Introduction
Endometrial cancer (EC) is a common gynecological
malignancy in developed countries. Unfortunately, the etiology of
EC remains unclear. Traditionally, ‘unopposed estrogen’ is
considered responsible for the carcinogenesis of EC (1,2).
According to this hypothesis, without enough progestin, estrogen
urges the malignant transformation of the endometrium by
stimulating the proliferation and inhibiting the apoptosis of
endometrial cells. Research supporting this hypothesis has revealed
that postmenopausal women receiving hormone replacement therapy
(HRT) containing estrogen alone, face increased risk of developing
EC, but, this risk decreased when progestin was added (3). However, ‘unopposed estrogen’ alone,
cannot totally explain the pathogenesis of EC. Recently,
accumulating evidence has revealed that insulin resistance is
probably an important risk factor of EC (4,5). In
the state of insulin resistance, elevated levels of insulin
stimulate the development of EC by activating phosphoinositide
3-kinase/protein kinase B (PI3K/PKB) and mitogen-activated protein
kinase/extracellular regulated kinase (MAPK/ERK) signaling pathways
(6,7). Furthermore, insulin was found to be an
independent risk factor of EC for both pre- and postmenopausal
women (8,9). Since estrogen and insulin are
established risk factors for the development of EC, they appear to
be targets for the treatment of EC.
To date, medical treatment for EC consists primarily
of surgery, including total abdominal hysterectomy, bilateral
salpingo-oophorectomy and additional lymph node dissection
(10). However, the surgical
operations add a potential threat to approximately 20% of patients
with EC who are premenopausal early-phase patients wishing to
maintain their fertility (11). In
addition, for some patients with EC, surgical treatment appears to
be unsuitable because of morbid obesity and other serious
complications. Recently, several studies on the non-surgical
treatment of EC have revealed new insights into this malignancy
(12,13). Progestin and metformin are promising
candidates as therapeutic agents for EC treatment (14). Progestin has been employed in the
treatment of EC for many years. However, the effect of progestin on
the treatment of EC is poor (15).
Metformin is a popular insulin-sensitizing agent used in the
treatment of type II diabetes mellitus. Furthermore, the antitumor
effects of metformin have attracted scientific attention (16). In addition, the results provided by
some studies reported that metformin plays an important role in the
treatment of several malignant diseases including EC (17).
However, there is limited knowledge about the
synergistic effect of progestin and metformin on EC. In the present
study, we examined the combined effects of progestin-MPA, a
synthetic progestin- and metformin on EC in vitro and in
vivo. Furthermore, the mechanism of the effect was also
explored.
Materials and methods
Cell line and reagents
The well-differentiated human EC cell line Ishikawa
(a kindly gift from Professor Fengxia Xue), expressing estrogen and
progestin receptors was used in the present study. The cells were
maintained in phenol red-free DMEM/F12 with 10% fetal bovine serum
(FBS) at 37°C in 5% CO2. We passaged the Ishikawa cells
every 3 to 5 days. Metformin, medroxyprogesterone 17-acetate (MPA)
and MTT dye were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Caspase-3 ELISA kit was purchased from Cell Signaling Technology
(Beverly, MA, USA). All primary and secondary antibodies were
purchased from Santa Cruz Biotechnology (Dallas, TX, USA): β-actin
monoclonal antibody (sc-47778), cyclin D1 monoclonal antibody
(sc-450), cyclin E monoclonal antibody (sc-247), phosphor-Akt
monoclonal antibody (sc-514032), phosphor-ERK monoclonal antibody
(sc-7383) and secondary antibody (mouse IgGκ light chain binding
protein) (sc-516102).
Proliferation assay
The effects of metformin and (or) MPA treatment on
Ishikawa cell proliferation were determined by an MTT assay. EC
cells were plated and grown in 96-well plates at a concentration of
about 8,000 cells/µl for 24 h. The EC cells were then treated with
metformin and (or) MPA for 72 h at a concentration of 5 and 1 µM,
respectively. The cells treated with phosphate-buffered saline
(PBS) were considered as the control group. Cell densities at
different time-points (1, 12, 24, 48 and 72 h) were determined by
metabolic conversion of the MTT dye. We added MTT (5 mg/ml) to the
96-well plates at 10 µl in every well, and then incubated those
plates for an additional hour. Finally, we ended the MTT reaction
through the addition of 100 µl of dimethyl sulfoxide (DMSO).
Subsequently, the MTT assay results were examined by determining
absorption at 595 nm. The effect of metformin and (or) MPA on the
proliferation of Ishikawa cells was assessed as a percentage of the
control cell-growth obtained from the PBS-treated cells grown in
the same 96-well plates. All experiments were performed in
triplicate and repeated three times.
Apoptosis assay
Cells were cultured in 6-well plates at a
concentration of ~2×105 cells/well for 24 h and then
treated with metformin (5 µM) and (or) MPA (1 µM) in 0.5% stripped
serum for an additional 72 h. Cells treated with PBS were
considered as the control group. Caspase-3 kit was used to
determine the apoptosis rate of the Ishikawa cells at different
time-points (1, 12, 24, 48 and 72 h). Reagents were added according
to the manufacturer's instructions and the ELISA plate was examined
by assessing absorption at 450 nm. All experiments were performed
in triplicate and repeated three times.
Flow cytometry
In order to explore the mechanism of the effects of
metformin and (or) MPA on Ishikawa cells, we examined the cell
cycle profile. The cells were plated at a concentration of
2×105 cells/well in 6-well plates for 24 h.
Subsequently, the cells were starved overnight and then, treated
with 15% serum for 72 h with metformin (5 µM) and (or) MPA (1 µM).
The cells were then collected and washed with PBS, fixed in a 90%
methanol solution and stored at −20°C until the flow cytometric
analysis was performed. In the analysis, the Ishikawa cells were
firstly washed and centrifuged with cold PBS, suspended in 100 µl
PBS and 10 µl of RNase. Then the solution (250 µg/ml) was incubated
at 37°C for 30 min. After incubation, 110 µl of propidium iodide
(PI) stain (100 µg/ml) were added to each tube and incubated at 4°C
for at least 30 min before the examination. Flow cytometric
analysis was performed on a CyAn machine (Beckman Coulter, Inc.,
Miami, FL, USA). ModFit (Verity Software House, Topsham, ME, USA)
was used for the analysis for dead cells and cell debris.
Animal study
Four-week old female Balb/C nude mice with a mean
body mass of 15–18 g were purchased from Charles River Laboratories
(Beijing, China). After one week to adapt to the new environment,
the mice were injected subcutaneously into the right flank with
~5×106 Ishikawa cells. One week after cell implantation,
all the tumors became palpable. The mice were randomly divided into
four groups (n=8 for each group). One group received no
treatment and served as the control group. The other three groups
were treated with metformin (250 mg/kg, daily, per os) and
(or) MPA (1 mg in 0.1 ml volume, weekly, intramuscular). Tumor
dimensions were measured twice a week. Tumor volume was calculated
using the following formula: V=a × b ×
b/2 (a is the longest axis and b the
shortest axis of the tumor). On day 60, the animals were
sacrificed. The tumors were excised and frozen for further
analysis. All procedures involving animals in the present study
were approved by the Animal Care and Use Committee of Yantai
Yuhuangding Hospital Affiliated to Qingdao University. The welfare
of the animals was well-ensured in the present study (18).
Western blot analysis
In the present study, the expression of cyclin D1,
cyclin E and phosphorylated Akt and ERK was examined by western
blot analysis. Frozen tumor tissues were thawed and lysed in RIPA
buffer (1% NP 40, 0.5 sodium deoxycholate and 0.1% SDS). Lysates
(10 µg of protein) were separated by gel electrophoresis and
transferred onto the nitrocellulose membranes. Subsequently, the
membranes were blocked by 5% non-fat dry milk and 0.1% Tween-20 to
saturate non-specific sites. The primary antibodies were diluted
(1:1,000) and incubated overnight at 4°C. The secondary antibody
was diluted (1:4,000) and incubated at room temperature for 60 min.
The signals were detected using the ECL reagent (GE Healthcare,
Chicago, IL, USA) and the ImageQuant LAS 4000 system (GE
Healthcare). The gray value of each band in the imaging data was
analyzed using Quantity One software (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The ratios of the gray value of the target band
and β-actin used in the analysis are listed in Table I.
 | Table I.Gray values in the western blot
analysis. |
Table I.
Gray values in the western blot
analysis.
|
| Gray value |
|---|
|
|
|
|---|
| Proteins | Control | Metformin | MPA | Metformin+MPA |
|---|
| Cyclin D1 | 1.21±0.11 | 0.88±0.08 | 0.95±0.08 | 0.55±0.10 |
| Cyclin E | 0.98±0.12 | 0.76±0.06 | 0.80±0.07 | 0.48±0.09 |
| p-Akt | 0.90±0.08 | 0.89±0.09 | 0.88±0.08 | 0.84±0.08 |
| p-ERK | 0.81±0.07 | 0.77±0.10 | 0.80±0.09 | 0.76±0.09 |
Statistical analysis
The data were analyzed using SAS software package
(version 9; SAS Institute, Cary, NC, USA). Significance of
difference between the variables, except for flow-cytometry data,
was examined by the Student's t test. Mann-Whitney U test was used
to analyze the data of flow cytometry. All P-values were two-sided
among which a value <0.05 was considered to indicate a
statistically significant difference.
Results
Effect of metformin and (or) MPA on
cell proliferation
The effect of metformin and (or) MPA on the
proliferation of cancer cells was determined at different
time-points in the present study. As displayed in Fig. 1, one hour after the treatment, there
was no significant difference among the proliferation rate of the
cells treated with PBS, metformin, MPA or metformin + MPA. At the
time-points of 12, 24, 48 and 72 h after treatment, the
proliferation rate of the cells treated with metformin
(P-value=0.047, 0.041, 0.026 and 0.020, respectively) MPA
(P-value=0.048, 0.043, 0.030 and 0.022, respectively) and metformin
+ MPA (P-value=0.044, 0.038, 0.019 and 0.016, respectively) was
significantly lower than that of the cells treated with PBS.
Furthermore, the proliferation inhibitory effect of metformin + MPA
was found significantly stronger than that of metformin
(P-value=0.045, 0.042 and 0.039, respectively) or MPA
(P-value=0.040, 0.037 and 0.026, respectively) used alone at 24, 48
and 72 h after treatment.
Effect of metformin and (or) MPA on
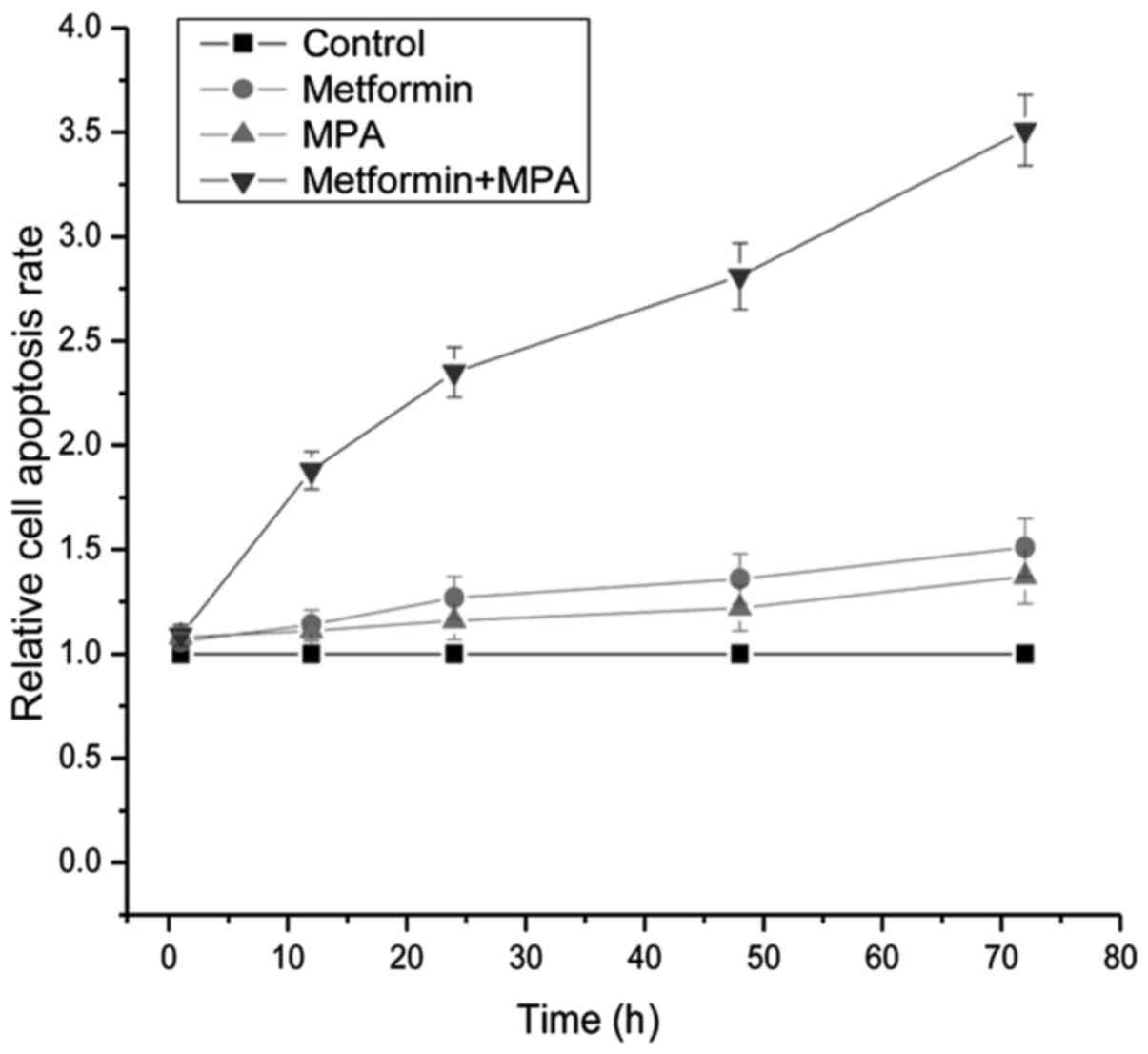
cell apoptosis
The apoptosis assay was performed by examining the
activity of caspase-3 which is a biomarker for cell apoptosis. As
displayed in Fig. 2, twelve hours
after treatment, the apoptosis rate of cells treated with metformin
+ MPA was significantly higher than that of the other three groups
(P=0.013 for the metformin group, 0.011 for the MPA group and 0.006
for the control group). At the time-points of 24, 48 and 72 h after
treatment, the apoptosis rate of the cells treated with metformin
(P=0.041, 0.037 and 0.033, respectively), MPA (P=0.048, 0.042 and
0.038, respectively) and metformin + MPA (P=0.002, <0.001 and
<0.001, respectively) were all significantly higher than that of
the cells treated with PBS. Furthermore, at 24, 48 and 72 h after
treatment, the proliferation inhibitory effect of metformin + MPA
was found significantly stronger than that of metformin
(P<0.001, <0.001 and <0.001, respectively) or MPA
(P<0.001, <0.001 and <0.001 respectively) used alone.
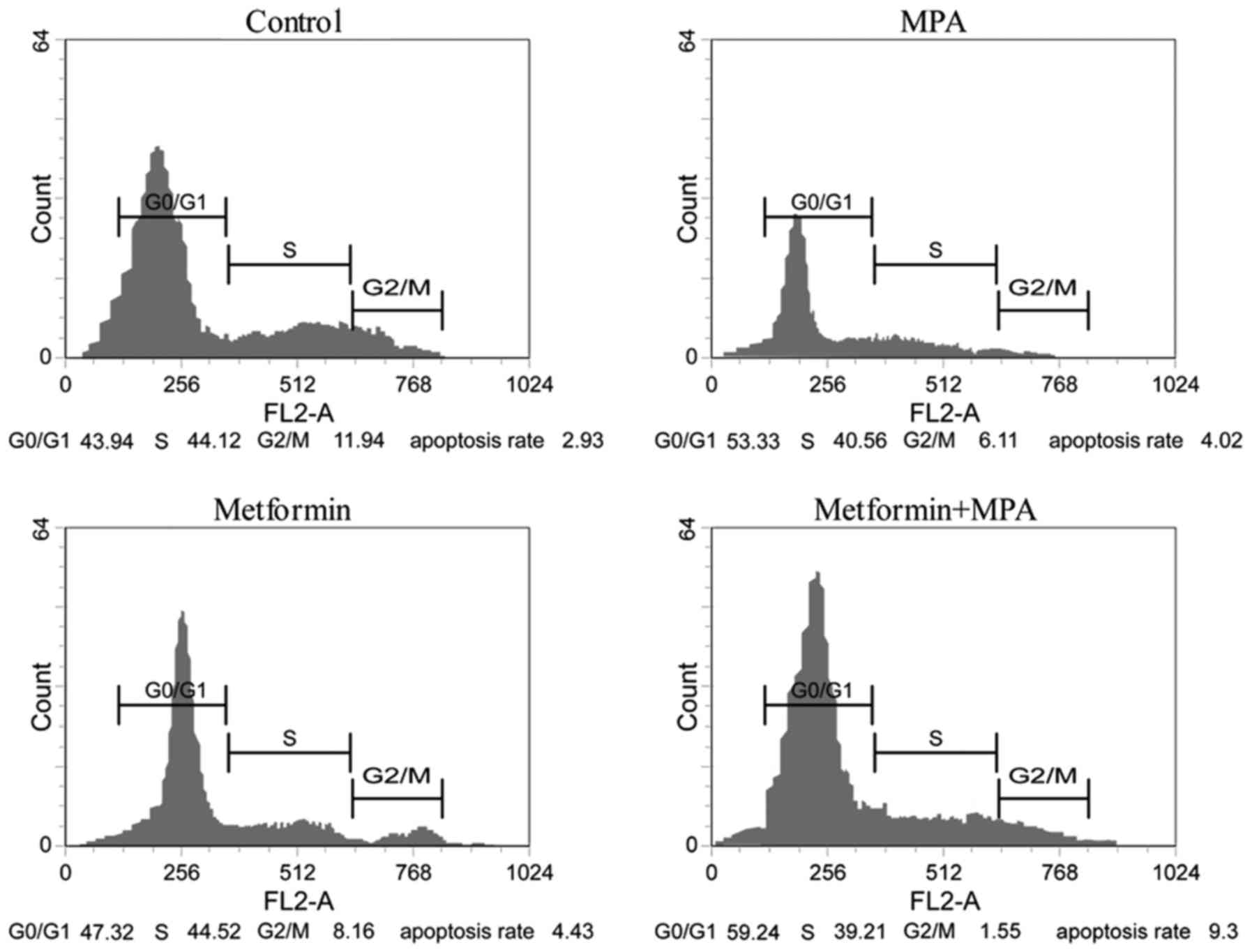
Effect of metformin and (or) MPA on
the cell cycle
As displayed in Fig.
3, metformin, MPA and combined application of them all
significantly inhibited the proliferation of the Ishikawa cells.
Compared with the control group, more cells were arrested in the
G0/G1 phase, whereas, less cells were found in the G2/M phase when
stimulated with metformin and (or) MPA. This synergistic arresting
effect of the two agents was significantly stronger than that of
metformin or MPA applied alone.
Tumor growth assay in vivo
In this section, we evaluated the effect of
metformin and (or) MPA on the growth of the xenograft tumor model.
As displayed in Fig. 4, between the
fifth to the eighth week, metformin (P=0.034, 0.032, 0.039 and
0.030, respectively), MPA (P=0.039, 0.035, 0.036 and 0.033
respectively) and metformin + MPA (P=0.014, 0.011, 0.019 and 0.018,
respectively) all significantly inhibited the growth of the
xenograft tumors. Furthermore, between the fifth to the eighth
week, the synergistic suppressive effect of metformin combined with
MPA on tumor growth was significantly stronger than metformin
(P=0.020, 0.028, 0.025 and 0.024, respectively) or MPA (P=0.017,
0.021, 0.019 and 0.020, respectively), alone. Although tumor growth
appeared more severely suppressed by metformin than MPA (Fig. 4B), the difference was not
statistically significant.
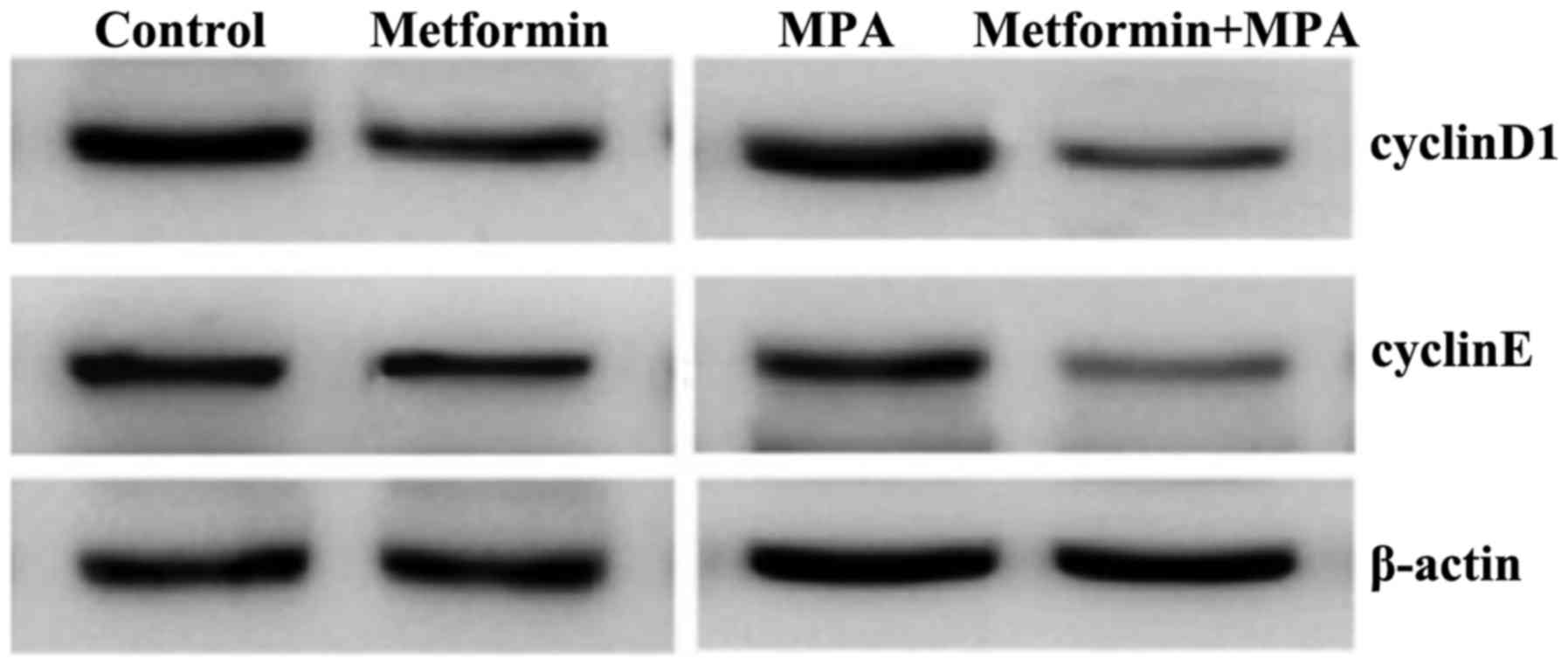
Western blot analysis of the xenograft
tumors
Cyclin D1 and cyclin E play important roles in
cell-cycle progression. The expression of these two proteins is
reported as being positively associated with cell proliferation.
The PI3K/Akt and MAPK/Ras signaling pathways are also significant
factors in regulating cell proliferation and apoptosis. Therefore,
the expression of Cyclin D1, cyclin E and phosphorylated Akt and
ERK in xenograft tumors was evaluated. As displayed in Fig. 5, the expression of cyclin D1 and
cyclin E was significantly decreased by metformin and (or) MPA
compared with the control group. Although the inhibitory effect of
metformin appeared stronger than that of MPA, the difference was
not statistically significant. Furthermore, the combined effect of
metformin and MPA was found significantly stronger than either of
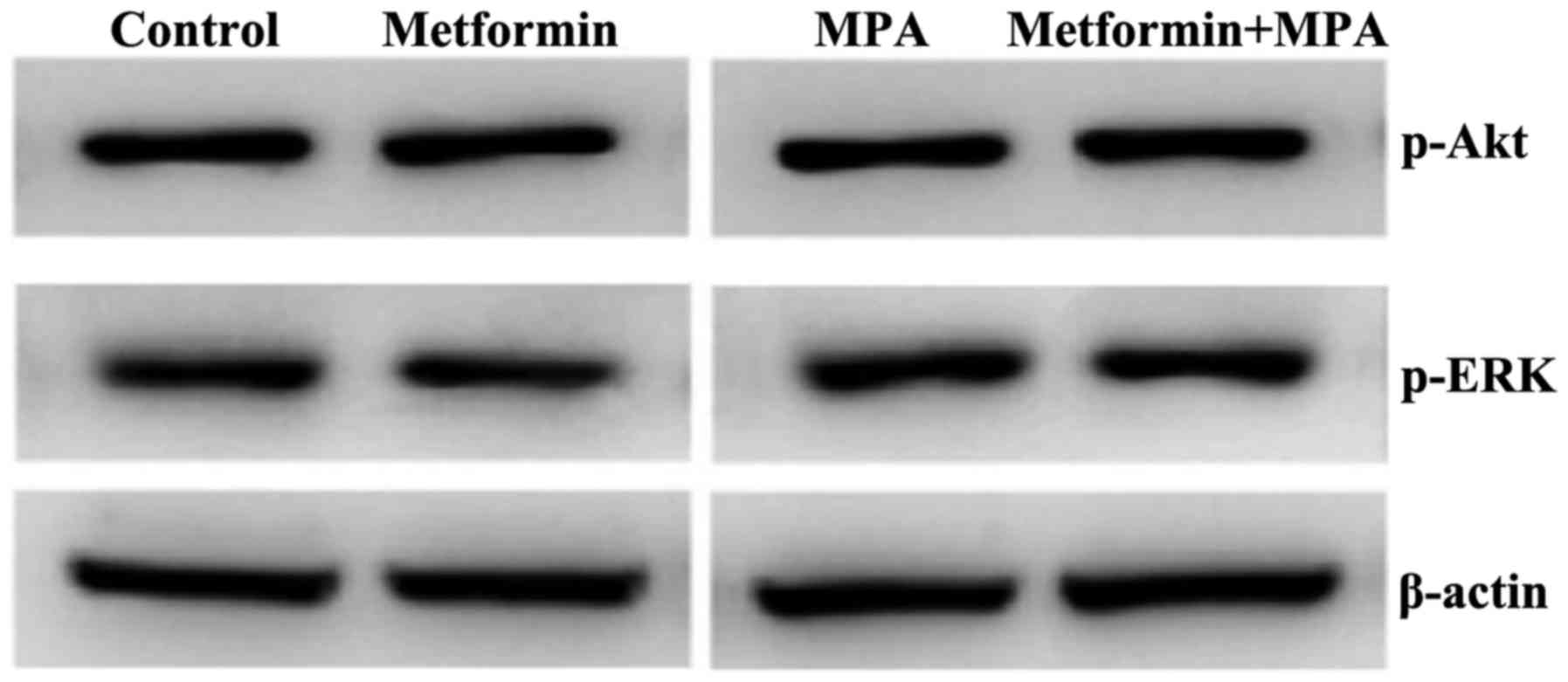
them used alone. Subsequently, we examined the phosphorylation of
Akt and ERK in the xenograft tumors. Notably, as dipslayed in
Fig. 6, we observed no difference
in the key-protein phosphorylation among the groups with different
treatments.
Discussion
The results provided by the present study
demonstrated that both metformin and MPA have potent inhibitory
effects on the development of EC cells. Furthermore, the
synergistic effect of these two agents was significantly stronger
than either of them used alone. Through arresting cancer cells in
the G0/G1 phase, the agents promoted the apoptosis and inhibited
the proliferation of the Ishikawa cells. Furthermore, these two
agents potently inhibited the development of the xenograft tumors
in vivo and the combined effect of them displayed greater
inhibitory effect. The inhibition of the expression of cyclin D1
and cyclin E is probably one of the mechanisms of the synergistic
effect of these two agents. Our findings indicated that the
combined use of metformin and MPA may be a more effective strategy
for the treatment of EC.
The anti-estrogen strategy had been applied in the
treatment of EC for many years. MPA is one of the most popular
agents clinically used in the treatment of EC. By binding to its
receptor, especially the B subtype, progestin regulates multiple
signaling pathways related to cell proliferation, apoptosis and
differentiation (19). This
inhibitory effect of MPA on cancer cells was revealed in a cell
cycle phase-specific mode (20).
The decreased expression and (or) inactivation of c-Myc, cyclin and
the associated cyclin-dependent kinases (CDKs) appeared to play
significant roles in the cell cycle-phase arresting effect
(21–24). Cyclin is a family of proteins
playing an important role in cell cycle progression. CDK family is
also a key factor in cell cycle progression the activation of which
is positively associated with the expression of cyclins. Enough
cyclin binding to CDK is pivotal for cells to pass through the
G1-phase. As indicated in our results, MPA effectively inhibited
the proliferation of the Ishikawa cells in vitro and the
G0/G1-phase arrest was probably one of the mechanisms.
Subsequently, the animal study revealed that the MPA management
significantly inhibited the growth of the xenograft tumors. The
expression of cyclin D1 and cyclin E of the xenograft tumors
treated with MPA was significantly lower than that of the controls.
The results of the current study were similar to previous studies
(25–27). Accordingly, it was indicated that
MPA inhibits the development of EC through the cell cycle
arrest.
Metformin is a common insulin-sensitizing agent used
to treat type II diabetes. However, accumulating evidence indicated
that metformin could be applied in the treatment of some malignant
diseases including EC (28–30). Firstly, as an insulin-sensitizing
agent, metformin decreased the circulating insulin levels by
inhibiting the hepatic glucose and lipid synthesis, as well as by
increasing muscle glucose uptake. As a result, the insulin induced
proliferation-promoting and apoptosis-inhibiting effects were
weakened by metformin. Sex hormone binding globulin (SHBG) is a
significant serum sex hormone-concentration regulator which tightly
binds to sex hormones. It was reported that insulin inhibited the
production of SHBG (31) leading
elevated free-estrogen levels to promote the development of EC.
Since metformin downregulates serum insulin levels, circulating
SHBG levels will be increased, resulting in less free-estrogen to
stimulate the pathogenesis of EC. Secondly, insulin acts as an
antitumor agent directly suppressing the development of EC. It is
well-known that metformin phosphralytes LKB-1 and then
AMP-activated protein kinase (AMPK) is activated which leads to the
inactivation of mammalian target of rapamycin (mTOR)-signaling
pathway (32). Furthermore,
metformin was found to exert cell cycle-inhibitory effect on cancer
cells by downregulating the expression of the related key proteins
such as cyclin D1 and cyclin E (33–36).
This cell cycle-arresting effect greatly impaired cell
proliferation. These results were in line with that of the present
study. In our study, metformin significantly inhibited the
proliferation of the Ishikawa cells and arrested cancer cells in
the G0/G1-phase. Furthermore, the growth of the xenograft tumors
was significantly inhibited by metformin management. The expression
of cyclin D1 and cyclin E were significantly lower. The data
provided by the present study indicated that cell cycle
phase-arrest can be used to explain the inhibitory effects of
metformin on the development of EC.
To date, only a small number of studies have
reported the combined application of metformin and progestin on the
treatment of EC. It was revealed that metformin inhibited the
expression of glyoxalase which is a key regulator of
progestin-resistance to strengthen the therapeutic effect of
progestin on EC cells (37).
Furthermore, the inhibitory effect of metformin on the mTOR
signaling pathway was associated with the upregulation of the
expression of the progestin receptor in EC cells (38). These studies indicated that the
combined application of metformin and progestin had stronger
inhibitory effect on the EC cells than using either of the agents
alone. However, the role played by metformin appeared adjuvant, but
not synergistic. Furthermore, the common targets of the two agents
were not provided. In the present study, the combined use of
metformin and progestin exhibited stronger inhibitory effect on the
development of EC than either of the two agents used alone.
Furthermore, we revealed that cyclin D1 and cyclin E were the
common targets of these two agents. The synergistic inhibitory
effect of metformin and progestin on the expression of cyclin D1
and cyclin E caused cancer cell-arrest in the G0/G1 phase. As a
result, the development of EC was severely delayed. In the present
study, the expression of phosphorylated Akt and ERK was also
evaluated. However, the differences were not significant among
different groups. The data presented in the study indicated that
G0/G1 phase-arrest induced by the downregulation of the expression
of cyclin D1 and cyclin E is at least partly responsible for the
synergistic effect of metformin and progestin on EC.
Although the present study provided interesting
results, there are still some weak points. Firstly, only one dose
of each agent was used in the present study, therefore it remains
unknown whether the synergistic inhibitory effect is
dose-dependent. Furthermore, the most optimal dose combination of
the two agents was not provided. Secondly, since the observation
time for the animal study was short, it is unknown whether there
are any adverse impacts of the combination use of these two agents.
Thirdly, only cyclin D1 and cyclin E were identified as the common
targets of metformin and MPA, however the whole signaling network
was not explored.
In conclusion, the present study provided novel
insights into the treatment of EC. The combined application of
metformin and MPA inhibited the development of EC in a synergistic
manner. The downregulation of cyclin D1 and cyclin E was identified
as one of the mechanisms of the synergistic inhibitory effect.
Future studies are warranted to further evaluate the combined
application of metformin and MPA and the mechanisms underlying this
synergistic inhibitory effect.
Acknowledgements
The present study was generously supported by the
Shandong Provincial Natural Science Foundation of China (no.
ZR2016HL38).
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Key TJ and Pike MC: The dose-effect
relationship between ‘unopposed’ oestrogens and endometrial mitotic
rate: Its central role in explaining and predicting endometrial
cancer risk. Br J Cancer. 57:205–212. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Henderson BE, Ross RK, Pike MC and
Casagrande JT: Endogenous hormones as a major factor in human
cancer. Cancer Res. 42:3232–3239. 1982.PubMed/NCBI
|
|
3
|
Sjogren LL, Morch LS and Lokkegaard E:
Hormone replacement therapy and the risk of endometrial cancer: A
systematic review. Maturitas. 91:25–35. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mu N, Zhu Y, Wang Y, Zhang H and Xue F:
Insulin resistance: A significant risk factor of endometrial
cancer. Gynecol Oncol. 125:751–757. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Berstein LM, Kvatchevskaya JO, Poroshina
TE, Kovalenko IG, Tsyrlina EV, Zimarina TS, Ourmantcheeva AF,
Ashrafian L and Thijssen JH: Insulin resistance, its consequences
for the clinical course of the disease, and possibilities of
correction in endometrial cancer. J Cancer Res Clin Oncol.
130:687–693. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang Y, Hua S, Tian W, Zhang L, Zhao J,
Zhang H, Zhang W and Xue F: Mitogenic and anti-apoptotic effects of
insulin in endometrial cancer are phosphatidylinositol 3-kinase/Akt
dependent. Gynecol Oncol. 125:734–741. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang Y, Zhu Y, Zhang L, Tian W, Hua S,
Zhao J, Zhang H and Xue F: Insulin promotes proliferation,
survival, and invasion in endometrial carcinoma by activating the
MEK/ERK pathway. Cancer Lett. 322:223–231. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shao Y, Cheng S, Hou J, Zuo Y, Zheng W,
Xia M and Mu N: Insulin is an important risk factor of endometrial
cancer among premenopausal women: A case-control study in China.
Tumour Biol. 37:4721–4726. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hernandez AV, Pasupuleti V, Benites-Zapata
VA, Thota P, Deshpande A and Perez-Lopez FR: Insulin resistance and
endometrial cancer risk: A systematic review and meta-analysis. Eur
J Cancer. 51:2747–2758. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Smith RA, Andrews KS, Brooks D, Fedewa SA,
Manassaram-Baptiste D, Saslow D, Brawley OW and Wender RC: Cancer
screening in the United States, 2017: A review of current American
Cancer Society guidelines and current issues in cancer screening.
CA Cancer J Clin. 67:100–121. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Garrett A and Quinn MA: Hormonal therapies
and gynaecological cancers. Best Pract Res Clin Obstet Gynaecol.
22:407–421. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hawkes AL, Quinn M, Gebski V, Armes J,
Brennan D, Janda M; feMME Trial Committee, ; Obermair A: Improving
treatment for obese women with early stage cancer of the uterus:
Rationale and design of the levonorgestrel intrauterine device ±
metformin ± weight loss in endometrial cancer (feMME) trial.
Contemp Clin Trials. 39:14–21. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nwanodi O: Progestin intrauterine devices
and metformin: Endometrial hyperplasia and early stage endometrial
cancer medical management. Healthcare (Basel). 5:E302017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Umene K, Banno K, Kisu I, Yanokura M,
Nogami Y, Tsuji K, Masuda K, Ueki A, Kobayashi Y, Yamagami W, et
al: New candidate therapeutic agents for endometrial cancer:
potential for clinical practice (review). Oncol Rep. 29:855–860.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fournier A, Dossus L, Mesrine S, Vilier A,
Boutron-Ruault MC, Clavel-Chapelon F and Chabbert-Buffet N: Risks
of endometrial cancer associated with different hormone replacement
therapies in the E3N cohort, 1992–2008. Am J Epidemiol.
180:508–517. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Decensi A, Puntoni M, Goodwin P, Cazzaniga
M, Gennari A, Bonanni B and Gandini S: Metformin and cancer risk in
diabetic patients: A systematic review and meta-analysis. Cancer
Prev Res (Phila). 3:1451–1461. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mu N, Wang Y and Xue F: Metformin: A
potential novel endometrial cancer therapy. Int J Gynecol Cancer.
22:1812012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Workman P, Aboagye EO, Balkwill F, Balmain
A, Bruder G, Chaplin DJ, Double JA, Everitt J, Farningham DA,
Glennie MJ, et al: Guidelines for the welfare and use of animals in
cancer research. Br J Cancer. 102:1555–1577. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim JJ and Chapman-Davis E: Role of
progesterone in endometrial cancer. Semin Reprod Med. 28:81–90.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Musgrove EA and Sutherland RL: Cell cycle
control by steroid hormones. Semin Cancer Biol. 5:381–389.
1994.PubMed/NCBI
|
|
21
|
Musgrove EA, Lee CS, Cornish AL, Swarbrick
A and Sutherland RL: Antiprogestin inhibition of cell cycle
progression in T-47D breast cancer cells is accompanied by
induction of the cyclin-dependent kinase inhibitor p21. Mol
Endocrinol. 11:54–66. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Watts CK, Brady A, Sarcevic B, DeFazio A,
Musgrove EA and Sutherland RL: Antiestrogen inhibition of cell
cycle progression in breast cancer cells in associated with
inhibition of cyclin-dependent kinase activity and decreased
retinoblastoma protein phosphorylation. Mol Endocrinol.
9:1804–1813. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Musgrove EA, Swarbrick A, Lee CS, Cornish
AL and Sutherland RL: Mechanisms of cyclin-dependent kinase
inactivation by progestins. Mol Cell Biol. 18:1812–1825. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang S, Thiel KW and Leslie KK:
Progesterone: The ultimate endometrial tumor suppressor. Trends
Endocrinol Metab. 22:145–152. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chang WT, Lin HL, Chang KL, Ker CG, Huang
MC, Chen JS, Kuo KK, Chen YL and Lee KT: Progesterone increases
epirubicin's apoptotic effects in HepG2 cells by S phase cell cycle
arrest. Hepatogastroenterology. 57:107–113. 2010.PubMed/NCBI
|
|
26
|
Hernández-Hernández OT, González-García TK
and Camacho-Arroyo I: Progesterone receptor and SRC-1 participate
in the regulation of VEGF, EGFR and Cyclin D1 expression in human
astrocytoma cell lines. J Steroid Biochem Mol Biol. 132:127–134.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rivas MA, Venturutti L, Huang YW,
Schillaci R, Huang TH and Elizalde PV: Downregulation of the
tumor-suppressor miR-16 via progestin-mediated oncogenic signaling
contributes to breast cancer development. Breast Cancer Res.
14:R772012. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Soliman PT, Zhang Q, Broaddus RR, Westin
SN, Iglesias D, Munsell MF, Schmandt R, Yates M, Ramondetta L and
Lu KH: Prospective evaluation of the molecular effects of metformin
on the endometrium in women with newly diagnosed endometrial
cancer: A window of opportunity study. Gynecol Oncol. 143:466–471.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zou J, Hong L, Luo C, Li Z, Zhu Y, Huang
T, Zhang Y, Yuan H, Hu Y, Wen T, et al: Metformin inhibits
estrogen-dependent endometrial cancer cell growth by activating the
AMPK-FOXO1 signal pathway. Cancer Sci. 107:1806–1817. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gadducci A, Biglia N, Tana R, Cosio S and
Gallo M: Metformin use and gynecological cancers: A novel treatment
option emerging from drug repositioning. Crit Rev Oncol Hematol.
105:73–83. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Plymate SR, Matej LA, Jones RE and Friedl
KE: Inhibition of sex hormone-binding globulin production in the
human hepatoma (Hep G2) cell line by insulin and prolactin. J Clin
Endocrinol Metab. 67:460–464. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kourelis TV and Siegel RD: Metformin and
cancer: New applications for an old drug. Med Oncol. 29:1314–1327.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Scherbakov AM, Sorokin DV, Tatarskiy VJ
Jr, Prokhorov NS, Semina SE, Berstein LM and Krasil'nikov MA: The
phenomenon of acquired resistance to metformin in breast cancer
cells: The interaction of growth pathways and estrogen receptor
signaling. IUBMB Life. 68:281–292. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cai X, Hu X, Tan X, Cheng W, Wang Q, Chen
X, Guan Y, Chen C and Jing X: Metformin induced AMPK activation,
G0/G1 phase cell cycle arrest and the inhibition of growth of
esophageal squamous cell carcinomas in vitro and in vivo. PLoS One.
10:e1333492015. View Article : Google Scholar
|
|
35
|
Sikka A, Kaur M, Agarwal C, Deep G and
Agarwal R: Metformin suppresses growth of human head and neck
squamous cell carcinoma via global inhibition of protein
translation. Cell Cycle. 11:1374–1382. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tosca L, Ramé C, Chabrolle C, Tesseraud S
and Dupont J: Metformin decreases IGF1-induced cell proliferation
and protein synthesis through AMP-activated protein kinase in
cultured bovine granulosa cells. Reproduction. 139:409–418. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang Z, Dong L, Sui L, Yang Y, Liu X, Yu
Y, Zhu Y and Feng Y: Metformin reverses progestin resistance in
endometrial cancer cells by downregulating GloI expression. Int J
Gynecol Cancer. 21:213–221. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xie Y, Wang YL, Yu L, Hu Q, Ji L, Zhang Y
and Liao QP: Metformin promotes progesterone receptor expression
via inhibition of mammalian target of rapamycin (mTOR) in
endometrial cancer cells. J Steroid Biochem Mol Biol. 126:113–120.
2011. View Article : Google Scholar : PubMed/NCBI
|