Introduction
Hepatocellular carcinoma (HCC) is one of the most
common malignant cancers and major health problem worldwide
(1). Although HCC treatments have
improved within recent years, patients with HCC often face an
unfavorable prognosis, mainly due to tumor recurrence and
metastasis after liver resection. Metastasis, the main risk to the
long-term survival of HCC patients, involves a complicated process
of invasion-metastasis cascades (2). Therefore, a better understanding of
the molecular mechanisms underlying HCC metastasis is necessary for
its prevention, diagnosis and treatment.
Never in mitosis gene-A (NIMA)-related expressed
kinase 2 (NEK2), a serine/threonine centrosomal kinase, is highly
expressed and activated during the S and G2 phases of the cell
cycle and plays a pivotal role in regulating centrosome separation
and mitotic progression (3).
Aberrant NEK2 expression can cause chromosome instability (CIN) as
well as abnormal chromosome content. Aberrant NEK2 expression has
been reported in cancer cells (4),
and NEK2 has recently been identified as a potential biomarker of
several cancers, such as non-small lung cancer (5) and pancreatic ductal adenocarcinoma
(6). Most studies have focused on
the function of NEK2 in centrosome regulation as well as spindle
formation (7), whereas little is
known about the other functions of NEK2 in cancer. Previous reports
with small patient populations have identified NEK2 as a biomarker
of HCC (8,9), but few have provided evidence that
NEK2 can promote HCC migration and invasion as well as the
mechanisms underlying this process.
In the present study, we demonstrated that NEK2
overexpression predicted poor prognoses in HCC patients after
hepatectomy. Moreover, NEK2 can promote HCC metastasis through
induction of the epithelial-mesenchymal transition (EMT). We
further analyzed the downstream signaling targets of NEK2 that were
related to HCC metastasis.
Materials and methods
Cell lines
The human liver cancer cell lines MHCC97H, MHCC97L,
SMMC-7721, HepG2, Huh7, BEL7402, and HCCLM3 and the normal human
liver cell line LO2 were purchased from the Typical Culture
Preservation Committee Cell Bank of the Chinese Academy of Science,
Shanghai, China. The liver cancer cell lines were cultured in DMEM
with 10% FBS (Thermo Fisher Scientific, MA, USA) at 37°C in a
humidified incubator with 5% CO2.
NEK2 overexpression and knockdown cell
lines
The NEK2 overexpression, knockdown lentivirus and
negative control lentivirus vectors were purchased from GeneChem
(GeneChem, Shanghai, China). The transfection was performed
according to the manufacturer's instructions. Briefly, the
full-length NEK2 overexpression lentivirus was transfected into
MHCC97L cells, and the knockdown virus was transfected into HCCLM3
cells. In the meantime, the negative control virus was transfected
into MHCC97L or HCCLM3 cells as controls. Puromycin (3 µg/ml) was
then used to select the stable clones. The cDNA clone and shRNA
sequences are listed in Table
I.
 | Table I.The sequence of cDNA clone and shRNA
of NEK2. |
Table I.
The sequence of cDNA clone and shRNA
of NEK2.
| Name | Sequence |
|---|
| NEK2-RNAi-1 |
TGTATTGAGTGAGCTGAAA |
| NEK2-RNAi-2 |
AGACGAGCAAAGAAGAAAT |
| NEK2-RNAi-3 |
TTACTCTGATGAATTGAAT |
| ORF nucleotide
sequence of NEK2 (transcript variant 3) |
TTTTTTGTTAGACGAAGCTTGGGCTGCAGGTCGACTCTA |
|
|
GAGGATCCCCGGGTACCGGTCGCCACCATGCCTTCCCG |
|
|
GGCTGAGGACTATGAAGTGTTGTACACCATTGGCACAG |
|
|
GCTCCTACGGCCGCTGCCAGAAGATCCGGAGGAAGAGT |
|
|
GATGGCAAGATATTAGTTTGGAAAGAACTTGACTATGG |
|
|
CTCCATGACAGAAGCTGAGAAACAGATGCTTGTTTCTG |
|
|
AAGTGAATTTGCTTCGTGAACTGAAACATCCAAACATC |
|
|
GTTCGTTACTATGATCGGATTATTGACCGGACCAATACA |
|
|
ACACTGTACATTGTAATGGAATATTGTGAAGGAGGGGA |
|
|
TCTGGCTAGTGTAATTACAAAGGGAACCAAGGAAAGGC |
|
|
AATACTTAGATGAAGAGTTTGTTCTTCGAGTGATGACTC |
|
|
AGTTGACTCTGGCCCTGAAGGAATGCCACAGACGAAGT |
|
|
GATGGTGGTCATACCGTATTGCATCGGGATCTGAAACC |
|
|
AGCCAATGTTTTCCTGGATGGCAAGCAAAACGTCAAGC |
|
|
TTGGAGACTTTGGGCTAGCTAGAATATTAAACCACGAC |
|
|
ACGAGTTTTGCAAAAACATTTGTTGGCACACCTTATTAC |
|
|
ATGTCTCCTGAACAAATGAATCGCATGTCCTACAATGA |
|
|
GAAATCAGATATCTGGTCATTGGGCTGCTTGCTGTATGA |
|
|
GTTATGTGCATTAATGCCTCCATTTACAGCTTTTAGCCA |
|
|
GAAAGAACTCGCTGGGAAAATCAGAGAAGGCAAATTC |
|
|
AGGCGAATTCCATACCGTTACTCTGATGAATTGAATGA |
|
|
AATTATTACGAGGATGTTAAACTTAAAGGATTACCATC |
|
|
GACCTTCTGTTGAAGAAATTCTTGAGAACCCTTTAATAG |
|
|
CAGATTTGGTTGCAGACGAGCAAAGAAGAAATCTTGAG |
|
|
AGAAGAGGGCGACAATTAGGAGAGCCAGAAAAATCGC |
|
|
AGGATTCCAGCCCTGTATTGAGTGAGCTGAAACTGAAG |
|
|
GAAATTCAGTTACAGGAGCGAGAGCGAGCTCTCAAAGC |
|
|
AAGAGAAGAAAGATTGGAGCAGAAAGAACAGGAGCTT |
|
|
TGTGTTCGTGAGAGACTAGCAGAGGACAAACTGGCTAG |
|
|
AGCAGAAAATCTGTTGAAGAACTACAGCTTGCTAAAGG |
|
|
AACGGAAGTTCCTGTCTCTGGCAAGTAATCCAGAGTCT |
|
|
CACTTTGTTGCCCAGGCTGGAATGCAGTGGTGTGATCAC |
|
|
AGCTCAATGTAGCTAGCCTGTGGAATGTGTGTCAGTTA |
|
|
GGGTGTGGAAAGTCCCCAGGCTCCCCAGCAGGC |
MTT
[3-(4,5)-dimethylthiazol(-z-y1)-3,5-di-phenyltetrazolium bromide]
assay
Cell growth was examined using MTT assays.
MHCC97L-control, MHCC97L-NEK2, HCCLM3-control, HCCLM3-shNEK2 cells
were added to 96-well plates at concentration of 1×103
cells/well and placed at 37°C with 5% CO2 incubator.
Then MTT reagent (0.5 mg/ml) (Sigma-Aldrich, St. Louis, MO, USA)
was added to each well and further incubated for 4 h after
indicated hours. Dimethyl sulfoxide (150 µl) was added. The plates
were read at wavelength of 490 nm.
Migration and invasion assay
Cell migration and invasion were tested using a
Transwell assay (Corning, NY, USA) as we have reported previously
(10). The cells were placed into
the upper chamber of the insert using Matrigel (Corning). After a
48-h incubation at 37°C, the cells adhering to the lower membrane
of the insert were counted after staining with 0.1% crystal violet
for 10 min. The number of the cells was observed using a Leica
microscope (Leica, Wetzlar, Germany).
Quantitative real-time PCR
Total RNA was isolated using TRIzol reagent (Life
Technologies, Carlsbad, CA, USA) from the frozen tissue samples or
HCC cell lines according to the manufacturer's protocol. Generation
of cDNA from RNA was carried out using a cDNA conversion kit
(Takara, Shiga, Japan) at 37°C for 15 min. The resultant products
were then amplified using the SYBR Green PCR kit (Toyobo, Tokyo,
Japan) for qRT-PCR analysis. The Ct values were measured during the
amplification phase while the amplification plots were analyzed
using Bio-Rad IQ5 software (Bio-Rad Laboratories Inc., CA, USA).
All quantifications were normalized to the level of endogenous
GAPDH as a control, and the procedure was performed as the
previously reported (11). The
primers were all purchased from Genecopia (Guangzhou, China).
Gene microarray
An Agilent Gene Expression array (Kangchen Biotech
Inc.), containing >41,000 transcripts (http://www.Kangchen.com.cn), was used to investigate
the transcriptional profiles of the MHCC97L-control and
MHCC97L-NEK2 cells. The microarray datasets were normalized in
GeneSpring GX using the Agilent FE one-color scenario.
Differentially expressed genes were identified via fold-change
screening.
Western blot analysis
The proteins of interest were obtained from lysed
cells, fractioned by SDS-PAGE, and subsequently transferred to PVDF
membranes (Roche Life Sciences, Switzerland). The membranes were
then blocked with 5% skim milk in TBST for 1 h at room temperature
and then incubated with the specific primary antibodies overnight
at 4°C. Tubulin was used as a loading control. After incubation,
the membranes were washed with TBST and incubated with
HRP-conjugated secondary antibody. The antigen-antibody complex was
detected with enhanced chemiluminescence regents (Merck Millipore,
MA, USA). The antibodies are listed in Table II.
| Table II.Primary antibodies. |
Table II.
Primary antibodies.
| Antibody name | Source |
|---|
| NEK2 | Abcam (55550) |
| β-tubulin | Abcam (6046) |
| E-cadherin | Cell Signaling
Technology (9782) |
| N-cadherin | Cell Signaling
Technology (9782) |
| α-catenin | Abcam
(ab51032) |
| Vimentin | Cell Signaling
Technology (5741) |
Immunofluorescence
The cells were seeded on coverslips to analyze
cellular immunofluorescence. When the cells reached a confluence of
60%, they were fixed with 4% paraformaldehyde in PBS for 15 min,
washed twice with PBS, and then incubated with primary antibodies
against E-cadherin (Proteintech Group, Chicago, IL, USA) or
N-cadherin (Cell Signaling Technology) overnight at 4°C. The cells
were then incubated with FITC-conjugated goat anti-mouse or
anti-rabbit IgG according to the source of the primary antibody
(Genecopia) and counterstained with DAPI (Genecopia) for nuclear
identification. A Leica DMRA fluorescence microscope (Leica) was
used to obtain the images.
Patients and follow-up
In total, 259 patients diagnosed with HCC after
hepatectomy were enrolled from the First Affiliated Hospital, Sun
Yat-Sen University, Guangdong, China, between 2006 and 2009. All
surgical specimens were histologically determined as HCC. Patients
under 18 years of age and those with incomplete clinical,
laboratory, or follow-up data were excluded. The last follow-up was
conducted in December 2012. The disease-free survival (DFS) and
overall survival (OS) were calculated from date of surgery to the
date of recurrence or HCC-associated death, respectively. Informed
consent was received from each enrolled patient, and the research
was carried out with approval from the Ethics Committee of the
First Affiliated Hospital of Sun Yat-Sen University (Guangdong,
China). All patients in this study were classified according to the
American Joint Committee on Cancer (AJCC) and tumor node metastasis
(TNM) classification system.
Immunochemistry analysis for HCC
patients
A tissue microarray containing tumor samples as well
as the matched adjacent non-cancerous tissue from HCC patients
enrolled in the study was constructed as previously described
(12). A Dako Real Envision kit
(K5007; Dako Denmark A/S, Denmark) was used for IHC staining. For
antigen retrieval, the slides were boiled in a pressure cooker at
maximum heat for 2 min, which contained 0.01 mol/l sodium citrate
(pH 6.0), and then cooled to room temperature. Primary antibodies
against NEK2 (1:100 dilution; Abcam, Cambridge, UK), E-cadherin
(1:200 dilution; Cell Signaling Technology, Inc., MA, USA), and
N-cadherin (1:200 dilution; Cell Signaling Technology, Inc.) were
used for this study. A five-point scoring system as follows was
used to assess staining: 0, no positive cells; 1, >0-25%
positive cells; 2, >25-50% positive cells; 3, >50-75%
positive cells; and 4, >75% positive cells. To maintain
objectivity, we also applied a four-point scoring system as follows
to describe the intensity of staining: 0, negative staining; 1,
weak staining/light yellow; 2, moderate staining/yellow-brown; and
3, strong staining/brown. The NEK2, E-cadherin and N-cadherin
immunoreactivity scores (IRSs) were calculated by adding the
staining score to the intensity score. Cases with an IRS >4 were
defined as high expression and cases with an IRS ≤4 were defined as
low expression. Three independent pathologists without access to
the clinicopathological data scored the staining. The IRS was
determined only when all of the examining pathologists assigned a
consistent score to the sample. When different scores were
obtained, a consensus score was reached by discussion.
Statistical analysis
Statistical analyses were performed using SPSS 17.0.
Data were expressed as the mean ± standard error of the mean (SEM)
from at least three independent experiments. Quantitative data were
compared between groups using Student's t-test. Categorical data
were analyzed using the χ2 test or Fisher's exact test.
Spearman's rank analysis was used to analyze the correlations
between different protein expression levels. The Kaplan-Meier
method and log-rank test were used to analyze the overall survival
and the disease-free survival curve and differences. The
independent factors that influenced survival and recurrence based
on the variables selected from the univariate analysis were
determined using the Cox proportional hazards model. Values of
p<0.05 were considered as statistically significant.
Results
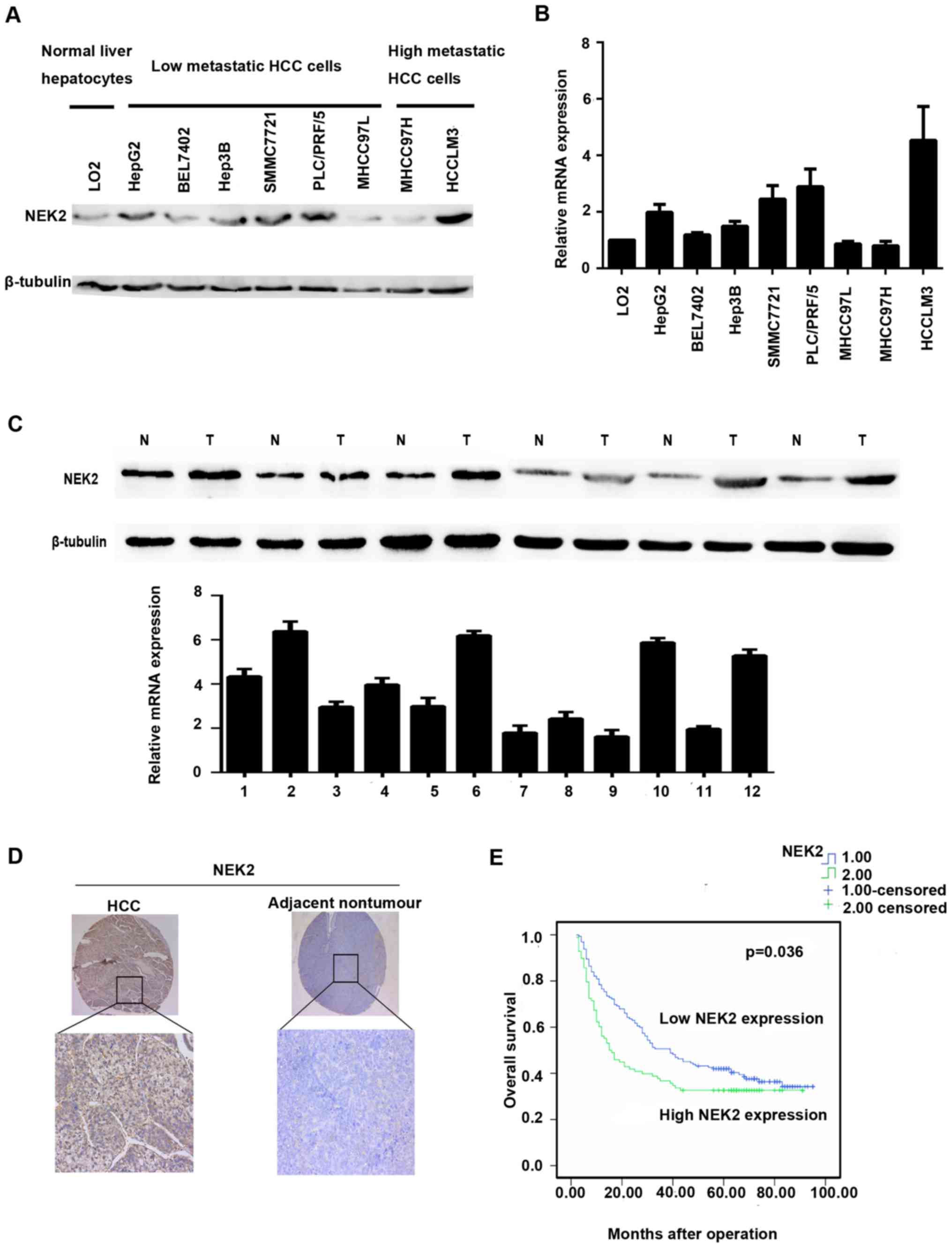
NEK2 expression is elevated in liver
cancer cell lines and tissues
Western blot and qRT-PCR analyses were used to
investigate the expression levels of NEK2 in liver cancer cell
lines and tissues. Compared with the LO2 cell line, NEK2 expression
was elevated in the low metastasis potential liver cancer cell
lines HepG2, BEL7402, Hep3B, SMMC7721, and PLC/PRF/5 and the high
metastasis potential HCC cell line HCCLM3 by qRT-PCR. However, NEK2
expression was downregulated in MHCC97L and MHCC97H cells (Fig. 1B). Western blot analysis
additionally confirmed such results (Fig. 1A).
NEK2 expression was further identified in 8 paired
fresh HCC tissues and adjacent non-cancerous liver tissues. qRT-PCR
analysis showed that mRNA levels of NEK2 were markedly elevated in
6 HCC tissues when compared with the adjacent non-cancerous liver
tissues (Fig. 1B). In accordance
with the qRT-PCR results, NEK2 protein expression was significantly
higher in HCC tissues than in adjacent non-cancerous tissues in 6/8
cases (Fig. 1C).
NEK2 expression is correlated with
poor HCC prognosis
To further evaluate the significance of NEK2
expression in HCC prognosis, we employed another HCC tissue
microarray with follow-up data from 259 pairs of HCC and adjacent
non-cancerous tissues. The IHC staining demonstrated that NEK2
expression was dramatically enhanced in 37.84% of the HCC samples
(98/259) when compared with the adjacent non-tumorous specimens
(Table III); HCC patients with
higher NEK2 expression exhibited shorter median OS time (15.5
months) compared with patients who had lower levels of NEK2
expression (39.0 months) (Fig. 1E).
In addition, HCC patients with higher NEK2 expression levels had
shorter overall survival than did those with lower NEK2 expression
levels (1-, 3- and 5-year OS: 76.5, 50.6 and 42.0% vs. 56.1, 36.7
and 32.7%, respectively). The 1-, 3- and 5-year disease-free
survival (DFS) rates for HCC patients with higher or lower NEK2
expression levels were 46.9, 30.6 and 26.5% and 50.3, 34.2 and
29.1% respectively, with no significant difference between the two
groups (p=0.171). Furthermore, univariate analysis revealed that
NEK2 overexpression was significantly correlated with tumor size
(p=0.031) and non-capsulation (p=0.019). However, no statistical
connections were found between NEK2 expression and other
clinicopathological parameters, such as age, sex, hepatitis B
surface antigen level, α-fetoprotein (AFP) level, tumor number,
liver cirrhosis, Edmondson grading, or vascular invasion (Table III). Notably, HCC patients with
NEK2-positive tumor exihibited shorter overall survival (p=0.036)
than did patients with NEK2-negative tumors. Moreover, multivariate
analysis revealed that higher expression of NEK2, bigger tumor
size, multiple tumors, non-capsulation, and vascular invasion were
independent risk factors associated with decreased survival
(Table IV). Collectively, the
clinical data indicated that NEK2 is correlated with poor prognosis
in HCC patients.
| Table III.Correlation between NEK2 expression
and clinicopathological characteristics of HCC patients. |
Table III.
Correlation between NEK2 expression
and clinicopathological characteristics of HCC patients.
|
|
| NEK2 |
|---|
|
|
|
|
|---|
| Category | n | Low expression | High
expression | p-value |
|---|
| Sex |
|
Female | 29 | 20 | 9 | 0.543 |
|
Male | 230 | 141 | 89 |
|
| Age |
|
<60 | 198 | 127 | 71 | 0.291 |
|
≥60 | 61 | 34 | 27 |
|
| HBsAg |
|
Negative | 31 | 18 | 13 | 0.695 |
|
Positive | 228 | 139 | 84 |
|
| AFP |
|
≥200 | 148 | 94 | 54 | 0.608 |
|
<200 | 111 | 67 | 44 |
|
| Size |
| ≥5 | 188 | 109 | 79 | 0.031 |
|
<5 | 71 | 52 | 19 |
|
| Tumor nos. |
|
>1 | 84 | 49 | 35 | 0.413 |
| 1 | 175 | 112 | 63 |
|
| Liver
cirrhosis |
|
Present | 206 | 125 | 81 | 0.347 |
|
Absent | 53 | 36 | 17 |
|
| Capsulation |
|
Capsulated | 96 | 55 | 41 | 0.019 |
|
Non-capsulated | 163 | 106 | 57 |
|
| Vascular
invasion |
|
Absent | 208 | 132 | 76 | 0.422 |
|
Present | 51 | 29 | 22 |
|
| Edmondson
grade |
|
I–II | 201 | 126 | 75 | 0.760 |
|
III–IV | 58 | 35 | 23 |
|
| Table IV.Univariate and multivariate analysis
of risk factors associated with overall survival of HCC
patients. |
Table IV.
Univariate and multivariate analysis
of risk factors associated with overall survival of HCC
patients.
|
|
| Overall
survival |
|---|
|
|
|
|
|---|
| Category | Subcategory | Univariate
analysis | HR (95%) | Multivariate
analysis | HR (95%) |
|---|
| Sex | Male | 0.037 | 1.037–3.221 | NA | NA |
|
| Female |
|
|
|
|
| Age | <60 | 0.927 | NA | NA | NA |
|
| ≥60 |
|
|
|
|
| HBs-Ag | Negative | 0.252 | NA | NA | NA |
|
| Positive |
|
|
|
|
| AFP (ng/ml) | <200 | 0.008 | 1.122–2.118 | NA | NA |
|
| ≥200 |
|
|
|
|
| Tumor size
(cm) | <5 |
<0.001 |
1.703–3.446 |
<0.001 | 1.324–2.941 |
|
| ≥5 |
|
|
|
|
| Tumor no. | Single |
<0.001 |
0.332–0.622 |
0.009 | 0.462–0.896 |
|
| Multiple |
|
|
|
|
| Liver
cirrhosis | Absent | 0.375 | NA | NA | NA |
|
| Present |
|
|
|
|
| Capsulation | Capsulated |
<0.001 |
0.367–0.678 |
0.034 | 0.479–0.971 |
|
| Non-capsulated |
|
|
|
|
| Vascular
invasion | Absent |
<0.001 |
2.160–4.315 |
0.002 | 1.095–2.076 |
|
| Present |
|
|
|
|
| Edmondson
grade | I–II | 0.004 | 1.174–2.310 | NA | NA |
|
| III–IV |
|
|
|
|
| ELMO1 | Lower
expression | 0.039 |
1.017–1.897 | 0.012 | 0.457–0.835 |
|
| Higher
expression |
|
|
|
|
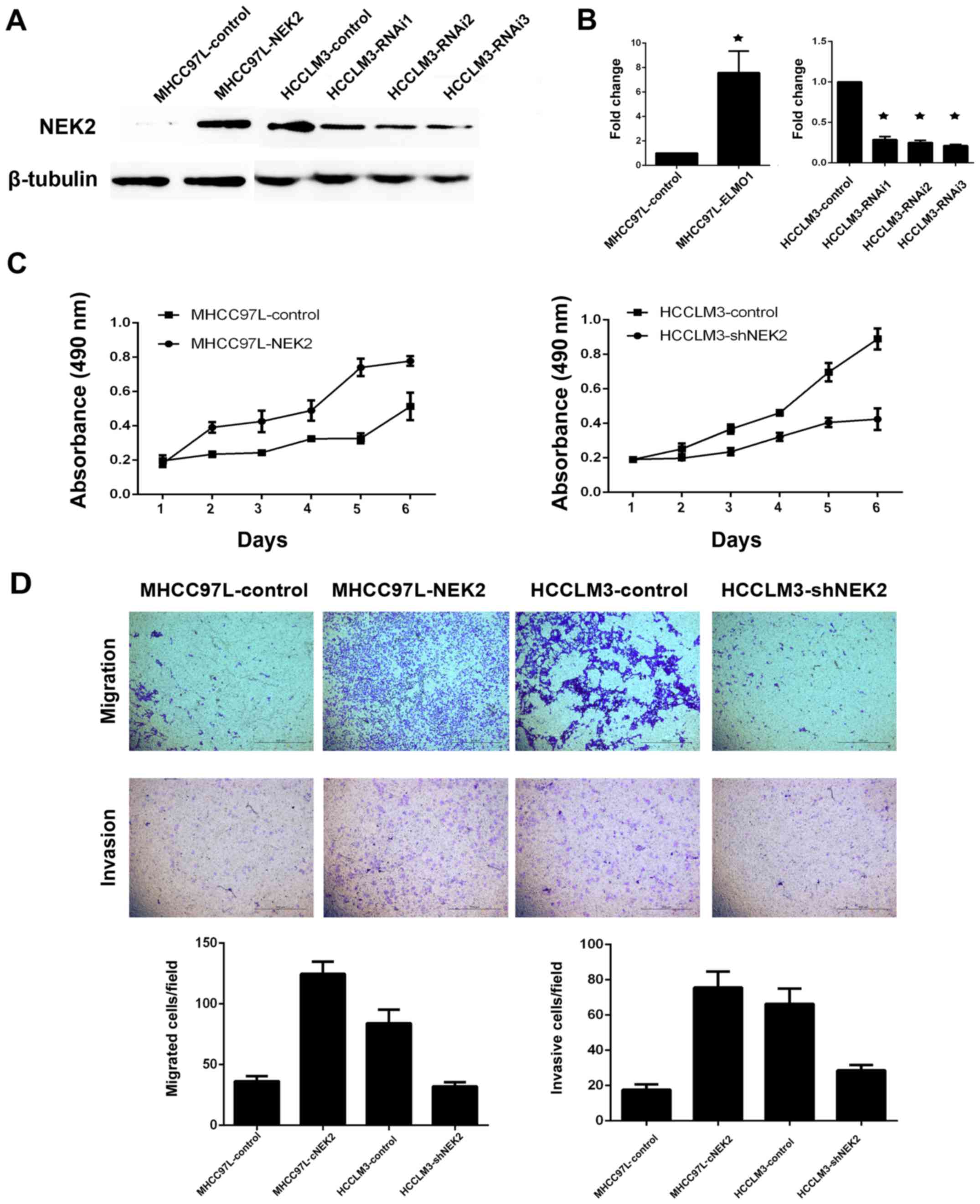
NEK2 can promote cell migration and
invasion
As tumor non-capsulation is a significant risk
factor for tumor metastasis, higher NEK2 expression was correlated
with tumor non-capsulation in our study. Therefore, we determined
whether NEK2 plays a role in HCC cell proliferation or invasion. As
NEK2 expression levels were upregulated in HCCLM3 cells (high
metastasis liver cancer cell line) and downregulated in MHCC97L
cells (low metastasis liver cancer cell line), we established two
stable cell lines (MHCC97L-NEK2 and HCCLM3-shNEK2) using
lentivirus. MTT assay showed that overexpression of NEK2 in MHCC
97L cells (MHCC97L-NEK2) can promote cell proliferation, whereas
knockdown of NEK2 in highly metastatic HCCLM3 cells (HCCLM3-shNEK2)
resulted in a remarkable suppression of cell proliferation
(Fig. 2C). Furthermore, Transwell
migration and Matrigel invasion assays revealed that overexpression
of NEK2 dramatically inhibited cell migration and invasion when
compared with control cells (Fig.
2D). Our results demonstrated that NEK2 can promote cell
proliferation, migration and invasion.
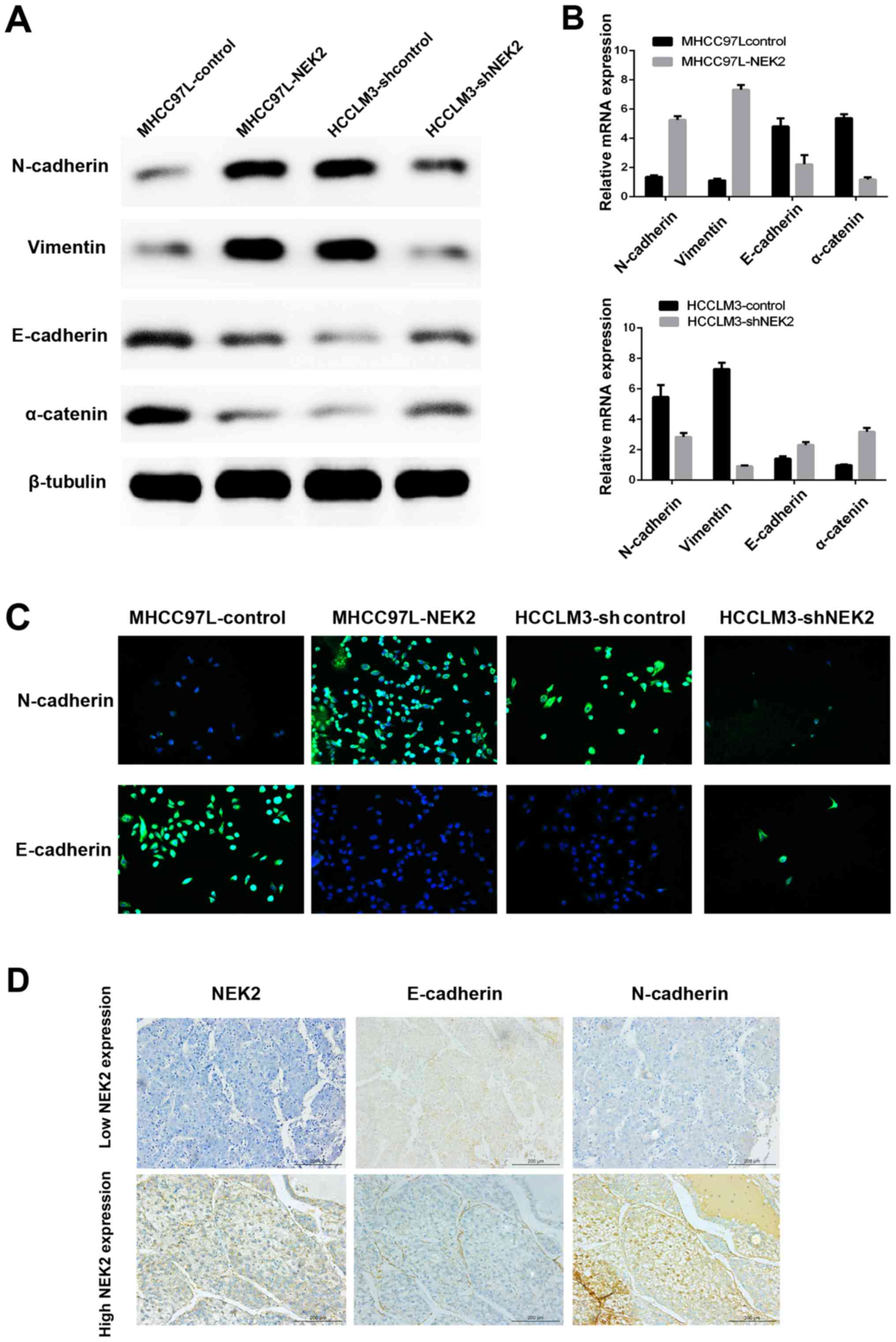
NEK2 induces epithelial mesenchymal
transition (EMT)
A previous study indicated that NEK2 contributed to
altered β-catenin localization from the intercellular adherens
junction to the cytoplasm and nucleus, which is a key process
during EMT and an invasive phenotype typical of HCC. Here, we
confirmed that NEK2 can induce EMT by immunofluorescence (IF),
qRT-PCR, western blot analysis and IHC of EMT molecular marker
expression. We showed that NEK2 overexpression increased the levels
of mesenchymal marker (N-cadherin and vimentin) expression and
decreased epithelial marker (E-cadherin and α-catenin) expression
by qRT-PCR and western blot analyses. Conversely, knocking down
NEK2 had the opposite effect (Fig. 3A
and B). Moreover, IF showed that ectopic expression of NEK2
suppressed E-cadherin expression, while inducing N-cadherin
expression in MHCC97L-NEK2 cells. In contrast, E-cadherin
expression was enhanced, whereas N-cadherin expression was
inhibited, in HCCLM3-shNEK2 cells compared with the parental HCCLM3
cells (Fig. 3C). Furthermore, IHC
of tissue microarray data showed that the levels of E-cadherin and
N-cadherin were strikingly altered in HCC tumor samples expressing
high levels of NEK2 (Fig. 3D).
N-cadherin was upregulated, whereas E-cadherin was downregulated
(Fig. 3D). These results suggested
that NEK2 can affect the expression of epithelial and mesenchymal
markers and may induce EMT in HCC cells.
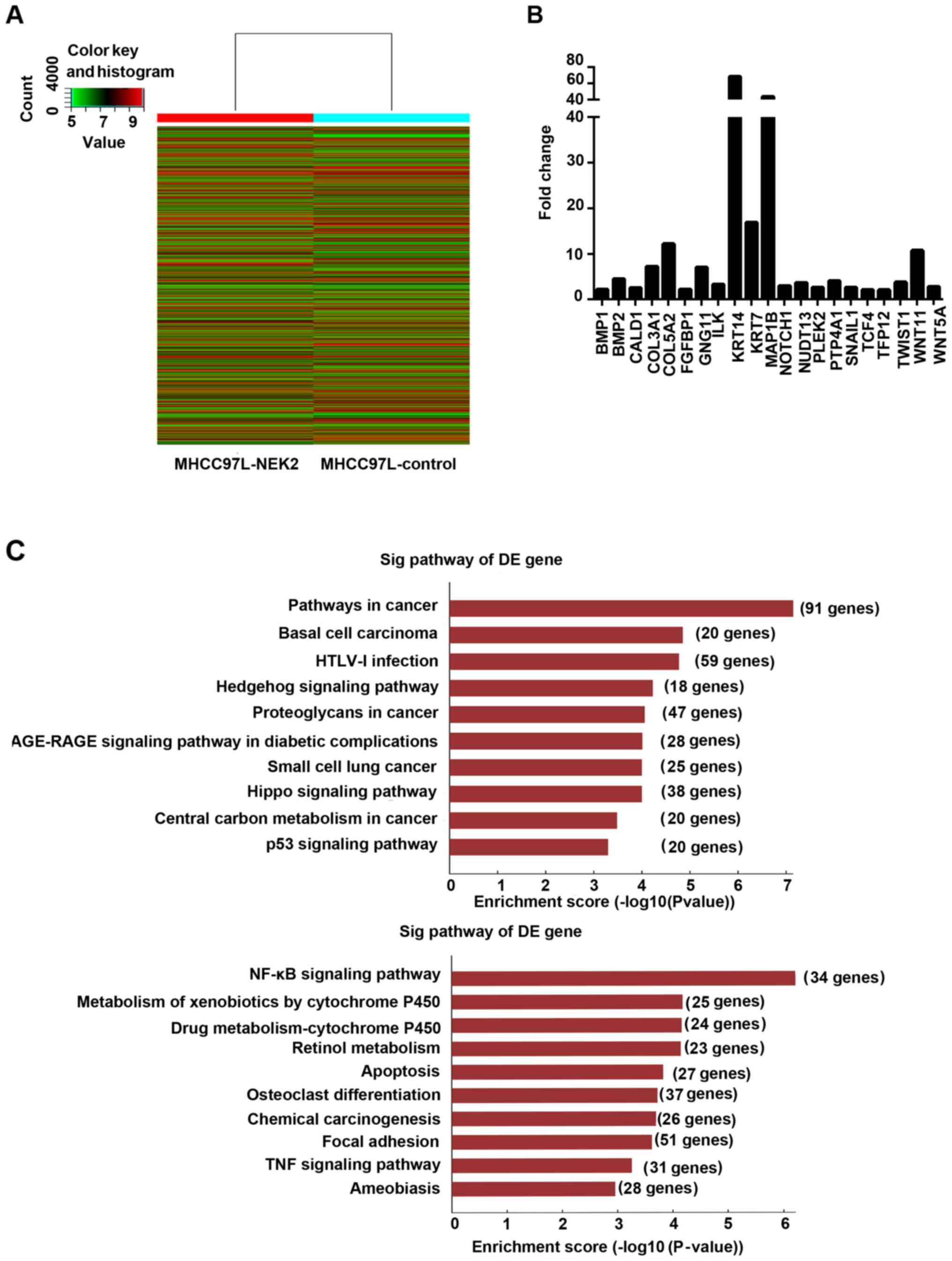
NEK2 can regulate metastasis-related
pathways
To study the genes and signaling pathways regulated
by NEK2 in HCC, we subjected MHCC97L-control and MHCC97L-NEK2 cells
to gene expression microarray analysis. EMT-related gene analysis
revealed that NOTCH1, SNAIL1, TWIST1, WNT11, and WNT5A were
upregulated in MHCC97L-NEK2 cells compared with the MHCC97L-control
(Fig. 4B). Then, signaling pathways
influenced by NEK2 were analyzed. Notably, the Wnt, NF-κB, focal
adhesion, and VEGF signaling pathways were activated by NEK2
overexpression. Interestingly, tumor suppression pathways, such as
the Hippo and p53 pathways, were downregulated when NEK2 was
overexpressed (Fig. 4C). Our data
provided novel insights into NEK2 regulation of HCC cells and
suggested that NEK2 regulates gene expression and signaling
involved in EMT.
Discussion
In this study, we identified that overexpression of
NEK2 predicted poor prognosis for HCC patients after hepatectomy.
We then demonstrated that overexpression of NEK2 led to EMT in HCC
cells and promoted HCC cell migration and invasion. Moreover, we
discovered that Wnt or NF-κB signaling may be involved in the
NEK2-induced EMT process in HCC using gene expression
microarray.
Aberrant NEK2 activities resulted in failed
regulation of centrosome duplication, causing aneuploidy and,
therefore, oncogenic effects (13).
Recently, accumulating evidence has demonstrated that NEK2 is
overexpressed in several neoplastic diseases, such asbreast
carcinoma, testicular seminomas, diffuse large B cell lymphomas and
prostate cancer (14–17). Further studies should focus on the
expression levels of NEK2 in HCC. Zhang et al (18) reported that NEK2 expression was
higher in HepG2 cells than in Huh7, SMMC-7721 and BEL7402 cells.
However, these authors did not compare the expression levels of
NEK2 between cancer cell lines and normal liver cell lines. Li
et al (9) found that the
NEK2 expression level is higher in SMMC-7721 cells than in the
normal liver cell line HL-7702. Neither study could identify
differences in NEK2 expression levels in high or low potential
metastasis HCC cell lines. In our study, we found that the NEK2
expression level was elevated in cancer cell lines relative to the
normal liver cell line LO2, and we also found NEK2 expression was
higher in the high metastasis potential HCC cell line (HCCLM3) than
in the low metastasis potential HCC cell line (MHCC97L).
Furthermore, we revealed that NEK2 expression levels were much
higher in HCC tissues when compared with the adjacent non-cancerous
tissues. Collectively, such data suggest that NEK2 is overexpressed
in HCC.
Emerging evidence has shown that NEK2 is an
independent risk for cancer patients prognosis and identified NEK2
as a putative oncogene. NEK2 overexpression is a frequent event in
cancer cells. For instance, NEK2 was found to be upregulated in
breast tissue when compared with adjacent non-cancerous tissue, and
its upregulation was closely linked to poor prognosis and high
recurrence in patients (14). Zhou
et al (19) investigated the
levels of 56 genes related to drug resistance using clinical data,
and found that NEK2 was the gene most strongly associated with
overall survival in myeloma. Further study also identified NEK2 as
an effective tumor proliferation marker of poor prognosis for
non-small cell lung cancer (5).
Recently, several studies have highlighted the potential role of
NEK2 in predicting HCC patient prognosis. One report indicated that
NEK2 was a promising biomarker for HCC recurrence by analyzing 50
HCC patients who underwent hepatectomy. They showed that NEK2 had a
statistically significant association with DFS although not with
OS, which was completely contradictory to our results. The study
population was relatively small and the background hepatitis C
virus infection was present in most cases, which might explain the
big difference compared with our results.
Another report revealed that NEK2 was related with
diolame complete, tumor nodule number and recurrence (9). However, like the above-mentioned
study, this study concerned only 63 patients, which might not
provide strong evidence to determine the relationship between NEK2
status and HCC characteristics. These authors then verified the
relationship of NEK2 expression with p-AKT and MMP-2 by IHC, but
they did not explore the underlying mechanism as to how NEK2
promotes cell migration and invasion. Consistent with previous
studies, our study confirmed the clinical significance of NEK2 as
an independent prognostic marker for HCC patients after hepatectomy
in more specimens, and confirmed that NEK2 expression level was
related to tumor size and tumor non-capsulation. Taken together,
our data revealed that NEK2 may serve as an independent risk factor
for HCC. Unlike previous reports concerning NEK2 expression level
in HCC, we enrolled more HCC patients (259 cases), which could
provide more exact evidence for the function of NEK2 in HCC and
further performed experiments in vitro to explore the role
of NEK2 expression in cell migration and invasion as well as its
underlying mechanism.
Notably, as we discovered that the NEK2 expression
level is related to tumor non-capsulation in clinical specimens, of
interest, we employed functional experiments to confirm our
suspicion. Interestingly, upregulated NEK2 expression fosters the
migration and invasion ability in HCC cells, whereas downregulated
NEK2 expression results in decreasing migration and invasion
ability. This upregulation pattern and its invasion and metastasis
activator function as characterized here in HCC cells were similar
to those functions as previously demonstrated in pancreatic cancer
(20). Nevertheless, the underlying
biological mechanism by which overexpression of NEK2 promotes
cancer cell invasion remains poorly understood. Currently, little
information underlying the molecular mechanism of NEK2 regulation
of cancer cell invasion is available. A previous report indicated
that NEK2 may contribute to HCC metastasis, which revealed that
NEK2 may mediate liver cancer cell migration via pAKT signaling and
matrix metalloproteinase (MMP) activation (21), however, such signaling might not be
the unique mechanism by which NEK2 promotes HCC metastasis. One
recent report demonstrated that NEK2 contributed to altered
β-catenin localization from the intercellular adherens junction to
the cytoplasm and nucleus (22), a
key process of EMT and an invasive phenotype typical of HCC
(23). Therefore, we speculated
that NEK2 is involved mainly in the events that trigger EMT.
Notably, the present study provides further evidence that
overexpressed NEK2 result in upregulated N-cadherin and
downregulated E-cadherin in HCC cells; conversely, silenced NEK2
expression showed contrary results. Furthermore, these EMT
process-related markers all showed similarly correlated staining
patterns for NEK2 in HCC clinical tissue microarray. Based on our
results, we speculated that NEK2 can induce EMT in HCC.
Our findings provided evidence that NEK2 is a
critical mediator of HCC cell invasion and that activation of EMT
is its novel regulating mechanism. However, how NEK2 induces EMT
remains unclear. One recent report indicated that overexpression of
NEK2 can activate AKT and Jnk pathways and upregulate Wnt/Wingless
signaling and alter the expression of Rho1, Rac1 and E-cadherin
(24). Consistently, our study
identified that upregulation of NEK2 can alter Wnt, NF-κB, focal
adhesion and VEGF signaling. All of the above signaling pathways
are classical signaling pathways that are involved in cancer cell
invasion and metastasis (25–28).
Interestingly, overexpression of NEK2 could suppress the Hippo
signaling pathway and p53 signaling pathway, which were reported as
cancer progression suppressors by numerous studies (29,30).
Collectively, our data highlight the potential role of NEK2 in
regulating several cancer-related signaling pathways. Nevertheless,
future studies to assess how NEK2 regulation of such pathways
promotes HCC cell invasion will be warranted, and whether these
pathways take part in NEK2 induction of EMT is required.
In conclusion, our results revealed an oncogenic
role for NEK2 in HCC and as an independent prognosis indicator for
HCC patients. Forced expression of NEK2 in HCC cells promoted cell
proliferation, migration and invasion, at least partly via EMT
activation, and provided evidence that Wnt, NF-κB, focal adhesion,
VEGF, Hippo and p53 signaling pathways may be downstream of NEK2.
Therefore, NEK2 may be a novel target for the treatment of HCC.
Acknowledgements
This study was supported by National Natural Science
Foundation of China (81302142; 81172039), the Natural Science
Foundation of Guangdong Province (S2011010005864; 2014A030313108),
and the Young Teacher Training Program of Sun Yat-Sen University
(15ykpy15).
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
El-Serag HB: Hepatocellular carcinoma. N
Engl J Med. 365:1118–1127. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hu CM, Zhu J, Guo XE, Chen W, Qiu XL, Ngo
B, Chien R, Wang YV, Tsai CY, Wu G, et al: Novel small molecules
disrupting Hec1/Nek2 interaction ablate tumor progression by
triggering Nek2 degradation through a death-trap mechanism.
Oncogene. 34:1220–1230. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fang Y and Zhang X: Targeting NEK2 as a
promising therapeutic approach for cancer treatment. Cell Cycle.
15:895–907. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhong X, Guan X, Liu W and Zhang L:
Aberrant expression of NEK2 and its clinical significance in
non-small cell lung cancer. Oncol Lett. 8:1470–1476. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ning Z, Wang A, Liang J, Liu J, Zhou T,
Yan Q and Wang Z: Abnormal expression of Nek2 in pancreatic ductal
adenocarcinoma: A novel marker for prognosis. Int J Clin Exp
Pathol. 7:2462–2469. 2014.PubMed/NCBI
|
|
7
|
Jeong AL, Lee S, Park JS, Han S, Jang CY,
Lim JS, Lee MS and Yang Y: Cancerous inhibitor of protein
phosphatase 2A (CIP2A) protein is involved in centrosome separation
through the regulation of NIMA (never in mitosis gene A)-related
kinase 2 (NEK2) protein activity. J Biol Chem. 289:28–40. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wubetu GY, Morine Y, Teraoku H, Yoshikawa
M, Ishikawa D, Yamada S, Ikemoto T, Saito YU, Imura S and Shimada
M: High NEK2 expression is a predictor of tumor recurrence in
hepatocellular carcinoma patients after hepatectomy. Anticancer
Res. 36:757–762. 2016.PubMed/NCBI
|
|
9
|
Li G, Zhong Y, Shen Q, Zhou Y, Deng X, Li
C, Chen J, Zhou Y and He M: NEK2 serves as a prognostic biomarker
for hepatocellular carcinoma. Int J Oncol. 50:405–413. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Guo Y, Wang J, Zhang L, Shen S, Guo R,
Yang Y, Chen W, Wang Y, Chen G and Shuai X: Theranostical
nanosystem-mediated identification of an oncogene and highly
effective therapy in hepatocellular carcinoma. Hepatology.
63:1240–1255. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang Y, Zhang JX, Huang LL, He LJ, Liao
YJ, Lai YR, Deng HX, Tian XP, Kung HF, Xie D, et al: Low expression
of BARX2 in human primary hepatocellular carcinoma correlates with
metastasis and predicts poor prognosis. Hepatol Res. 45:228–237.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang LL, Zhang Y, Zhang JX, He LJ, Lai
YR, Liao YJ, Tian XP, Deng HX, Liang YJ, Kung HF, et al:
Overexpression of NKX6.1 is closely associated with progressive
features and predicts unfavorable prognosis in human primary
hepatocellular carcinoma. Tumour Biol. 36:4405–4415. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cappello P, Blaser H, Gorrini C, Lin DC,
Elia AJ, Wakeham A, Haider S, Boutros PC, Mason JM, Miller NA, et
al: Role of Nek2 on centrosome duplication and aneuploidy in breast
cancer cells. Oncogene. 33:2375–2384. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Marina M and Saavedra HI: Nek2 and Plk4:
Prognostic markers, drivers of breast tumorigenesis and drug
resistance. Front Biosci (Landmark Ed). 19:352–365. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Barbagallo F, Paronetto MP, Franco R,
Chieffi P, Dolci S, Fry AM, Geremia R and Sette C: Increased
expression and nuclear localization of the centrosomal kinase Nek2
in human testicular seminomas. J Pathol. 217:431–441. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
de Vos S, Hofmann WK, Grogan TM, Krug U,
Schrage M, Miller TP, Braun JG, Wachsman W, Koeffler HP and Said
JW: Gene expression profile of serial samples of transformed B-cell
lymphomas. Lab Invest. 83:271–285. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zeng YR, Han ZD, Wang C, Cai C, Huang YQ,
Luo HW, Liu ZZ, Zhuo YJ, Dai QS, Zhao HB, et al: Overexpression of
NIMA-related kinase 2 is associated with progression and poor
prognosis of prostate cancer. BMC Urol. 15:902015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang MX, Xu XM, Zhang P, Han NN, Deng JJ,
Yu TT, Gan YY, He XQ and Long ZX: Effect of silencing NEK2 on
biological behaviors of HepG2 in human hepatoma cells and MAPK
signal pathway. Tumour Biol. 37:2023–2035. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou W, Yang Y, Xia J, Wang H, Salama ME,
Xiong W, Xu H, Shetty S, Chen T, Zeng Z, et al: NEK2 induces drug
resistance mainly through activation of efflux drug pumps and is
associated with poor prognosis in myeloma and other cancers. Cancer
Cell. 23:48–62. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kokuryo T, Hibino S, Suzuki K, Watanabe K,
Yokoyama Y, Nagino M, Senga T and Hamaguchi M: Nek2 siRNA therapy
using a portal venous port-catheter system for liver metastasis in
pancreatic cancer. Cancer Sci. 107:1315–1320. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wu SM, Lin SL, Lee KY, Chuang HC, Feng PH,
Cheng WL, Liao CJ, Chi HC, Lin YH, Tsai CY, et al: Hepatoma cell
functions modulated by NEK2 are associated with liver cancer
progression. Int J Cancer. 140:1581–1596. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Neal CP, Fry AM, Moreman C, McGregor A,
Garcea G, Berry DP and Manson MM: Overexpression of the Nek2 kinase
in colorectal cancer correlates with beta-catenin relocalization
and shortened cancer-specific survival. J Surg Oncol. 110:828–838.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang ZC, Gao Q, Shi JY, Guo WJ, Yang LX,
Liu XY, Liu LZ, Ma LJ, Duan M, Zhao YJ, et al: Protein tyrosine
phosphatase receptor S acts as a metastatic suppressor in
hepatocellular carcinoma by control of epithermal growth factor
receptor-induced epithelial-mesenchymal transition. Hepatology.
62:1201–1214. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Das TK, Dana D, Paroly SS, Perumal SK,
Singh S, Jhun H, Pendse J, Cagan RL, Talele TT and Kumar S:
Centrosomal kinase Nek2 cooperates with oncogenic pathways to
promote metastasis. Oncogenesis. 2:e692013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pedersen EA, Menon R, Bailey KM, Thomas
DG, Van Noord RA, Tran J, Wang H, Qu PP, Hoering A, Fearon ER, et
al: Activation of Wnt/β-catenin in Ewing sarcoma cells antagonizes
EWS/ETS function and promotes phenotypic transition to more
metastatic cell states. Cancer Res. 76:5040–5053. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shi W, Ye Z, Zhuang L, Li Y, Shuai W, Zuo
Z, Mao X, Liu R, Wu J, Chen S, et al: Olfactomedin 1 negatively
regulates NF-κB signalling and suppresses the growth and metastasis
of colorectal cancer cells. J Pathol. 240:352–365. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu C, Li Y, Xing Y, Cao B, Yang F, Yang
T, Ai Z, Wei Y and Jiang J: The interaction between cancer stem
cell marker CD133 and Src protein promotes focal sdhesion kinase
(FAK) phosphorylation and cell migration. J Biol Chem.
291:15540–15550. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lin X, Li HR, Lin XF, Yu ME, Tu XW, Hua
ZD, Lin M, Xu NL, Han LL and Chen YS: Silencing of Livin inhibits
tumorigenesis and metastasis via VEGF and MMPs pathway in lung
cancer. Int J Oncol. 47:657–667. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang X, Liu X, Luo J, Xiao W, Ye X, Chen
M, Li Y and Zhang GJ: Notch3 inhibits epithelial-mesenchymal
transition by activating Kibra-mediated Hippo/YAP signaling in
breast cancer epithelial cells. Oncogenesis. 5:e2692016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Adriaens C, Standaert L, Barra J, Latil M,
Verfaillie A, Kalev P, Boeckx B, Wijnhoven PW, Radaelli E, Vermi W,
et al: p53 induces formation of NEAT1 lncRNA-containing
paraspeckles that modulate replication stress response and
chemosensitivity. Nat Med. 22:861–868. 2016. View Article : Google Scholar : PubMed/NCBI
|