Introduction
Thyroid cancer is the most common endocrine tumor
and the incidence of thyroid cancer is rising by 4% every year,
approximately 80% of which accounts for papillary thyroid carcinoma
(PTC) (1). Although PTC is a highly
curable disease and its mortality rate is low, nearly 10–20% of
PTCs are progressive with aggressive tumor behavior and high
disease recurrence (2). The
underlying factors and mechanisms for the aggressiveness of PTC
remain unclear.
Recently, research has shed light on the importance
of fatty acid metabolism in cancer progression (3). The alteration of fatty acid metabolism
influences the energy stores, affects drug resistance, modulates
cell proliferation and survival and stimulates the extracellular
environment (4,5). Some studies have revealed that
transformed cells and malignant tumors exhibit overexpression of
elements involved in de novo lipid metabolism (6,7). This
increased lipid biosynthesis in tumor tissue may offer a new
insight into the molecular mechanisms that manage the progression
of malignant tumors.
KMT5A, known as SET8, PR-Set7/9 and SETD8, a member
of the SET domain containing methyltransferase family specifically
targeting H4K20 for monomethylation, has been reported to play key
roles in diverse biological processes, including gene
transcriptional control, replication origin modulation, genome
integrity maintenance and cell cycle progression and development
(8–13). Accumulating evidence revealed that
SET8 may participate in tumor development and progression (14,15).
However, whether KMT5A is involved in the oncogenesis of PTC is
currently not known.
In the present study, we found that KMT5A was
overexpressed in PTC tissues as well as in PTC cells. Knockdown
functional analysis revealed that KMT5A participated in the
proliferation, apoptosis, cell cycle, migration and invasion of PTC
cells. Upon further research we found that downregulation of KMT5A
also attenuated fatty acid metabolism in in vitro models of
PTC. These results provide mounting evidence of KMT5A as an
oncogenic mediator in fatty acid metabolism of PTC.
Materials and methods
Tissue specimens
Fifty pairs of papillary thyroid cancer samples
consisting of 50 PTCs and 50 matched normal thyroid tissues (1 cm
away from the edge of the tumor tissue) were obtained between 2012
and 2015 from patients who underwent thyroid cancer surgery at the
Shanghai Cancer Center in Fudan University. Tissue specimens were
frozen in liquid nitrogen immediately after surgical resection and
stored at −80°C. Final histological classification was based on
paraffin-embedded sections. The use of clinical materials for
research purposes in the present study was approved by the
Institutional Research Ethics Committee of Fudan University prior
to the obtained written consent of patients.
The KMT5A gene expression and clinical data from 455
papillary thyroid carcinomas in the TCGA database are available
from the website www.cbioportal.org/. The relationship between KMT5A
and the clinical parameters was analyzed using the TCGA data
(Table I).
 | Table I.Relationship between the KMT5A
expression and the clinicopathological parameters of the PTC
patients. |
Table I.
Relationship between the KMT5A
expression and the clinicopathological parameters of the PTC
patients.
|
| KMT5A expression |
|
|---|
|
|
|
|
|---|
| Clinicopathological
parameters | Low, n (%) | High, n (%) | P-value |
|---|
| Age (years) |
|
| 0.769 |
|
<45 | 77 (36.5) | 134 (63.5) |
|
|
≥45 | 85 (35.0) | 158 (65.0) |
|
| Sex |
|
| 0.259 |
|
Male | 38 (40.9) | 55 (59.1) |
|
|
Female | 92 (34.5) | 179 (65.5) |
|
| Tumor size
(cm) |
|
| 0.29625 |
| ≤1 | 23 (41.8) | 32 (58.2) |
|
|
>1 | 137 (62.1) | 260 (39.7) |
|
| Extrathyroidal
extension |
|
| 0.0001b |
|
Positive | 25 (18.9) | 107 (81.1) |
|
|
Negative | 130 (42.3) | 177 (57.7) |
|
| Lymph node
metastasis |
|
| 0.0057a |
|
Positive | 89 (42.2) | 122 (57.8) |
|
|
Negative | 71 (29.3) | 171 (70.7) |
|
| TNM stage |
|
| 0.0001b |
|
I+II | 135 (6.9) | 183 (12.7) |
|
|
III+IV | 25 (93.1) | 101 (87.3) |
|
Cell lines and transfections
Human thyroid follicular epithelial cell line
Nthy-ori 3-1 and human papillary thyroid cancer cell lines K1 and
TPC-1 were grown in RPMI-1640 media (Gibco, Carlsbad, CA, USA)
supplemented with 10% fetal bovine serum (FBS; Invitrogen,
Carlsbad, CA, USA) and 1% penicillin-streptomycin (10,000 U/ml
penicillin and 10 mg/ml streptomycin in 0.9% NaCl; Sigma-Aldrich,
St. Louis, MO, USA) at 37°C with 5% CO2. We constructed
a lentiviral system to knock down KMT5A: HEK-293T cells were
co-transfected with pLKO.1-KMT5A 1 and 2 (Biotend, Shanghai, China)
using Lipofectamine 2000 (Invitrogen, Shanghai, China). Forty-eight
hours after transfection, the virus-containing medium collected
from HEK-293T cells was used to infect PTC cells K1 and TPC-1 to
establish KMT5A-shRNA cells, followed by 2 µg/ml puromycin
selection (Sigma-Aldrich). Human thyroid follicular epithelial cell
line Nthy-ori 3-1 was obtained from Sigma-Aldrich. Human papillary
thyroid cancer cell lines K1 and TPC-1 were purchased from the
University of Colorado Cancer Center Cell Bank.
RNA extraction and RT-PCR
Total RNA of tissue or cell lines was extracted
using TRIzol reagent (Invitrogen). Total RNA (1 µg) was used for
cDNA synthesis employing PrimeScript™ RT Reagent kit (Takara Bio,
Inc., Shiga, Japan). RT-PCR was performed in triplicate with SYBR
Premix Ex Taq™ kit (Takara Bio) according to the manufacturer's
instructions. The primers for the target genes were synthesized by
Sangon Biotech Co., Ltd. (Shanghai, China) (Table II). The thermal cycling parameters
were: 95°C for 30 sec, 95°C for 5 sec and 60°C for 30 sec. β-actin
was used as an internal control. The threshold cycle (Ct) values
were analyzed using the comparative Ct (2−ΔΔCt) method.
The level of target genes was calculated by normalizing to the
endogenous reference and relative to a control (16).
| Table II.qRT-PCR primers of the target
genes. |
Table II.
qRT-PCR primers of the target
genes.
| Target genes | Forward sequence
(5′-3′) | Reverse sequence
(5′-3′) |
|---|
| KMT5A |
CGCAAACTTACGGATTTCT |
CGATGAGGTCAATCTTCATT |
| SREBP |
GCGGAGCCATGGATTGCAC |
CTCTTCCTTGATACCAGGCCC |
| SCD |
TTCCTACCTGCAAGTTCTACACC |
CCGAGCTTTGTAAGAGCGGT |
| FASN |
AGCTGCCAGAGTCGGAGAAC |
TGTAGCCCACGAGTGTCTCG |
| ACC |
TAGATCCGTCAGAAAATGGGC |
TACGGATATATTCTGCGTTGGC |
| β-actin |
TGACGTGGACATCCGCAAAG |
CTGGAAGGTGGACAGCGAGG |
Immunohistochemical staining
Immunohistochemical staining (IHC) was carried out
according to the manufacturer's protocol. Briefly, formalin-fixed
and paraffin-embedded tissue sections were deparaffinized in xylene
and hydrated through descending concentrations of ethanol before
being placed in a blocking solution to inhibit endogenous
peroxidase activity. The slides were incubated with primary
antibodies (rabbit anti-human KMT5A; dilution, 1:200; Cell
Signaling Technology, Shanghai, China) at 4°C overnight.
HRP-conjugated rabbit secondary antibody (dilution, 1:100; Cell
Signaling Technology) was added for 60 min at room temperature,
followed by DAB development (DAB Substrate Chromogen system; Dako,
Agilent Technologies, Shanghai, China) following standard staining
protocol. The slides were fixed and images obtained with the
Olympus IX71 inverted microscope using the DP2-BSW Olympus image
acquisition software system. The results were confirmed by two
experienced pathologists who were blinded to the
clinicopathological data of the patients. The staining results were
scored based on the percentage of stained tumor cell nuclei (0, no
staining; 1, ≤10%; 2, >10–50%; and 3, >50%).
Western blotting
The cells were lysed in RIPA buffer and boiled at
100°C for 10 min to obtain the protein lysates. Approximately 20 µg
protein lysates was extracted from each sample and separated by 10%
SDS-PAGE. After being blocked in 5% non-fat milk at room
temperature for 1 h, the protein concerned was probed using the
primary antibodies: KMT5A (dilution, 1:1,000), p53 (dilution,
1:1,000; Santa Cruz Biotechnology, Shanghai, China), β-actin
(dilution, 1:1,000; Cell Signaling Technology) at 4°C overnight,
and then incubated with goat anti-rabbit IgG or goat anti-mouse IgG
(dilution, 1:5,000; Cell Signaling Technology) at room temperature
for 1 h and detected with enhanced chemiluminescence reagents
(Thermo Fisher Scientific, Shanghai, China). The bands were
visualized using 1-step™ NBT/BCIP reagents (Thermo Fisher
Scientific, Rockford, IL, USA) and detected by the AlphaImager
(Alpha Innotech, San Leandro, CA, USA).
Cellular viability assays
Cell viability was determined using Cell Counting
Kit-8 (CCK-8) assay according to the manufacturer's protocol.
Briefly, the cells were seeded into 96-well plates at
4×103 cells/well. An aliquot of 10 µl CCK-8 solution was
added to each well and the plate was incubated for 3 h at 37°C. At
the indicated time-points, the absorbance at 450 nm was assessed
using a spectrophotometer. For each group, data from five wells
were pooled. Each experiment was performed in triplicate.
Apoptosis assay
The apoptotic cells were assayed by flow cytometric
analysis using the Annexin V-FITC/propidium iodide (PI) staining
kit (BD Biosciences, Shanghai, China). The suspended cells were
stained with 5 µl Annexin V-FITC and 10 µl PI at room temperature
for 15 min. Flow cytometry was then used to immediately analyze the
cells. The apoptotic cells were marked based on the Annexin V
expression (Annexin V+/PI− and Annexin
V+/PI+), quantified and compared to the
controls from each group. The experiment was performed in
triplicate.
Migration and invasion assays
The migration of cancer cells was assayed using
6.5-mm diameter chambers with 8-µm pore filters (Transwell, 24-well
cell culture; BD Biosciences), while the invasion of cancer cells
was assayed using chambers precoated with 20 µg Matrigel (BD
Biosciences). Both cell lines were suspended at 3×104
cells/ml in serum-free media and then 0.1 ml cell suspension was
added to the upper chamber. Subsequently 0.6 ml complete medium was
added to the lower chamber. The chambers were incubated for 24 h at
37°C with 5% CO2. After incubation, the filters were
fixed and stained. The upper surface of the filters was scraped
with cotton swabs to remove non-migrating cells. The experiments
were repeated in triplicate wells and the number of migrating cells
in five high-power fields per filter was counted microscopically at
×200 magnification.
Cell cycle
In order to synchronize the cell cycle, the cells
were harvested by trypsinization, washed in ice-cold
phosphate-buffered saline (PBS) and fixed in 70% ice-cold ethanol
in PBS. After being resuspended in cold PBS, RNAase (2 µg/ml; BD
Biosciences) was added and the cells were incubated at 37°C for 30
min, followed by incubation in 400 µl PI (10 µg/ml; BD Biosciences)
for 40 min at room temperature. The DNA content was analyzed by
flow cytometry and the cell population in the G0, G1, S, G2 and M
phase was analyzed. All experiments were performed in
triplicate.
Lipid peroxidation (MDA) assay
Malondialdehyde (MDA) in the cells was determined
using a lipid peroxidation MDA kit purchased from Beyotime
Institute of Biotechnology (Shanghai, China) according to the
manufacturer's instructions. Lysates from 2×106 cultured
cells were collected with a mixture of ProteoJET Mammalian Cell
Lysis Reagent (Fermentas, Shanghai, China). Afterwards the cells
were homogenized in lysis solution on ice and then centrifuged at
12,000 × g for 10 min. TBA reagent was added into each well
containing 200 µl standard and 200 µl sample and incubated at 95°C
for 60 min. Subsequently the mix was cooled at room temperature in
an ice bath for 10 min. The activity of SOD was determined using a
spectrophotometer at Ex/Em = 532/553 nm for fluorometric assay.
Flow cytometry
The reactive oxygen species (ROS) level in the K1
and TPC-1 cells after the knockdown of KMT5A was assessed by flow
cytometry using Reactive Oxygen Species Assay kit (Beyotime
Institute of Biotechnology) following the manufacturer's
instructions. After being stained as described in the kit, ROS
generation of cells was determined by flow cytometry (FC500;
Beckman Coulter, Inc., Brea, CA, USA).
Statistical analysis
Statistical analyses were performed using GraphPad
Prism 5.1 (GraphPad Software, Inc., La Jolla, CA, USA). Student's
t-test and one-way ANOVA were performed to analyze the data.
P<0.05 was considered to indicate a statistically significant
difference. The data are expressed as mean ± SEM. All the
experiments were performed at least three times.
Results
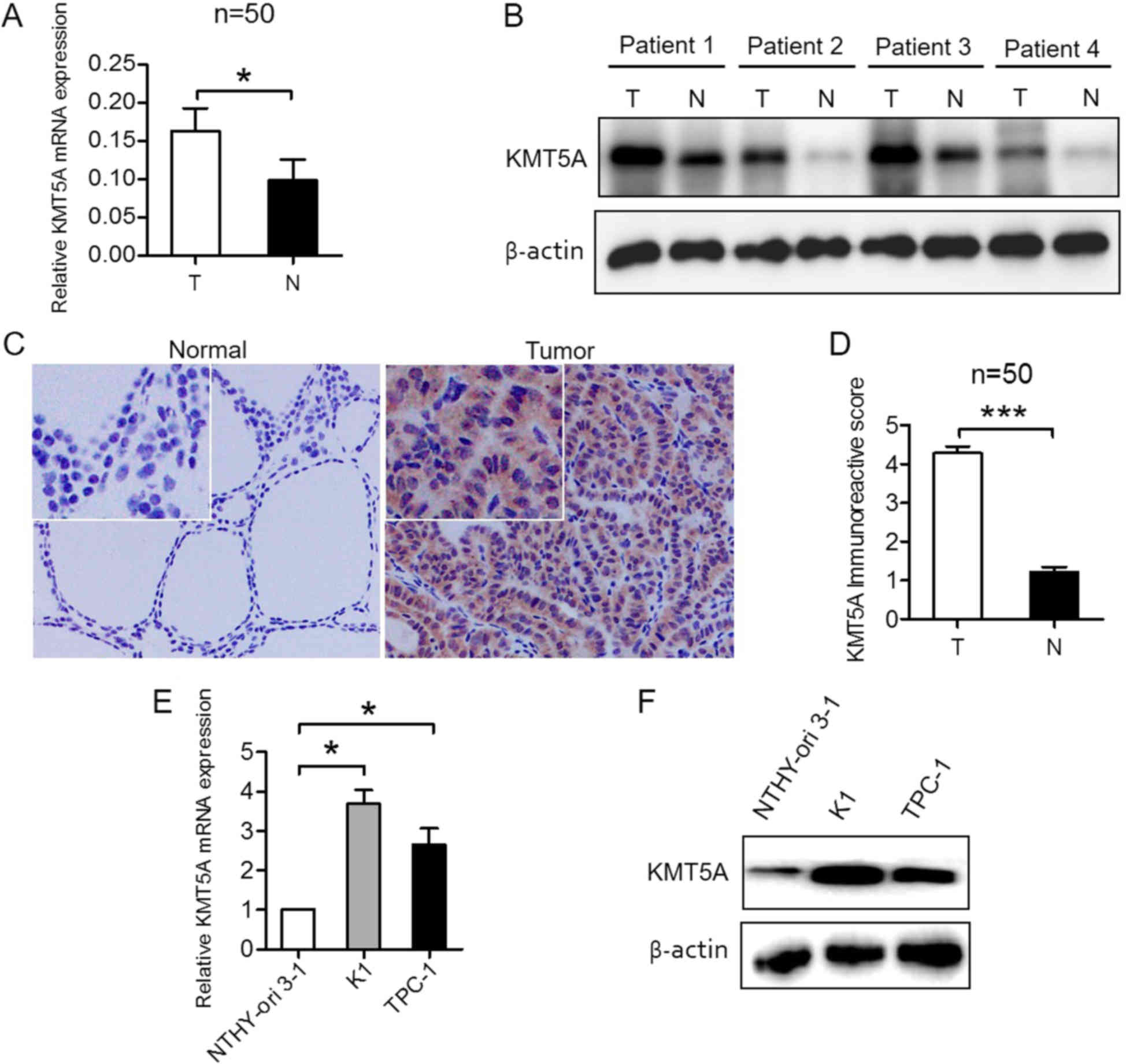
KMT5A is upregulated in the PTC
samples and cell lines
To evaluate KMT5A expression in PTC tissue, qRT-PCR
and western blotting, as well as IHC were performed in PTC and
matched normal thyroid tissue specimens. The results revealed that
KMT5A was transcriptionally upregulated in PTC samples (P<0.05;
Fig. 1A). Western blotting
(Fig. 1B) and IHC (P<0.001;
Fig. 1C and D) analysis confirmed
elevated KMT5A protein expression in PTC and low KMT5A protein
expression in normal thyroid tissue specimens. To investigate
whether KMT5A expression in PTC cell lines recapitulated its
expression patterns observed in PTC patient tissues, human normal
thyroid epithelial NTHY-ori 3-1 cells and PTC K1 and TPC-1 cells
were tested. KMT5A was found to be overexpressed in K1 (P<0.05)
and TPC-1 cells (P<0.05) compared to NTHY-ori 3-1 cells, both
transcriptionally (Fig. 1E) and at
protein level (Fig. 1F), which was
consistent with the results in the PTC tissue samples.
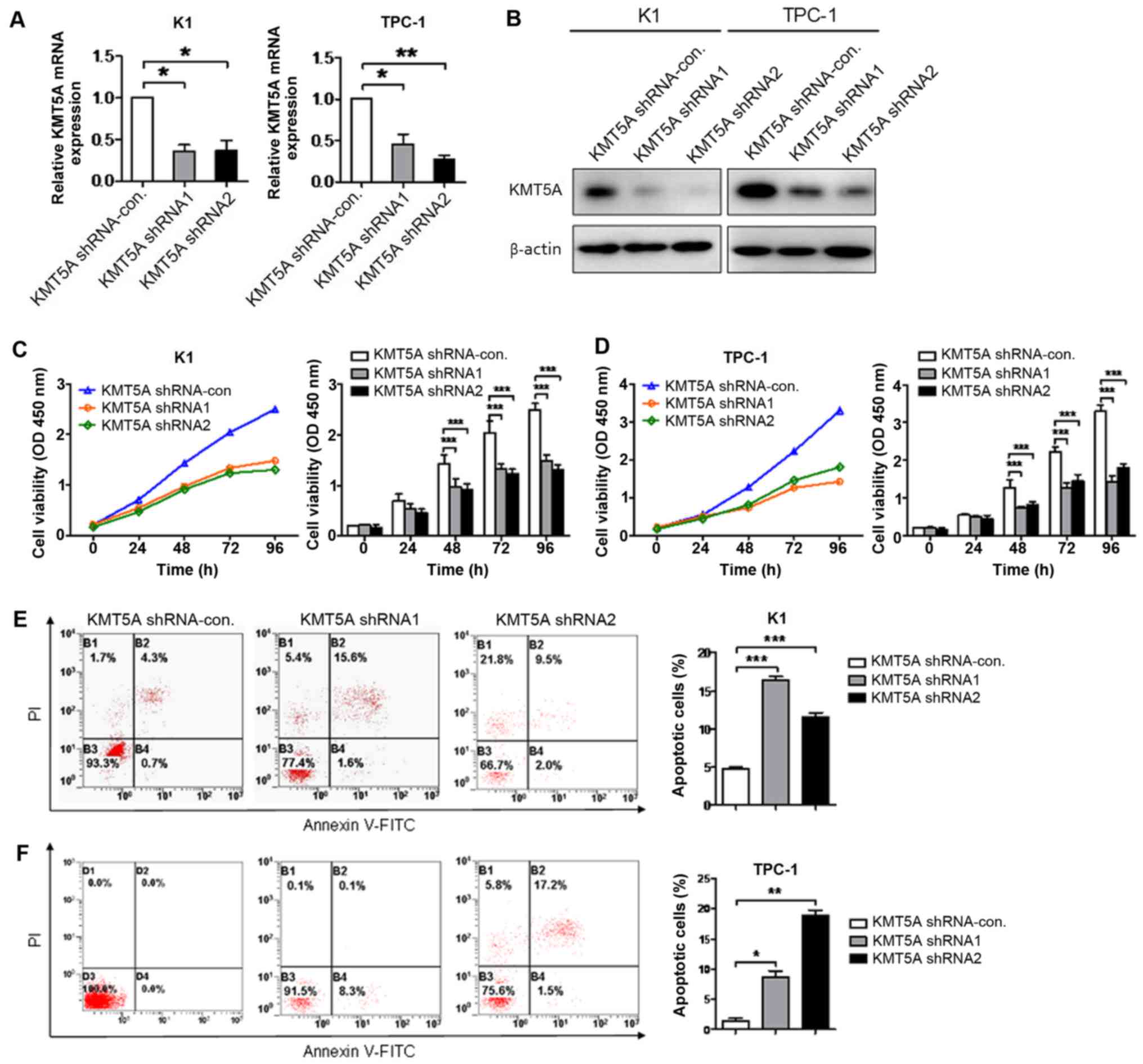
Knockdown of KMT5A inhibits
proliferation and induces apoptosis in PTC cells
To illuminate the efficacy of KMT5A on tumor cell
proliferation and apoptosis, the expression of KMT5A was knocked
down using targeted lentiviruses in K1 and TPC-1 cell lines.
Transcriptional and protein knockdown was achieved using two
separate lentiviral construct shRNA1 and shRNA2 as compared with
shRNA-control-infected cells in both K1 (shRNA1, P<0.05; shRNA2,
P<0.05) and TPC-1 (shRNA1, P<0.05; shRNA2, P<0.01) cells
(Fig. 2A and B). Targeted knockdown
of KMT5A resulted in attenuated proliferation in both K1 (shRNA1,
P<0.001; shRNA2, P<0.001; Fig.
2C) and TPC-1 (shRNA1, P<0.001; shRNA2, P<0.001; Fig. 2D) cells after 48 h. Moreover, a
significant increase in cell apoptosis measured by flow cytometry
of PI/Annexin V-stained cells was observed in both K1 (shRNA1,
P<0.001; shRNA2, P<0.001; Fig.
2E) and TPC-1 (shRNA1, P<0.05; shRNA2, P<0.01; Fig. 2F) cells using both shRNA1 and shRNA2
constructs.
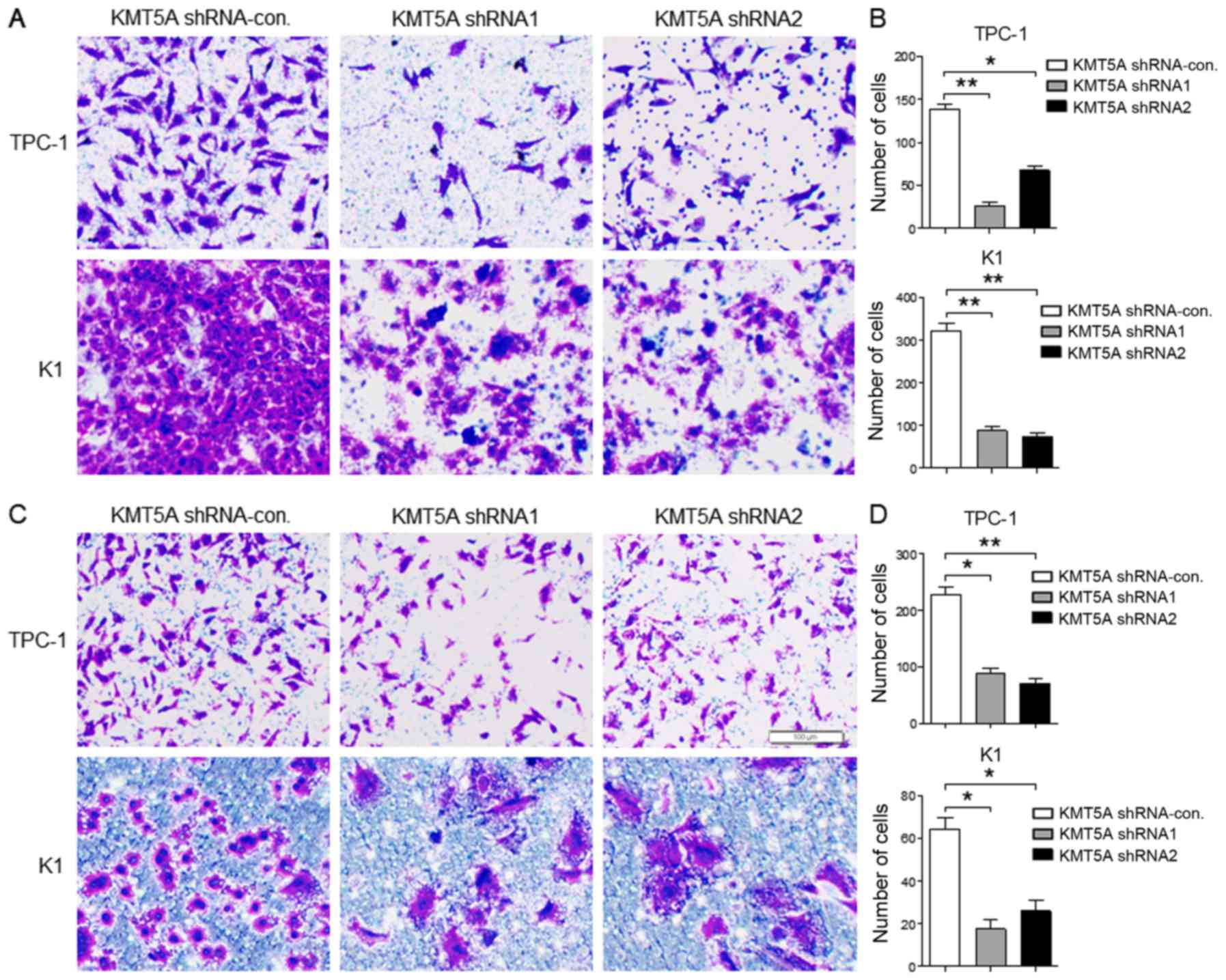
Downregulated KMT5A suppresses
migration and invasion in PTC cells
Next we evaluated the protumorigenic role of KMT5A
in the migration and invasion of PTC cells. The results revealed
that both TPC-1 (shRNA1, P<0.01; shRNA2, P<0.05) and K1
(shRNA1, P<0.01; shRNA2, P<0.01) cells transfected with KMT5A
shRNA1 or shRNA2 displayed significantly decreased cell migration
capacity than that of the shRNA-control vector (Fig. 3A and B). As shown in Fig. 3C and D, the invasion capacity was
examined by Transwell assay and the invasive rate was markedly
decreased in the KMT5A shRNA1- or shRNA2-transfected TPC-1 (shRNA1,
P<0.05; shRNA2, P<0.01) and K1 cells (shRNA1, P<0.05;
shRNA2, P<0.05) than in the shRNA-control transfected cells.
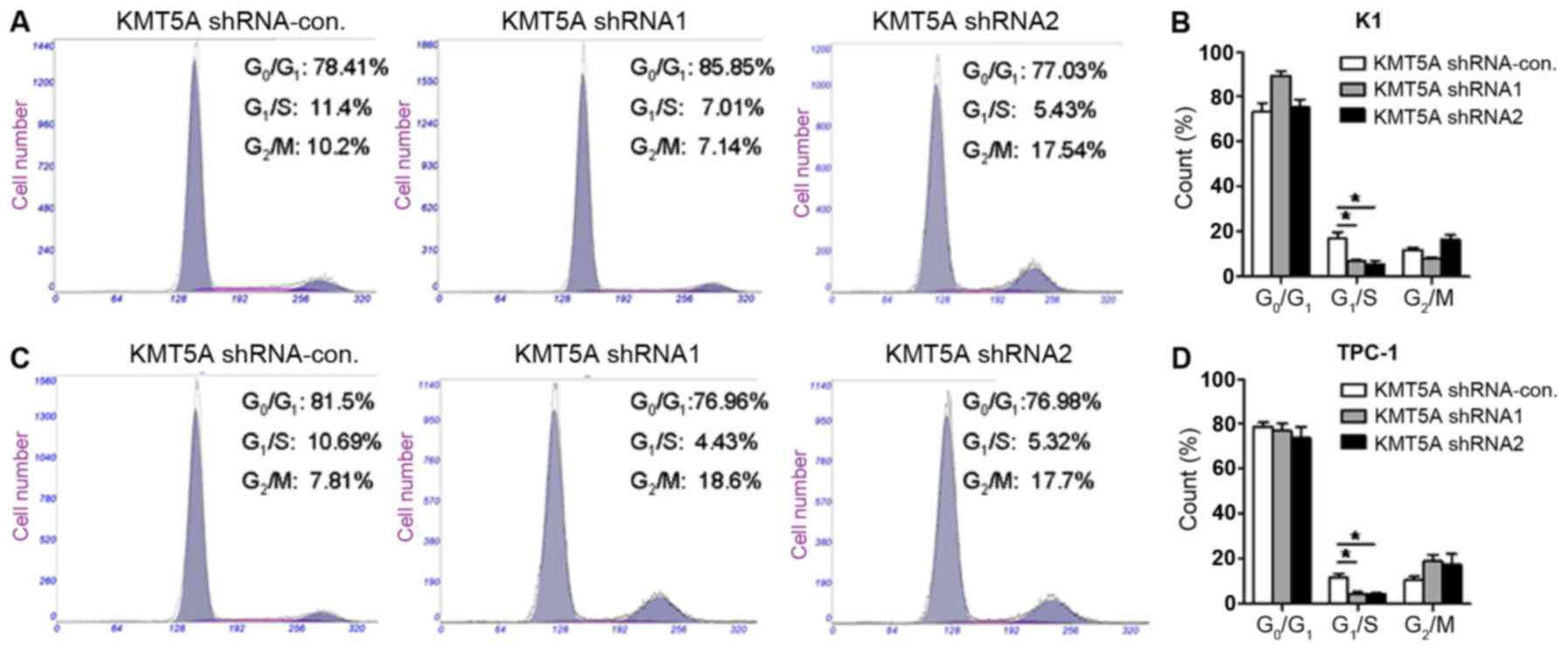
Loss of KMT5A arrests the G1/S phase
in PTC cells
Several lines of evidence have revealed that KMT5A
exerts crucial biological functions in modulating cell cycle and
DNA damage response (17). We
further verified the effect of KMT5A on the cell cycle of PTC cells
by flow cytometry assay. As shown in Fig. 4, both K1 (Fig. 4A and B) (shRNA1, P<0.05; shRNA2,
P<0.05) and TPC-1 (Fig. 4C and
D) (shRNA1, P<0.05; shRNA2, P<0.05) cells transfected
with KMT5A shRNA1 or shRNA2 had a significantly lower percentage of
cells in the G1/S phase compared with the control group. The data
revealed that the downregulation of KMT5A resulted in the cell
cycle arrest of the PTC cells in the G1/S phase.
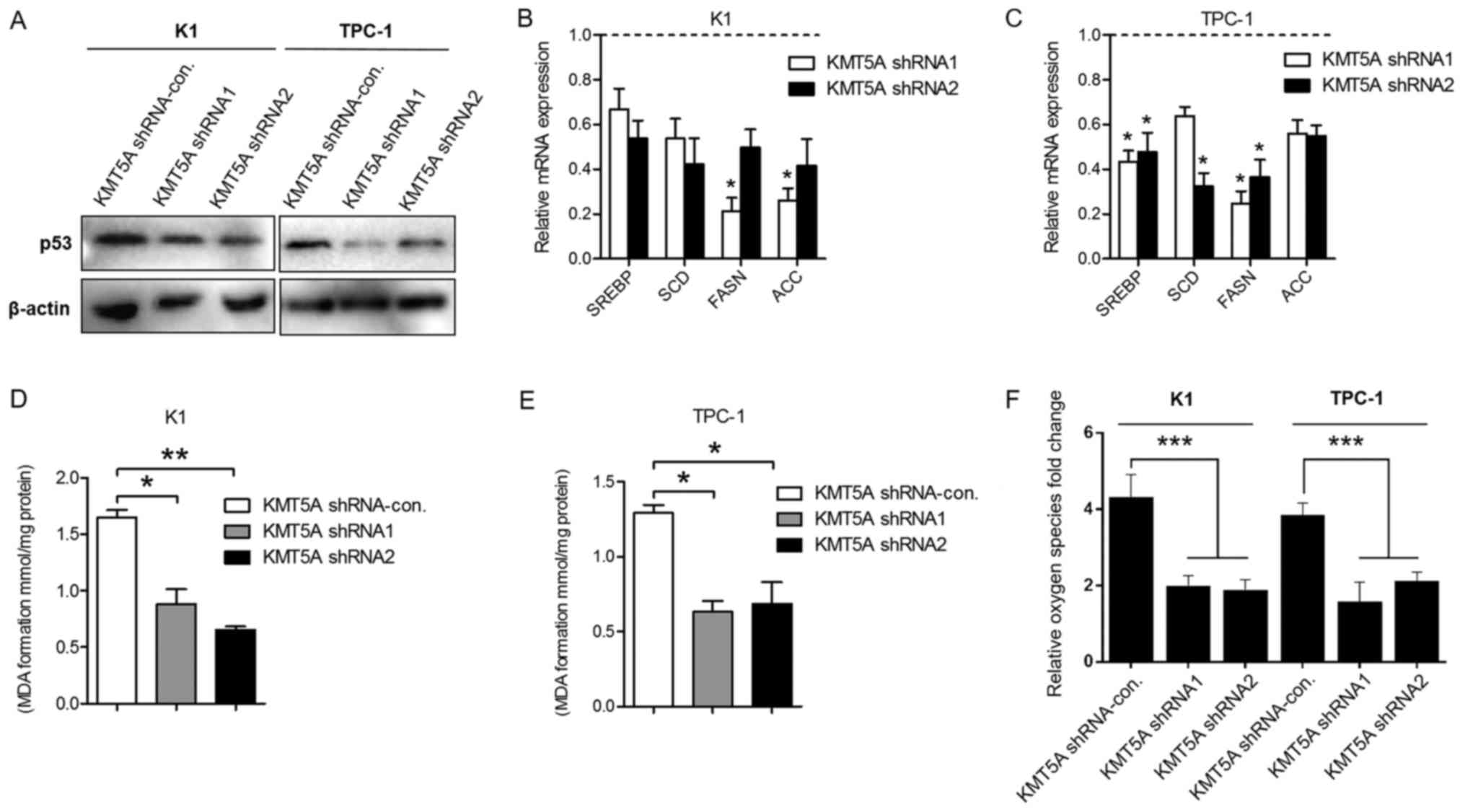
Targeting KMT5A reduces lipid
metabolism of PTC cells
As a lysine methyltransferase enzyme, KMT5A
regulates posttranslational modification of p53 at lysine 382 and
cell cycle arrest functions of p53 (18). Our results revealed that KMT5A
silencing led to the downregulation of p53 in the K1 and TPC-1
cells (Fig. 5A), which is
consistant with previous studies. Recently, p53 has been reported
to regulate metabolic pathways of breast cancer via
sterol-regulatory element-binding factor (SREBP) transcription
factors, a key modulator gene of the lipid/cholesterol metabolism
(19–22). To investigate the impact of KMT5A
silencing on the lipid metabolism, we examined the expression of
SREBP1 in KMT5A knockdown K1 and TPC-1 cells. As shown in Fig. 5B and C, the inhibition of KMT5A
reduced SREBP1 in K1 and TPC-1 when compared with control
transfected cells. Several lipid metabolism-related genes are
controlled by SREBP1, such as stearoyl-CoA desaturase (SCD), fatty
acid synthase (FASN) and acetyl-CoA carboxylase (ACC) (23). Subsequently, we revealed that KMT5A
inhibition also decreased SCD, FASN and ACC transcriptional
expression in the PTC cell lines (Fig.
5B and C).
Lipid peroxidation is the degradation of lipids that
occurs as a result of oxidative damage. Polyunsaturated lipids are
susceptible to oxidative attack, typically by ROS, resulting in a
well-defined chain reaction with the production of end products
such as MDA and ROS level. To further identify the effect of KMT5A
on lipid metabolism, we assessed the lipid peroxidation level using
MDA assay kit and the ROS generation level using Reactive Oxygen
Species assay kit. We demonstrated that silencing of KMT5A markedly
decreased both the level of MDA and ROS in the K1 and TPC-1 cells
compared with the shRNA-control-transfected cells (Fig. 5D-F).
Elevated KMT5A correlates with poor
prognosis of PTC patients
To investigate the relationship between KMT5A and
prognosis of PTC patients, we analyzed the KMT5A expression and
clinical data in the TCGA database. Our results revealed that
elevated KMT5A was significantly correlated with extrathyroidal
extension (P=0.0001), lymph node metastasis (P=0.0057) and advanced
pathological stage (P=0.0001) in PTC tissues. However, KMT5A
expression had no correlation with other clinicopathological
paremeters, such as age (P=0.769), sex (P=0.259) or tumor size
(P=0.29625) (Table I). These data
reveal that high KMT5A expression is associated with poor outcome
in PTC patients.
Discussion
KMT5A, also known as SET8, is a
monomethyltransferase required in cell cycle, replication,
transcription and chromosome segregation (24). Previous studies have revealed that
it plays an extensive role in multiple solid tumors. For example,
it is related with the prognosis of breast cancer (25), it induces epithelial-mesenchymal
transition and promotes metastasis of prostate cancer cells
(26) and is associated with
overall survival of gastric cancer patients (27). However, whether KMT5A has a function
on PTC is still unknown.
In the present study we demonstrated that KMT5A is
overexpressed in PTC tissue and cell lines K1 and TPC-1. To
investigate the biological role in PTC, we used a lentivirus system
to downregulate KMT5A expression in PTC cells. We found that
knockdown of KMT5A inhibited the proliferation, migration and
invasion of K1 and TPC-1 cells. In addition the inhibition of KMT5A
induced apoptosis and arrested the G1 phase of the cell cycle in
PTC cells. It is reported that KMT5A is almost undetectable in the
S phase of the cell cycle and its impact on cell cycle progression
is mainly through H4K20me1. Thus, the disruption of endogenous
KMT5A may be accompanied by the suppression of H4K20
monomethylation and lead to cell cycle defects, chromatin
decondensation and enlarged nuclei, indicating the essential role
of KMT5A in DNA replication. Our results revealed that KMT5A may
take part in the oncogenesis of PTC as a tumor gene. The
relationship between KMT5A gene expression and extrathyroidal
extension, lymph node metastasis, advanced pathological stage of
papillary thyroid cancer patients from TCGA was in line with our
in vitro data, indicating that KMT5A can be a novel marker
for the prognosis of PTC patients.
Another finding in the present study was that the
knockdown of KMT5A resulted in the decrease of p53, a crucial tumor
suppressor maintaining metabolic homeostasis and orchestrating
cellular stress responses. Recent research has reported that p53
and Hippo pathway cooperate on multiple levels to fine-tune SREBP
activity and regulate cholesterol/lipid levels (28). The interaction between KMT5A and
p53, p53 and SREBP raises the question of whether KMT5A regulates
SREBP-mediated lipid metabolism. As expected, our results indicated
that inhibition of KMT5A reduced the level of SREBP1 and its target
genes (SCD, FASN and ACC), as well as suppressed the level of MDA
and ROS generation in PTC cells, suggesting that KMT5A may regulate
the lipid metabolism of PTC. However, whether the effect of KMT5A
on the lipid metabolism is implemented via p53 needs more evidence
to be elucidated.
In conclusion, a key issue we addressed for the
first time is that histone methyltransferase KMT5A regulates the
lipid metabolism of PTC. However, the underlying mechanism needs to
be further explored. Additional investigation into other
constituents of lipid metabolism may provide fresh insight into
papillary thyroid carcinogenesis as well as reveal other potential
therapeutic targets.
Acknowledgements
We would like to thank the University of Colorado
Cancer Center Cell Bank for providing the PTC cell lines K1 and
TPC-1. This study was partly supported by funds from the project
sponsored by the Scientific Research Foundation for the Returned
Overseas Chinese Scholars, State Education Ministry (to T.L.), the
National Natural Science Foundation of China (nos. 81272934,
81572622 and 81772854 to Q.-H.J. and no. 81702753 to T.L.) and the
Shanghai Science and Technology Commission Western Guide project
(no. 14411962402 to D.-S.L.).
References
|
1
|
Morris LG, Tuttle RM and Davies L:
Changing trends in the incidence of thyroid cancer in the United
States. JAMA Otolaryngol Head Neck Surg. 142:709–711. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xing M, Alzahrani AS, Carson KA, Shong YK,
Kim TY, Viola D, Elisei R, Bendlová B, Yip L, Mian C, et al:
Association between BRAF V600E mutation and recurrence of papillary
thyroid cancer. J Clin Oncol. 33:42–50. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Carracedo A, Cantley LC and Pandolfi PP:
Cancer metabolism: Fatty acid oxidation in the limelight. Nat Rev
Cancer. 13:227–232. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Santos CR and Schulze A: Lipid metabolism
in cancer. FEBS J. 279:2610–2623. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Baenke F, Peck B, Miess H and Schulze A:
Hooked on fat: The role of lipid synthesis in cancer metabolism and
tumour development. Dis Model Mech. 6:1353–1363. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
von Roemeling CA, Marlow LA, Pinkerton AB,
Crist A, Miller J, Tun HW, Smallridge RC and Copland JA: Aberrant
lipid metabolism in anaplastic thyroid carcinoma reveals stearoyl
CoA desaturase 1 as a novel therapeutic target. J Clin Endocrinol
Metab. 100:E697–E709. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Uddin S, Siraj AK, Al-Rasheed M, Ahmed M,
Bu R, Myers JN, Al-Nuaim A, Al-Sobhi S, Al-Dayel F, Bavi P, et al:
Fatty acid synthase and AKT pathway signaling in a subset of
papillary thyroid cancers. J Clin Endocrinol Metab. 93:4088–4097.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li Z, Nie F, Wang S and Li L: Histone H4
Lys 20 monomethylation by histone methylase SET8 mediates Wnt
target gene activation. Proc Natl Acad Sci USA. 108:3116–3123.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Abbas T, Shibata E, Park J, Jha S, Karnani
N and Dutta A: CRL4Cdt2 regulates cell proliferation and
histone gene expression by targeting PR-Set7/Set8 for degradation.
Mol Cell. 40:9–21. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Centore RC, Havens CG, Manning AL, Li JM,
Flynn RL, Tse A, Jin J, Dyson NJ, Walter JC and Zou L:
CRL4Cdt2-mediated destruction of the histone
methyltransferase Set8 prevents premature chromatin compaction in S
phase. Mol Cell. 40:22–33. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Houston SI, McManus KJ, Adams MM, Sims JK,
Carpenter PB, Hendzel MJ and Rice JC: Catalytic function of the
PR-Set7 histone H4 lysine 20 monomethyltransferase is essential for
mitotic entry and genomic stability. J Biol Chem. 283:19478–19488.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Oda H, Hübner MR, Beck DB, Vermeulen M,
Hurwitz J, Spector DL and Reinberg D: Regulation of the histone H4
monomethylase PR-Set7 by CRL4Cdt2-mediated
PCNA-dependent degradation during DNA damage. Mol Cell. 40:364–376.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu S, Wang W, Kong X, Congdon LM, Yokomori
K, Kirschner MW and Rice JC: Dynamic regulation of the PR-Set7
histone methyltransferase is required for normal cell cycle
progression. Genes Dev. 24:2531–2542. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shi X, Kachirskaia I, Yamaguchi H, West
LE, Wen H, Wang EW, Dutta S, Appella E and Gozani O: Modulation of
p53 function by SET8-mediated methylation at lysine 382. Mol Cell.
27:636–646. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang F, Sun L, Li Q, Han X, Lei L, Zhang H
and Shang Y: SET8 promotes epithelial-mesenchymal transition and
confers TWIST dual transcriptional activities. EMBO J. 31:110–123.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liao T, Wen D, Ma B, Hu JQ, Qu N, Shi RL,
Liu L, Guan Q, Li DS and Ji QH: Yes-associated protein 1 promotes
papillary thyroid cancer cell proliferation by activating the
ERK/MAPK signaling pathway. Oncotarget. 8:11719–11728. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang Z, Dai X, Zhong J, Inuzuka H, Wan L,
Li X, Wang L, Ye X, Sun L, Gao D, et al: SCFβ−TRCP
promotes cell growth by targeting PR-Set7/Set8 for degradation. Nat
Commun. 6:101852015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Swain JF, Dinler G, Sivendran R,
Montgomery DL, Stotz M and Gierasch LM: Hsp70 chaperone ligands
control domain association via an allosteric mechanism mediated by
the interdomain linker. Mol Cell. 26:27–39. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cook KL, Soto-Pantoja DR, Clarke PA, Cruz
MI, Zwart A, Wärri A, Hilakivi-Clarke L, Roberts DD and Clarke R:
Endoplasmic reticulum stress protein GRP78 modulates lipid
metabolism to control drug sensitivity and antitumor immunity in
breast cancer. Cancer Res. 76:5657–5670. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sorrentino G, Ruggeri N, Specchia V,
Cordenonsi M, Mano M, Dupont S, Manfrin A, Ingallina E, Sommaggio
R, Piazza S, et al: Metabolic control of YAP and TAZ by the
mevalonate pathway. Nat Cell Biol. 16:357–366. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Freed-Pastor WA, Mizuno H, Zhao X,
Langerød A, Moon SH, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li
W, Polotskaia A, et al: Mutant p53 disrupts mammary tissue
architecture via the mevalonate pathway. Cell. 148:244–258. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li X, Wu JB, Chung LW and Huang WC:
Anti-cancer efficacy of SREBP inhibitor, alone or in combination
with docetaxel, in prostate cancer harboring p53 mutations.
Oncotarget. 6:41018–41032. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Horton JD, Goldstein JL and Brown MS:
SREBPs: Activators of the complete program of cholesterol and fatty
acid synthesis in the liver. J Clin Invest. 109:1125–1131. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dulev S, Tkach J, Lin S and Batada NN:
SET8 methyltransferase activity during the DNA double-strand break
response is required for recruitment of 53BP1. EMBO Rep.
15:1163–1174. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu B, Zhang X, Song F, Liu Q, Dai H,
Zheng H, Cui P, Zhang L, Zhang W and Chen K: A functional single
nucleotide polymorphism of SET8 is prognostic for breast cancer.
Oncotarget. 7:34277–34287. 2016.PubMed/NCBI
|
|
26
|
Hou L, Li Q, Yu Y, Li M and Zhang D: SET8
induces epithelial mesenchymal transition and enhances prostate
cancer cell metastasis by cooperating with ZEB1. Mol Med Rep.
13:1681–1688. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shi XL, Guo ZJ, Wang XL, Liu XL and Shi
GF: SET8 expression is associated with overall survival in gastric
cancer. Genet Mol Res. 14:15609–15615. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Aylon Y and Oren M: The Hippo pathway, p53
and cholesterol. Cell Cycle. 15:2248–2255. 2016. View Article : Google Scholar : PubMed/NCBI
|