism
underlying endoplasmic reticulum (ER) stress remains to be
elucidated.
Apoptosis is a crucial mechanism for cell clearance.
It is now accepted that cell apoptosis has mainly three apoptotic
signalling pathways, including the mitochondrial, the death
receptor- and the ER-associated pathway (5–7). In
recent years, researchers have discovered that ER stress-mediated
apoptosis is a new signalling pathway for apoptosis (8). The endoplasmic reticulum (ER), a
membrane-bound organelle, plays an essential role in eukaryotic
cell processes, including synthesizing, sorting, assembling,
modifying and trafficking proteins, and maintaining intracellular
Ca2+ homeostasis (9).
The normal functioning of the ER signalling cascades requires high
concentrations of free calcium ions within the ER lumen. Impairment
of Ca2+ homeostasis or other noxious stimuli may trigger
an ER stress response that is manifested by either an unfolded
protein response (UPR) or an ER overload response (EOR) within the
ER lumen (10–13). However, if the stress persists on
the ER for a longer period of time, the ER stress triggers cell
death (typically apoptosis) mainly through three pathways,
including CHOP (14), JNK (15) and caspase-12 (16–18).
Although caspase-12 has been implicated in ER stress-induced
apoptosis in rodents, it is controversial whether similar
mechanisms operate in humans. Recent research has indicated that
caspase-4, which is one of the closest paralogs of rodent
caspase-12, plays an important role in cell death (19). Thus, the present study investigated
the ER stress effects of cytisine on HepG2 cells in relation to
CHOP, JNK and caspase-4.
Additional studies have indicated that cytisine can
be used as an nAChR partial agonist in clinical use as a smoking
cessation aid. Therefore, cytisine is considered to be superior to
nicotine-replacement therapy for smoking cessation (20–22).
Nicotinic acetylcholine receptors (nAChRs) consist of various types
of subunits, such as α2-α10, β2-β4, which are a complex of five
subunits forming hetero- or homo-pentamers that form a central ion
channel. nAChRs were originally identified to be only expressed in
the nervous system and at neuromuscular junctions (muscle type
nAChRs). However, the discovery of widespread expression of nAChRs
in the cell membrane of all mammalian cells, including cancer
cells, suggested its direct role in the development and progression
of cancers (23). Therefore, nAChRs
are viewed as a novel drug target for the prevention and treatment
of various forms of cancers. Recently, increasing evidence has
shown that α-7 nicotinic receptors exhibit the highest permeability
for Ca2+, promoting cell death via a release of
Ca2+ from intracellular stores and intracellular
Ca2+ overloading (24,25).
The major aim of the present study was to elucidate
the the effects of cytisine on ER stress in HepG2 cells in terms of
the intracellular calcium concentration and the three pathways
involved in ER stress, including CHOP, JNK and caspase-4.
Additionally, the present study investigated the effect of cytisine
on the expression of nAchR.
Materials and methods
Instruments and reagents
Cytisine (no. 20111101C; Shanxi River Pharmaceutical
Co., Ltd., Xi'an, China); human hepatocellular carcinoma cell line,
HepG2 (Life Sciences and Environmental Sciences Development Centre
of Harbin University of Commerce, Harbin, China); RPMI-1640 medium
(no. 20130507; Gibco Co., Carlsbad, CA, USA); fetal calf serum
(FCS) (no. 20140206; Zhejiang Tianhang Biotechnology Co., Ltd.,
Zhejiang, China); propidium iodide (PI) (no. P4170; Sigma Co., St.
Louis, MO, USA); fluorescence microscope (Olympus Corporation,
Tokyo, Japan); HCPT (no. H109197; Aladdin Reagent Co. Ltd.,
Shanghai, China); monoclonal antibody of β-actin (no. 141125),
p-JNK (no. A1514) (both from ZSGB-BIO Co., Ltd., Beijing, China);
monoclonal antibody of GADD153 (no. 980787W; Beijing Biosynthesis
Biotechnology Co., Ltd., Beijing, China), JNK (no. 150116; Wanleibo
Co., Ltd., Shanghai, China); caspase-4 test kit (no. 20140526;
Beyotime Institute of Biotechnology, Haimen, China); JEM-1220
transmission electron microscope (JEOL Ltd.); EPICS XL-MCL flow
cytometry (Beckman Coulter, Inc., Brea, CA, USA); 680 enzyme-linked
analyser (Bio-Rad Laboratories, Inc., Hercules, CA, USA);
peroxidase-conjugated AffiniPure goat anti-rabbit IgG (H+L; cat.
no. ZB-2301) and peroxidase-conjugated AffiniPure goat anti-mouse
IgG (H+L; cat. no. ZB-2305) (both from ZSGB-BIO Co., Ltd.).
Effect of cytisine on the
morphological appearance of HepG2 cells
Transmission electron microscopy was used to observe
the morphological appearance of the HepG2 cells after drug
treatment (26). Cells were gently
harvested after trypsin digestion and washed twice with PBS.
Approximately 3×105 cells were treated in single wells
of 6-well plates. The cultured HepG2 cells were exposed to cytisine
at concentrations of 2.5, 5 and 10 mM, or they were exposed to HCPT
at concentrations of 60 µM for 24 h at 37°C with 5% CO2.
Since cytisine and HCPT both belong to the same family of
alkaloids, HCPT was used as the positive drug in the present study
to verify that the experimental method was correct. A blank control
group was added with the same volume of RPMI-1640 culture medium,
and the cells were collected after 24 h. Next, 2% glutaraldehyde
was used to fix the cells for >2 h, and they were immobilized
with osmium acid. After alcohol gradient dehydration, all groups
were treated with epoxy resin embedding, ultrathin sectioning,
uranyl acetate and lead citric acid double staining and then imaged
under the transmission electron microscope.
Apoptosis assays
Quantitative analysis of the apoptosis was evaluated
via flow cytometry with PI staining according to prior guidelines
(27). Cells were gently harvested
after trypsin digestion and washed twice with PBS. The cells were
then centrifuged at 1,000 rpm for 10 min. Cells (1×106)
were treated approximately into a single cell suspension with PBS
solution and fixed with 70% ice-cold ethanol at 4°C overnight.
Afterwards, the cells were washed twice with PBS and stained with a
solution containing 800 µl of PI (PI staining contains sodium
citrate 33.4 mg, PI 5 mg, RNase A 1 mg, joined Triton X-100 0.5 ml)
for 30 min in the dark at room temperature. A minimum of 10,000
cells were maintained for all of the samples. The cells were later
analysed for their DNA content using flow cytometry. By analysing
the DNA histograms, the percentage of the cells in different cell
cycle phases was evaluated using MyltiCycle for Windows 32-bit
software (Beckman Coulter, Inc.). Finally, the cells with
sub-G0/G1 DNA (sub-G1) were
calculated as apoptotic cells. Apoptosis was recorded using the
laser line of 488/630 nm.
Confocal laser scanning microscope
(CLSM) analysis
The accumulation of Ca2+ ions in the
cytoplasm was studied using the fluorescent dye, Fluo3 AM (28). Approximately, 3×105 cells
were seeded per well in 6-well plates and incubated for 24 h before
treatment with various concentrations of cytisine and a further 24
h of incubation at 37°C with 5% CO2. Cytisine at
concentrations of 2.5, 5 and 10 mM or HCPT at a concentration of 60
µM was added, and the plate was incubated at 37°C in 5%
CO2. The HepG2 cells were resuspended with PBS at 1,500
rpm for 10 min after trypsin digestion. After fixation, the cells
were incubated with 200 µl of Fluo3 AM (4 µg/ml) to detect the
intracellular Ca2+ at 37°C at ~30 min. Next, the cells
were washed twice with PBS. The fluorescence emitted by the Fluo3
AM when bound to Ca2+ ions was recorded using the laser
line of 488/570 nm.
Western blot analysis
Cells were harvested after the treatment with test
drugs for 24 h. After the indicated treatments, the HepG2 cells
were harvested and lysed with a cell lysis solution at 4°C for 30
min. Whole cellular proteins were extracted and cytosolic fractions
were prepared following the procedure described by the
manufacturer. Total proteins were quantitatively assayed using the
BCA method. A loading buffer was added to the cytosolic extracts,
and it was boiled for 10 min. Afterwards, 50 µg of protein was
loaded onto a 15% sodium dodecyl sulphate-polyacrylamide gel
electrophoresis (SDS-PAGE) gel and run at 80 V for 30 min and 120 V
for 1 h. Proteins were transferred onto nitrocellulose membranes.
Afterwards, incubation was performed for 1 h in a blocking solution
(5% non-fat dry milk in 20 mM of TBS with 0.1% Tween) at room
temperature. Next, the membranes were gently washed with diluted
TBS-T and subsequently incubated for 24 h with the primary antibody
of GADD153 (1:500), JNK (1:500), p-JNK (1:500) and α7-nAChR (1:500)
at 4°C, respectively. The secondary antibody was added at 1:2,000
dilutions and incubated at room temperature for 2 h. Cell membranes
were removed and coloured with DAB agent (29,30).
Membranes were examined via chemiluminescence detection using a
photographic film. The image was captured using a Tannon gel
imaging system, and the hybrid band was quantitatively analysed
with Gel-Pro Analyzer 3.1 density analysis software.
Caspase activity assay
The enzymatic activity of caspase-4 was determined
using a caspase colourimetric assay kit according to the
manufacturer's protocol. Briefly, the cells were lysed in a lysis
buffer for 15 min in an ice bath. The lysed cells were centrifuged
at 20,000 × g for 10 min, and 50 µl of the protein was incubated
with 40 µl of a reaction buffer and 10 µl of the colourimetric
tetrapeptides, Ac-DEVD-pNA for caspase-4 at 37°C for 1 h.
The optical density of the reaction mixture was quantified
spectrophotometrically at a wavelength of 405 nm. The caspase
enzymatic activity in the cell lysate is directly proportional to
the colour reaction.
Statistical analysis
Differences in proliferation between different cell
lines were analysed using one-way ANOVA. Statistical analysis was
performed using SPSS 19.0 software. (SPSS, Inc., Chicago, IL, USA).
P<0.01 was considered to be a statistically significant
difference.
Results
Effect of cytisine on the
morphological appearance of HepG2 cells
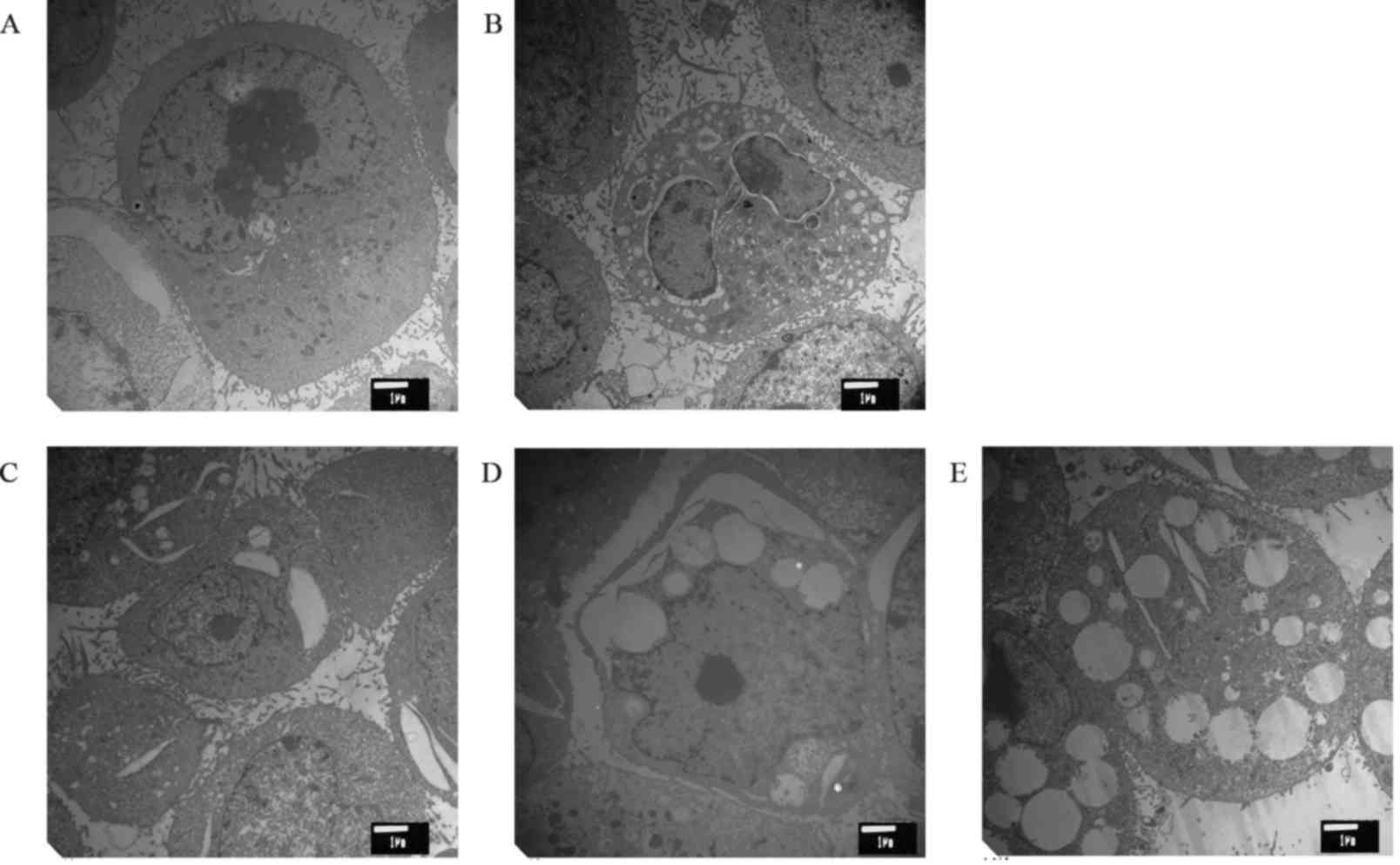
The effect of cytisine on the ultra-structure of the
HepG2 cells is shown in Fig. 1.
Cells from the control group had the following characteristics: a
clear structure, complete cell structure, a small number of
microvilli on the cell surface, large nuclei, abundant euchromatin
and obvious nucleolus. In addition, we also observed many free
ribosomes, and the rough ER and mitochondria were scattered in the
cytoplasm. In the positive control group, the mitochondria were
reduced, and the cell processes were obviously decreased. The ER
swelled and expanded, and typical apoptotic bodies appeared. The
cell surface microvilli decreased or disappeared after different
concentrations of cytisine. Under electron microscopy, the
apoptosis of the cells with cytoplasmic vacuolization was found to
be significant. Retraction of the cytoplasm, chromatin
condensation, expansion of the perinuclear space, rupture of the
membrane, expansion of the ER, and swelling of the mitochondria
were observed in HepG2 cells after drug treatment.
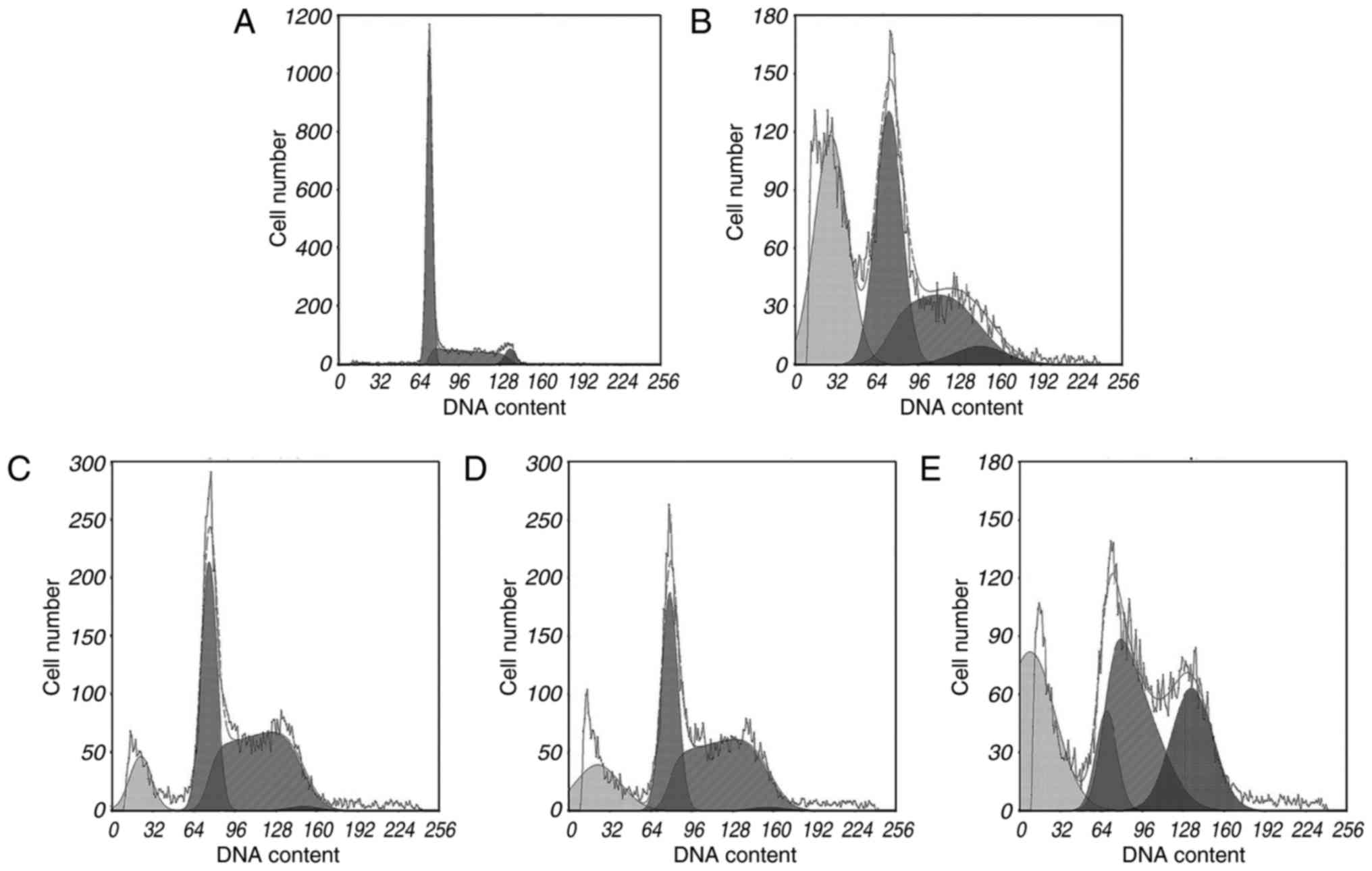
Apoptosis analysis
During apoptosis, activation of certain nucleases
results in DNA degradation. The sub-G1 method relies on
the fact that after DNA fragmentation, there are small fragments of
DNA capable of being eluted following washing in either PBS or a
specific phosphate-citrate buffer. This finding means that after
staining with a quantitative DNA-binding dye, cells that have lost
DNA may take up less stain and may appear to the left of the
G1 peak. Rates of apoptosis were analysed using PI
staining and flow cytometric analysis. The FCM results indicated
that cytisine was capable of inducing apoptosis at concentrations
of 2.5, 5 and 10 mM (Table I). In
particular, HCPT induced a higher percentage (37.79±1.55%) of
apoptosis at 60 mM compared to the control group (P<0.01). As
shown in Fig. 2, treatment with
cytisine increased the percentage of cells in the sub-G1
phase (P<0.01). The preincubation of HepG2 cells with cytisine
(2.5, 5 and 10 mM) significantly increased the sub-G1
cell population (P<0.01). The data demonstrated (Table I and Fig. 2) that cytisine increased HepG2 cell
apoptosis rates in a dose-dependent manner.
 | Table I.Apoptosis rate of HepG2 cells
following treatment with cytisine as detected by flow cytometry
(mean ± SD, n=3). |
Table I.
Apoptosis rate of HepG2 cells
following treatment with cytisine as detected by flow cytometry
(mean ± SD, n=3).
| Groups | Concentration | Number of
cells | Apoptosis rate
(%) |
|---|
| Control | – |
1×106 |
0.97±0.29 |
| HCPT | 60 µmol/l |
1×106 |
37.79±1.55a |
| Cytisine | 2.5 mmol/l |
1×106 |
10.83±0.45a |
|
| 5 mmol/l |
1×106 |
16.98±1.42a |
|
| 10 mmol/l |
1×106 |
34.28±1.11a |
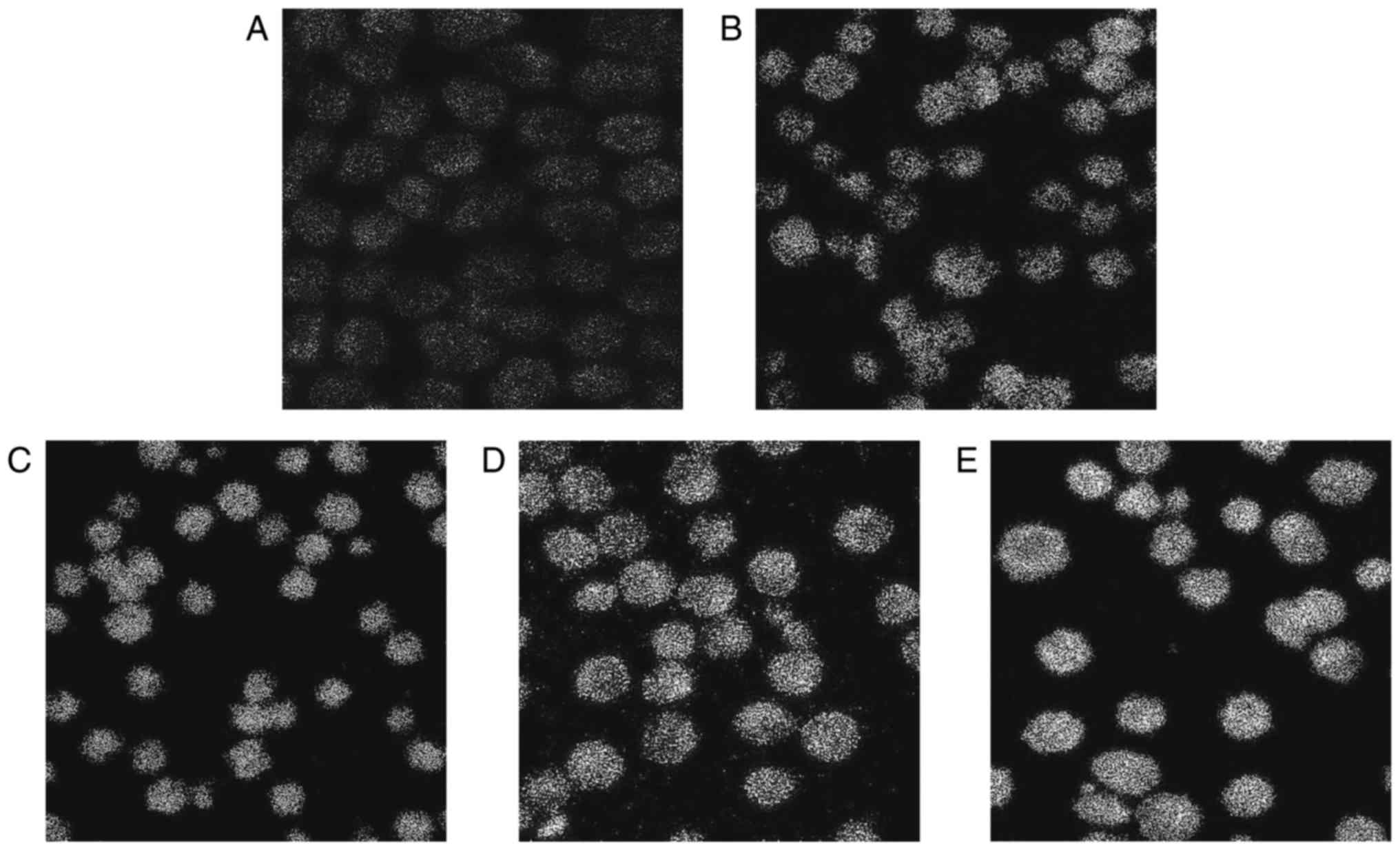
Laser confocal fluorescence microscopy
analysis
To determine the contribution of Ca2+
released from intracellular stores to the cytisine-increased
[Ca2+]i content, the HepG2 cells were treated
with cytisine for 24 h. Our results clearly indicated that cytisine
caused an increase in the cytoplasmic Ca2+ levels in a
dose-dependent manner (Table II).
Cytisine (10 mM) significantly increased the cytisine-increased
[Ca2+]i content by 30.69±0.80, and cytisine
(2.5 mM) slightly increased the cytisine-increased
[Ca2+]i content by 17.98±0.56 (P<0.01).
The green fluorescence intensity increased with the increase in
drug concentration, as shown in Fig.
3.
| Table II.Effects of cytisine on the
concentration of [Ca2+]i in the HepG2 cells
(mean ± SD, n=3). |
Table II.
Effects of cytisine on the
concentration of [Ca2+]i in the HepG2 cells
(mean ± SD, n=3).
| Groups | Concentration | Variation in
[Ca2+]i (fluorescent intensity) |
|---|
| Control | – |
7.39±0.17 |
| HCPT | 60 µmol/l |
27.17±0.76a |
| Cytisine | 2.5 mmol/l |
17.98±0.56a |
|
| 5 mmol/l |
21.54±0.26a |
|
| 10 mmol/l |
30.69±0.80a |
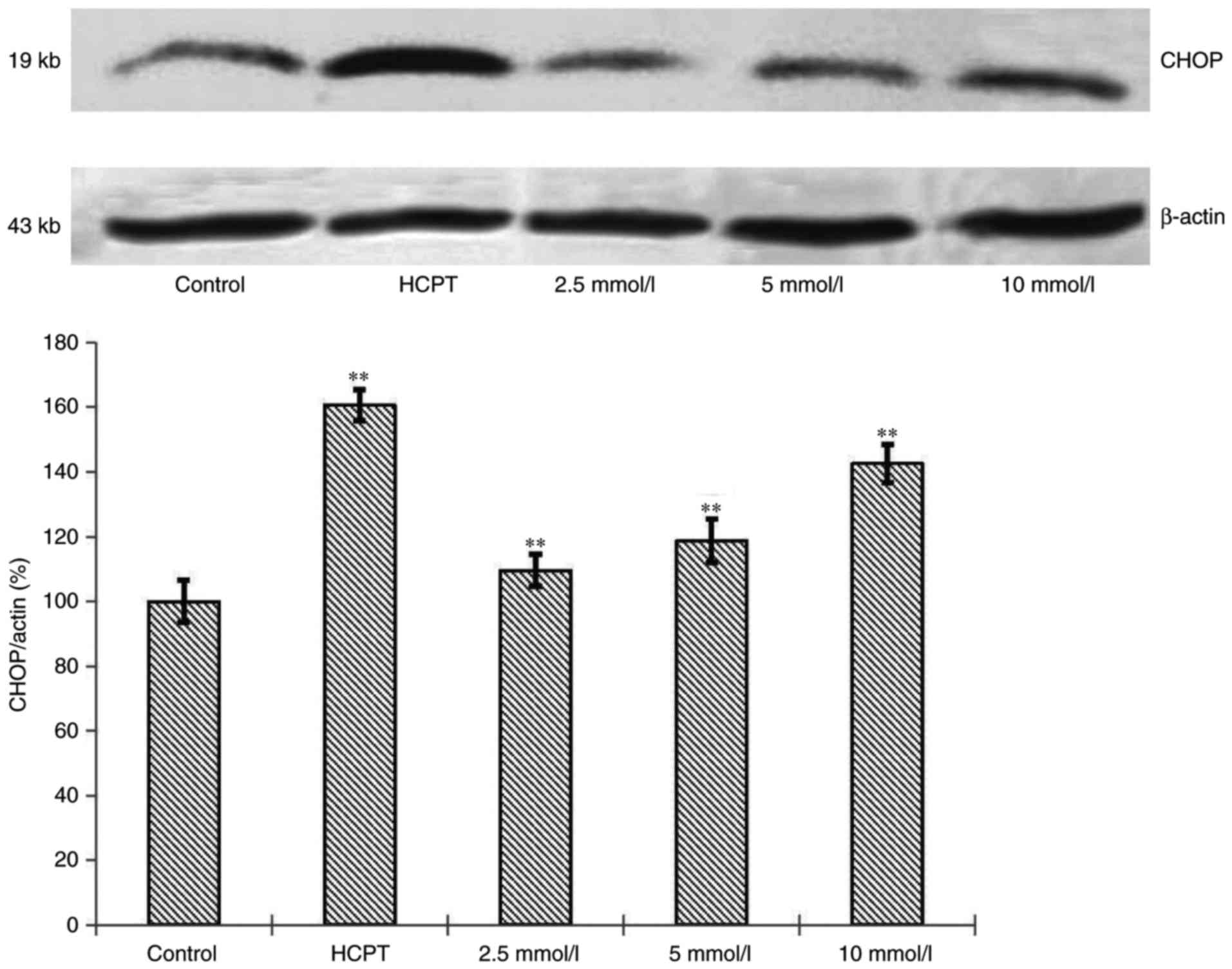
Effects of cytisine on the expression
of CHOP protein
To confirm our observation that cytisine induced ER
stress, we performed immunoblotting analyses on ER-regulated
protein, CHOP. CHOP level in the HepG2 cells in the cytisine group
was increased compared with the normal group. Significant
differences were noted (P<0.01, Fig.
4).
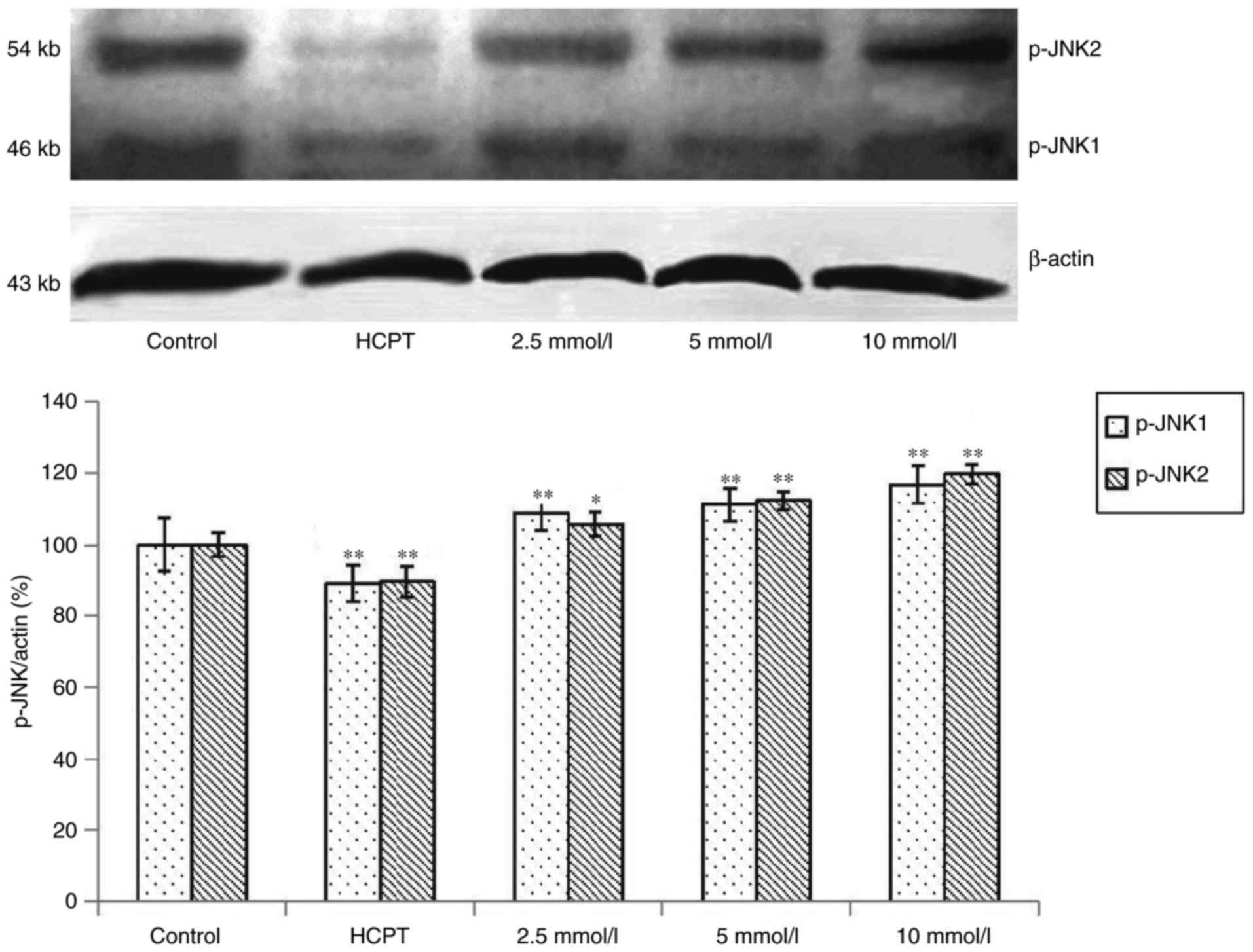
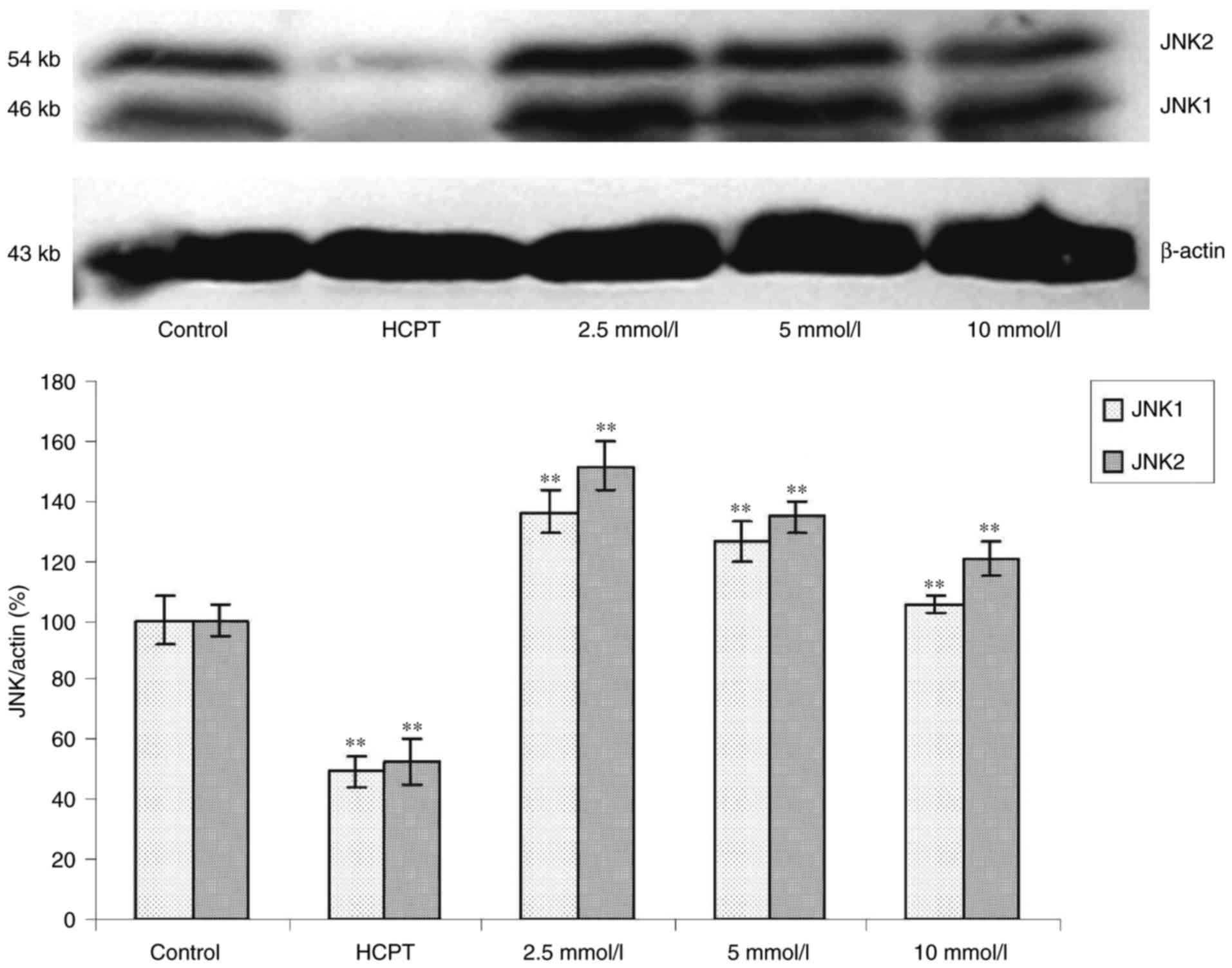
Effects of cytisine on the expression
of JNK protein
Western blot analysis revealed that exposure of the
HepG2 cells to cytisine for 24 h induced a slight upregulation of
expression of the p-JNK1 and p-JNK2 proteins (P<0.01, Fig. 5). In contrast, the expression levels
of JNK1 and JNK2 were significantly increased in the HepG2 cells in
a dose-dependent manner (P<0.01, Fig. 6).
Effect of cytisine on the activation
of caspase-4
As shown in Table
III, after treatment of the HepG2 cells with cytisine for 24 h,
the activities of caspase-4 were significantly increased compared
with the control group (P<0.01). Cytisine led to a significant
increase in the level of caspase-4 and significant upregulation of
caspase-4 expression.
| Table III.Effects of cytisine on caspase-4
activity in the HepG2 cells (mean ± SD, n=3). |
Table III.
Effects of cytisine on caspase-4
activity in the HepG2 cells (mean ± SD, n=3).
| Groups | Concentration | OD value
(x±s) | pNA (µM) | Caspase-4 activity
(%) |
|---|
| Control | – |
0.1093±0.006 | 29.25 | – |
| HCPT | 60 µmol/l |
0.3177±0.008a | 81.35 | 178.12 |
| Cytisine | 2.5 mmol/l |
0.1500±0.325a | 39.43 |
34.79 |
|
| 5
mmol/l |
0.2337±0.007a | 60.35 | 106.32 |
|
| 10 mmol/l |
0.3247±0.007a | 83.10 | 184.10 |
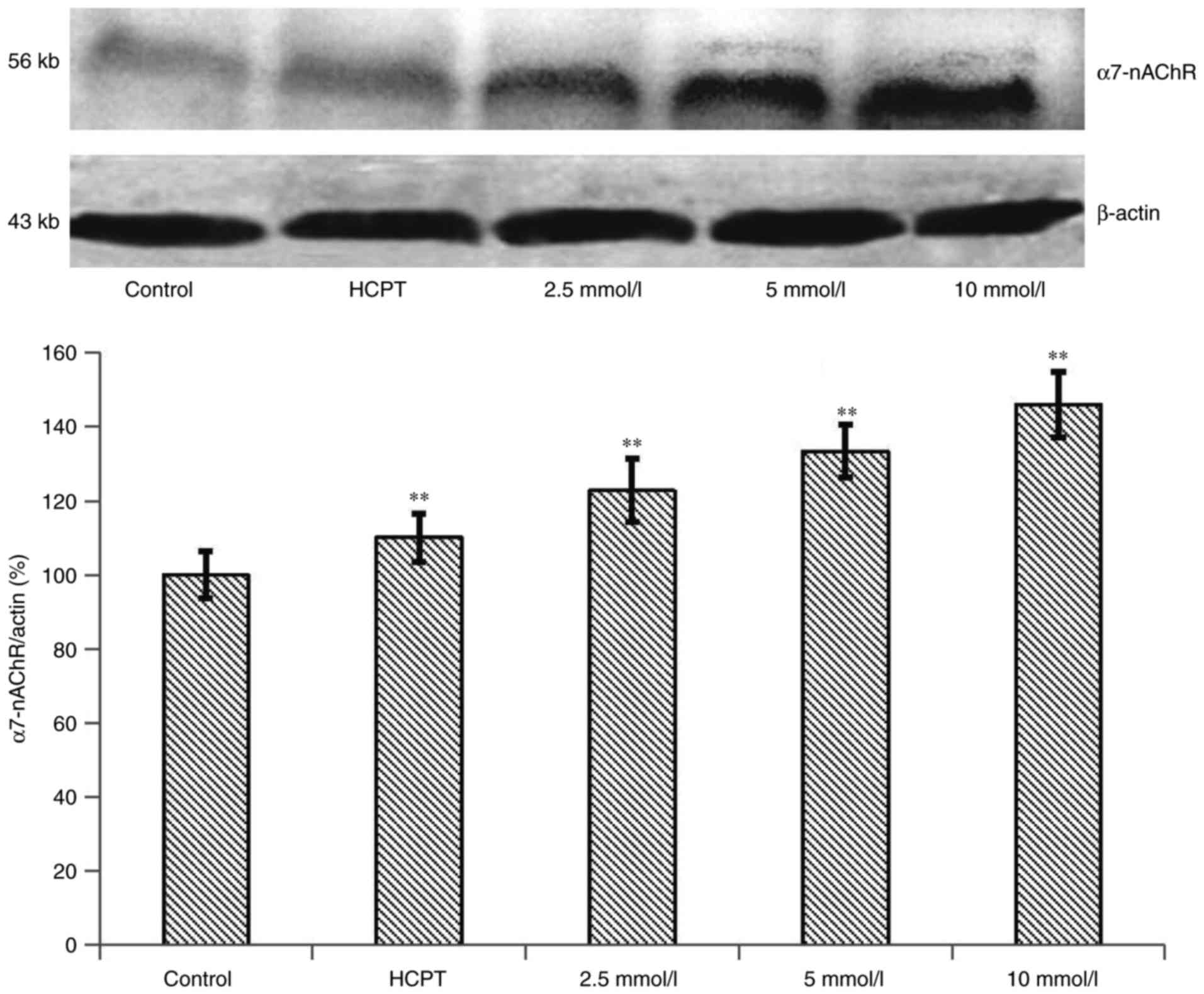
Effects of the expression of α7-nAChR
protein in HepG2 cells
To examine the α7-nAChR expression induced by the
overloading of Ca2+, western blot experiments were
conducted. As shown in Fig. 7, the
α7-nAChR expression level was significantly increased by treatment
with 10 mM of cytisine for 24 h compared with the control group
(P<0.01). Western blot analysis revealed that the exposure of
the HepG2 cells to HCPT for 24 h induced a slight upregulation of
the expression of the α7-nAChR protein compared with the control
group. The expression levels of α7-nAChR were significantly
upregulated in the HepG2 cells in a dose-dependent manner
(P<0.01).
Discussion
Apoptosis induction is one of the mechanisms
proposed for the anticancer therapeutic effects of cytisine. Our
team previously confirmed that cytisine could induce apoptosis in
HepG2 cells via a mitochondrial pathway (31). However, the molecular mechanisms of
the effects of ER stress due to cytisine have not been thoroughly
elucidated to date. ER stress, a newly defined signalling pathway,
initiates apoptosis (32–34). In the present study, we demonstrated
that Ca2+ overload induced by cytisine promoted
endoplasmic reticulum (ER) stress, consequently leading to HepG2
cell apoptosis. In a previous study, we showed that cytisine, which
is a naturally occurring quinolizidine alkaloid, had an anticancer
effect. Our prior MTT assay investigation proved that cytisine
could inhibit the proliferation of human HepG2 cells in a
dose-dependent manner (31).
In the present study, morphological changes of
apoptotic cells were observed after treatment with different
concentrations of cytisine. Cell surface microvilli decreased and
disappeared with an increase in drug concentration. Different
amounts of spherical protrusions, apoptotic bodies, cell membrane
rupture and cytoplasm overflow were observed in the apoptotic cells
with transmission electron microscopy. ER dilatation and
mitochondrial swelling were observed in the HepG2 cells after a
high dose of cytisine. In the positive control group, the
mitochondria were reduced, and the cell processes were obviously
decreased. Typical apoptotic cells and apoptotic bodies were
observed in the HepG2 cells. These results suggested that cytisine
induces the apoptosis of the HepG2 cells (Fig. 1).
The ER is associated with protein synthesis,
post-translational modification, and absorption and release of
calcium ions. However, when the quality control system is
overloaded under various stresses, including disruption of
Ca2+ homeostasis, ER functions are impaired, and
unfolded proteins are accumulated in the ER lumen. This situation
is called ER stress. The ER is the primary site of intracellular
Ca2+ storage (35–37).
Calcium homeostasis is central to all cellular functions and has
been studied for decades (38–40).
Ca2+ is a critical second messenger that mediates many
physiological cellular signalling pathways (41). The ER controls the calcium ion
through the ryanodine receptor, inositol triphosphate receptor and
sarco/endoplasmic reticulum Ca2+-ATPase (41). Moreover, research has clearly
confirmed that [Ca2+]i accumulation
contributes to cell death (42).
Studies have shown that cytisine can give rise to Ca2+
overload (43). Whether
Ca2+ overload induced by cytisine activates apoptosis in
HepG2 cells remained undetermined. We hypothesized that cytisine
stimulation would cause ER stress due to Ca2+ overload,
leading to apoptosis. To determine whether the effects of cytisine
on ER resulted in apoptosis, HepG2 cells were treated for 24 h with
various concentrations of cytisine. Flow cytometry with PI staining
revealed that the drug treatment increased the proportion of
apoptotic cells (Table I and
Fig. 2), confirming the
cytisine-induced apoptosis in HepG2 cells. Next, laser confocal
fluorescence microscopy was used to examine the intracellular
Ca2+ concentration. Our data showed that cytisine
increased the intracellular Ca2+ concentration in a
cytisine-dependent manner (Table
II and Fig. 3). These results
suggested that cytisine increased the intracellular Ca2+
concentration by releasing Ca2+ from the ER, thereby
causing ER stress. These findings revealed that calcium as a
starting factor could induce ER stress, resulting in HepG2 cell
apoptosis. It was concluded that HepG2 cellular exposure to
cytisine resulted in a direct and immediate consequence of
Ca2+ overload that led to disturbance in Ca2+
homeostasis in the ER, which caused ER stress leading to
apoptosis.
When ER stress is prolonged, pro-apoptotic signaling
pathways are activated, such as the CCAAT/enhancer-binding protein
(C/EBP) and homologous protein (CHOP/GADD153), caspase-12 and
JNK-dependent pathways (44,45).
The caspase family plays an important role in apoptosis. Previous
research has shown that caspase-12 is involved in ER-mediated
apoptosis (46). Overexpression of
caspase-12 only occurs during ER stress. It has been reported that
caspase-12 triggers cellular apoptosis through the
caspase-9/caspase-3 pathway. Cleaved caspase-12 reportedly
activates caspase-9, followed by activation of caspase-3 (47,48).
In a previous study, we found that human caspase-4, one of the
closest paralogs of rodent caspase-12, plays an important role in
cell death via ER stress. However, the relevance of caspase-12 to
ER-induced apoptosis has been questioned due to the absence of
caspase-12 in most humans (49).
Upregulation of caspase-4 in HepG2 cells had been demonstrated in
the present study. When cells were incubated with cytisine, the
expression of caspase-4 was significantly higher than that noted in
the control (Table III). Thus, we
concludes that caspase-4 plays an important role in
cytisine-induced apoptosis that is associated with ER stress. It is
suggested that Ca2+ overload, as an apoptosis-inducing
factor, may cause ER stress to increase caspase-4 expression, which
ultimately activates caspase-3 (this result has been confirmed by
our team) (31), leading to
apoptosis.
Another mediator of programmed cell death in
ER-stressed cells is the transcription factor, CHOP. In line with
previous reports, we found that significant overexpression of
CHOP/GADD153 promotes cell death or DNA damage (50). Elevated levels of CHOP can promote
the transcription of Bim, BAX and DR5 while suppressing the
induction of Bcl-2 in ER stress (51). In addition, upregulation of the
expression of CHOP is involved in caspase activation and
mitochondrial events (50). In the
present study, the expression of CHOP (Fig. 4) was continuously increased after
cytisine treatment in the HepG2 cells. In the present study,
calcium overload was hypothesized to result in a high expression of
the CHOP gene, which promoted the accumulation of Bim, BAX and DR5
while suppressing the induction of Bcl-2. Indeed, upregulation of
the expression of CHOP is involved in ER stress-induced apoptosis,
and although it is normally undetectable in proliferating cells, it
becomes highly synthesized in cells exposed to cytisine that
perturb the homeostasis of the ER, which is linked to the
development of apoptosis.
JNK, a member of the Ser/Thr protein kinase family,
is induced by stress conditions, such as ER stress (52). Additionally, JNK participates in
cell proliferation, survival, and apoptosis (53). It has been confirmed by previous
research that inhibition of JNK phosphorylation results in
decreased levels of ER stress-induced caspase-3/-7 (54). JNK, which has three isoforms, i.e.,
JNK1, JNK2 and JNK3, is also one of the signaling pathways of ER.
JNK1 and JNK2 are associated with cell death and play an important
role in apoptosis (55). To
investigate the role of JNK in ER stress that is induced by
cytisine, western blotting was used to examine expression levels of
JNK1/2 and p-JNK1/2. We found that the expression levels of p-JNK1
and p-JNK2 were significantly enhanced in HepG2 cells in a
dose-dependent manner (Fig. 5).
JNK1/2 was increased by cytisine treatment (Fig. 6). These results suggested that c-Jun
phosphorylation by Ca2+ overload in the HepG2 cells was
mainly associated with JNK1/2. Our data suggested that the ER
stress-JNK pathway is involved in Ca2+ overload and that
cytisine induced ER stress via Ca2+ overload, thereby
promoting cell death due to subsequent JNK activation.
nAChRs play an important role in synaptic
transmission; thus, the primary focus of nicotine and its receptors
has been its physiological effects within the nervous system.
However, there is growing evidence suggesting that nAChRs are not
only expressed in the nervous system but also in nonneuronal cells,
including cancer cells (23,56).
In support of our findings, gastric, bladder, colon and non-small
lung cancer cells have been shown to express nAChR subunits, and
nicotine has been shown to be mitogenic for vascular smooth muscle
cells (57). nAChRs are known to
play several important roles in cancer cells, such as
proliferation, inflammation, angiogenesis and invasion (58,59).
Thus, the expression of nAChRs in non-neuronal cells indicates that
they possess functions independent of neurotransmission. Therefore,
nAChRs are viewed as a novel drug target for the treatment of
cancer. Current research indicates that the α-7 nicotinic receptor,
which is one of the various types of subunits, is related to
intracellular Ca2+ overload (60). However, the actual role of the α-7
nicotinic receptor in HepG2 cells is unknown. Determining the
relationship between the calcium ion concentration and α7-nAChR in
HepG2 cells is crucial. Our data indicated that α7-nAChR was
expressed at a high level under cytisine conditions (Fig. 7). After the cells were treated with
cytisine, an increase in intracellular calcium concentration in the
HepG2 cells was noted suggesting that intracellular Ca2+
overload was related to the upregulation of the expression of
α7-nAChR. As a result, cytisine, which is a partial agonist to
nicotinic acetylcholine, has a high affinity to α7-nAChR. Cytisine
activates nAChRs, and the confocal laser scanning results indicated
that cytisine can induce HepG2 cell calcium overload. The results
of the present study showed that cytisine activates nicotinic
acetylcholine leading to calcium overload. However, the specific
relevance between nicotinic acetylcholine activation and
Ca2+ overload remains to be studied.
Our team previously confirmed that cytisine induced
apoptosis of HepG2 cells via the mitochondrial pathway. Following
treatment with cytisine, mitochondrial permeability may increase,
which may subsequently lead to mitochondrial matrix expansion,
outer membrane rupture and the release of cytochome c. In
addition, a previous study confirmed that increased cytochome
c release into the cytoplasm caused activation of caspase-3
(31). The present study
investigated whether cytisine induced apoptosis via the ER
pathway.
In summary, the results of the present study
indicate that cytisine induces the apoptosis of tumour cells via an
ER pathway. Our data clearly suggest that calcium overload promotes
ER stress-induced apoptosis in cytisine-induced HepG2 cells, which
occurs through modulating the CHOP/GADD153, JNK and caspase-4
pathways. Finally, the caspase cascade is activated to induce
apoptosis of HepG2 cells. Cytisine has affinity to nAChRs, and it
can activate α7-nAChR expression. In conclusion, cytisine induced
ER stress-mediated apoptotic pathway by activating CHOP, JNK and
caspase-4 in HepG2 cells.
Acknowledgements
The present study was supported in part by the Open
Research Program for the Key Laboratory of College of Heilongjiang
Province (China) (CPAT-2012003), the Natural Science Item of the
Department of Education of Heilongjiang Province (China)
(12541205), Innovation Talents Item of Science and Technology of
Harbin City (China) (2014RFQXJ154), the Doctoral Research Project
of Harbin University of Commerce (12DL008), the Graduate Students
Innovative Research Project of Harbin University of Commerce
(YJSCX2015-390HSD), 2016 Harbin University of Commerce Youth
Innovation Talent Support Program (grant no. 2016QN057), and the
Scientific Research Team Program of Harbin University of Commerce
(grant no. 2016TD002).
Glossary
Abbreviations
Abbreviations:
|
PI
|
propidium iodide
|
|
ER
|
endoplasmic reticulum
|
|
nAChRs
|
nicotinic acetylcholine receptors
|
|
UPR
|
unfolded protein response
|
|
EOR
|
endoplasmic reticulum overload
response
|
|
HCPT
|
10-hydroxycamptothecin
|
|
SDS-PAGE
|
sodium dodecyl sulphate-polyacrylamide
gel electrophoresis
|
References
|
1
|
Zhang PP, Wang PQ, Qiao CP, Zhang Q, Zhang
JP, Chen F, Zhang X, Xie WF, Yuan ZL, Li ZS and Chen YX:
Differentiation therapy of hepatocellular carcinoma by inhibiting
the activity of AKT/GSK-3β/β-catenin axis and TGF-β induced EMT
with sophocarpine. Cancer Lett. 376:95–103. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liang L, Wang XY, Zhang XH, Ji B, Yan HC,
Deng HZ and Wu XR: Sophoridine exerts an anti-colorectal carcinoma
effect through apoptosis induction in vitro and in vivo. Life Sci.
91:1295–1303. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li YJ, Yang Q, Zhang K, Guo YY, Li XB,
Yang L, Zhao MG and Wu YM: Cytisine confers neuronal protection
against excitotoxic injury by down-regulating GluN2B-containing
NMDA receptors. Neurtoxicology. 34:219–225. 2013. View Article : Google Scholar
|
|
4
|
Lin Z, Huang CF, Liu XS and Jiang J: In
vitro anti-tumour activities of quinolizidine alkaloids derived
from Sophora flavescens Ait. Basic Clin Pharmacolo Toxicol.
108:304–309. 2011. View Article : Google Scholar
|
|
5
|
Malhi H and Kaufman RJ: Endoplasmic
reticulum stress in liver disease. J Hepatol. 54:795–809. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lenna S and Trojanowska M: The role of
endoplasmic reticulum stress and the unfolded protein response in
fibrosis. Curr Opin Rheumatol. 24:2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Friedman SL: Mac the knife?
Macrophages-the double-edged sword of hepatic fibrosis. J Clin
Invest. 115:29–32. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tabas I and Ron D: Integrating the
mechanisms of apoptosis induced by endoplasmic reticulum stress.
Nat Cell Biol. 13:184–190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Urra H, Dufey E, Lisbona F, Rojas-Rivera D
and Hetz C: When ER stress reaches a dead end. Biochim Biophys
Acta. 1833:3507–3517. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Farrukh MR, Nissar UA, Afnan Q, Rafiq RA,
Sharma L, Amin S, Kaiser P, Sharma PR and Tasduq SA: Oxidative
stress mediated Ca2+ release manifests endoplasmic
reticulum stress leading to unfolded protein response in UV-B
irradiated human skin cells. J Dermatol Sci. 75:24–35. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhu M, Zhou S, Huang Z, Wen J and Li H:
Ca2+-dependent endoplasmic reticulum stress regulates
mechanical stress regulates mechanical stress-mediated cartilage
thinning. J Dent Res. 95:889–896. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Biagioli M, Pifferi S, Ragghianti M, Bucci
S, Rizzuto R and Pinton P: Endoplasmic reticulum stress and
alteration in calcium homeostasis are involved in cadmium-induced
apoptpsis. Cell Calcium. 43:184–195. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wan S and Jiang L: Endoplasmic reticulum
(ER) stress and the unfolded protein response (UPR) in plants.
Protoplasma. 253:753–764. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li K, Zhang L, Xiang X, Gong S, Ma L, Xu
L, Wang G, Liu Y, Ji X, Liu S, et al: Arsenic trioxide alleviates
airway hyperresponsiveness and promotes apoptosis of
CD4+ T lymphocytes: Evidence for involvement of the ER
stress-CHOP pathway. Ir J Med Sci. 182:573–583. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li J and Holbrook NJ: Elevated
gadd153/chop expression and enhanced c-Jun N-terminal protein
kinase activation sensitizes aged cells to ER stress. Exp Gerontol.
39:735–744. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Momoi T: Caspases involved in ER
stress-mediated cell death. J Chem Neuroanat. 28:101–105. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang Q, Liu J, Chen S, Liu J, Liu L, Liu
G, Wang F, Jiang W, Zhang C, Wang S and Yuan X: Caspase-12 is
involved in stretch-induced apoptosis mediated endoplasmic
reticulum stress. Apoptosis. 21:432–442. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hetz C, Russelakis-Carneiro M, Maundrell
K, Castilla J and Soto C: Caspase-12 and endoplasmic reticulum
stress mediate neurotoxicity of pathological prion protein. EMBO J.
22:5435–5445. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hitomi J, Katayama T, Eguchi Y, Kudo T,
Taniguchi M, Koyama Y, Manabe T, Yamagishi S, Bando Y, Imaizumi K,
et al: Involvement of caspase-4 in endoplasmic reticulum
stress-induced apoptosis and Abeta-induced cell death. J Cell Biol.
165:347–356. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Walker N, Howe C, Glover M, McRobbie H,
Barnes J, Nosa V, Parag V, Bassett B and Bullen C: Cytisine versus
nicotine for smoking cessation. N Engl J Med. 371:2353–2362. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ponzoni L, Braida D, Pucci L, Andrea D,
Fasoli F, Manfredi I, Papke RL, Stokes C, Cannazza G, Clementi F,
et al: The cytisine derivatives, CC4 and CC26, reduce
nicotine-induced conditioned place preference in zebrafish by
acting on heteromeric neuronal nicotinic acetylcholine receptors.
Psychopharmacology. 231:4681–4693. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Radchenko EV, Dravolina OA and Bespalov
AY: Agonist and antagonist effects of cytisine in vivo.
Neuropharmacology. 95:206–214. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wessler I and Kirkpatrick CJ:
Acetylcholine beyond neurons: The non-neuronal cholinergic system
in humans. Br J Pharmacol. 154:1558–1571. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Séguéla P, Wadiche J, Dineley-Miller K,
Dani JA and Patrick JW: Molecular cloning, functional properties,
and distribution of rat brain alpha 7: A nicotinic cation channel
highly permeable to calcium. J Neurosci. 13:596–604.
1993.PubMed/NCBI
|
|
25
|
Guerra-Álvarez M, Moreno-Ortega AJ,
Navarro E, Fernández-Morales JC, Egea J, López MG and Cano-Abad MF:
Positive allosteric modulation of alpha-7 nicotinic receptors
promotes cell death by inducing Ca2+ release from the
endoplasmic reticulum. J Neurochem. 133:309–319. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Levine DS, Rubin CE, Reid BJ and Haggitt
RC: Specialized metaplastic columnar epithelium in Barrett's
esophagus. A comparative transmission electron microscopic study.
Lab Invest. 60:418–432. 1989.PubMed/NCBI
|
|
27
|
Kawamoto K, Kawakami K, Kawamura Y,
Matsumura H and Ohyama A: New sensitivity test using flow cytomety.
J Neurooncol. 6:361–370. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lev-Ram V and Ellisman MH: Axonal
activation-induced calcium transients in myelinating Schwann cells,
cells, andmechanisms. J Neurosci. 15:2628–2637. 1995.PubMed/NCBI
|
|
29
|
Towbin H, Staehelin T and Gordon J:
Electrophoretic transfer of proteins from polyacrylamide gels to
nitrocellulose sheets: Procedure and some applications. Proc Natl
Acad Sci USA. 76:pp. 4350–4354. 1979; View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Burnette WN: ‘Western blotting’:
Electrophoretic transfer of proteins from sodium dodecyl
sulfate-polyacrylamide gels to unmodified nitrocellulose and
radiographic detection with antibody and radioiodinated protein A.
Anal Biochem. 112:195–203. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yu L, Wang X, Chen ZF, Jing B, Shang DY,
Sun YX, Yang JH, Zhang LF and Ji YB: Cytisine induces apoptosis of
HepG2 cells. Mol Med Rep. 16:3363–3370. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Winter E, Chiaradia LD, Silva AH, Nunes
RJ, Yunes RA and Creczynski-Pasa TB: Involvement of extrinsic and
intrinsic apoptotic pathways together with endoplasmic reticulum
stress in cell death induced by naphthylchalcones in a leukemic
cell line: Advantages of multi-target action. Toxicol In Vitro.
28:769–777. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kawaguchi T, Miyazawa K, Moriya S, Ohtomo
T, Che XF, Naito M, Itoh M and Tomoda A: Combined treatment with
bortezomib plus bafilomycin A enhances the cytocidal effect and
induces endoplasmic reticulum stress in U266 myeloma cells:
Crosstalk among proteasome, autophagy-lysosome and ER stress. Int J
Oncol. 38:643–654. 2011.PubMed/NCBI
|
|
34
|
Zhu J, Chen M, Chen N, Ma A, Zhu C, Zhao
R, Jiang M, Zhou J, Ye L, Fu H and Zhang X: Glycyrrhetinic acid
induces G1-phase cell cycle arrest in human non-small cell lung
cancer cells through endoplasmic reticulum stress pathway. Int J
Oncol. 46:981–988. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Isomura M, Kotake Y, Masuda K, Miyara M,
Okuda K, Samizo S, Sanoh S, Hosoi T, Ozawa K and Ohta S:
Tributyltin-induced endoplasmic reticulum stress and its
Ca2+-mediated mechanism. Toxicol Appl Pharmacol.
272:137–146. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nyberg WA and Espinosa A: Imiquimod
induces ER stress and Ca2+ influx independently of TLR7
and TLR8. Biochem Biophysl Res Commun. 473:789–794. 2016.
View Article : Google Scholar
|
|
37
|
Kaufman RJ and Malhotra JD: Calcium
trafficking integrates endoplasmic reticulum function with
mitochondrial bioenergetics. Biochim Biophys Acta. 1843:2233–2239.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ahn C, An BS and Jeung EB: Streptozotocin
induces endoplasmic reticulum stress and apoptosis via disruption
of calcium homeostasis in mouse pancreas. Mol Cell Endocrinol.
412:302–308. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Verkhratsky A and Toescu EC: Endoplasmic
reticulum Ca2+ homeostasis and neuronal death. J Cell
Mol Med. 7:351–361. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Song YF, Luo Z, Zhang LH, Hogstrand C and
Pan YX: Endoplasmic reticulum stress and disturbed calcium
homeostasis are involved in copper-induced alteration in hepatic
lipid metabolism in yellow cafish Pelteobagrus fulvidraco.
Chemosphere. 144:2443–2453. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ermak G and Davies KJ: Calcium and
oxidative stress: From cell signaling to cell death. Mol Immunol.
38:713–721. 2001. View Article : Google Scholar
|
|
42
|
Sun DP, Li XX, Liu XL, Zhao D, Qiu FQ, Li
Y and Ma P: Gypenosides induce apoptosis by Ca2+
overload mediated by endoplasmic-reticulum and store-operated
Ca2+ channels in human hepatoma cells. Cancer Biother
Radiopharm. 28:320–326. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sciamanna MA, Griesmann GE, Williams CL
and Lennon VA: Nicotinic acetylcholine receptors of muscle and
neuronal (alpha7) types coexpressed in a small cell lung carcinoma.
J Neurochem. 69:2302–2311. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lakshmanan AP, Thandavarayan RA,
Palaniyandi SS, Sari FR, Meilei H, Giridharan VV, Soetikno V,
Suzuki K, Kodama M and Watanabe K: Modulation of
AT-1R/CHOP-JNK-Caspase12 pathway by olmesartan treatment attenuates
ER stress-induced renal apoptosis in streptozotocin-induced
diabetic mice. Eur J Pharm Sci. 44:627–634. 2011.PubMed/NCBI
|
|
45
|
Huang Y, Li X, Wang Y, Wang H, Huang C and
Li J: Endoplasmic reticulum stress-induced hepatic stellate cell
apoptosis through calcium-mediated JNK/P38 MAPK and
calpain/caspase-12 pathways. Mol Cell Biochem. 394:1–12. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rao RV, Castro-Obregon S, Frankowski H,
Schuler M, Stoka V, del Rio G, Bredesen DE and Ellerby HM: Coupling
endoplasmic reticulum stress to the cell death program. An
Apaf-1-independent intrinsic pathway. J Biol Chem. 277:21836–21842.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chow SE, Kao CH, Liu YT, Cheng ML, Yang
YW, Huang YK, Hsu CC and Wang JS: Resveratrol induced ER expansion
and ER caspase-mediated apoptosis in human nasopharyngeal carcinoma
cells. Apoptosis. 19:527–541. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Nakagawa T, Zhu H, Morishima N, Li E, Xu
J, Yankner BA and Yuan J: Caspase-12 mediates
endoplasmic-reticulum-specific apoptosis and cytotoxicity by
amyloid-beta. Nature. 403:98–103. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Fischer H, Koenig U, Eckhart L and
Tschachler E: Human caspase 12 has acquired deleterious mutations.
Biochem Biophys Res Commun. 293:722–726. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Komatsu S, Miyazawa K, Moriya S, Takase A,
Naito M, Inazu M, Kohno N, Itoh M and Tomoda A: Clarithromycin
enhances bortezomib-induced cytotoxicity via endoplasmic reticulum
stress-mediated CHOP (GADD153) induction and autophagy in breast
cancer cells. Int J Oncol. 40:1029–1039. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wang SF, Yen JC, Yin PH, Chi CW and Lee
HC: Involvement of oxidative stress-activated JNK signaling in the
methamphetamine-induced cell death of human SH-SY5Y cells.
Toxicology. 246:234–241. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Junttila MR, Li SP and Westermarck J:
Phosphatase-mediated crosstalk between MAPK signaling pathways in
the regulation of cell survival. FASEB J. 22:954–965. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Rius B, López-Vicario C, González-Périz A,
Morán-Salvador E, García-Alonso V, Clária J and Titos E: Resolution
of inflammation in obesity-induced liver disease. Front Immunol.
3:2572012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Du K, Takahashi T, Kuge S, Naganuma A and
Hwang GW: FBVO6 attenuates cadmium toxicity in HEK293 cells by
inhibiting ER stress and JNK activation. J Toxicol Sci. 39:861–866.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Bierut LJ: Nicotine dependence and genetic
variation in the nicotinic receptors. Drug Alcohol Depend. 104
Suppl 1:S64–S69. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Catassi A, Servent D, Paleari L, Cesario A
and Russo P: Multiple roles of nicotine on cell proliferation and
inhibition of apoptosis: Implications on lung carcinogenesis. Mutat
Res. 659:221–231. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Hecht SS: Tobacco carcinogens, their
biomarkers and tobacco-induced cancer. Nat Rev Cancer. 3:733–744.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ng MK, Wu J, Chang E, Wang BY,
Katzenberg-Clark R, Ishii-Watabe A and Cooke JP: A central role for
nicotinic cholinergic regulation of growth factor-induced
endothelial cell migration. Arterioscler Thromb Vasc Biol.
27:106–112. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
del Barrio L, Egea J, León R, Romero A,
Ruiz A, Montero M, Alvarez J and López MG: Calcium signalling
mediated through α7 and non-α7 nAChR stimulation is differentially
regulated in bovine chromaffin cells to induce catecholamine
release. Br J Pharmacol. 162:94–110. 2011. View Article : Google Scholar : PubMed/NCBI
|