Introduction
Cholesteatoma is a pathologically benign chronic
middle ear disease, but potentially destructive epithelial lesion
caused by aberrant keratinocyte proliferation and migration which
can result in erosion of adjacent osseous structures, leading to
various clinical manifestations and serious complications (1). Bone resorption of the middle and inner
ear can cause hearing loss, tinnitus, dizziness, and facial
paralysis. Erosion of the tegmen may lead to many severe
intracranial complications, including sigmoid sinus
thrombophlebitis, extradural, subdural or brain abscess,
meningitis, and hydrocephalus (2–5). To
date, there is no drug therapy for cholesteatoma and the standard
treatment is surgical resection (2). In addition, postoperative recurrences
are very common; thus many patients undergo multiple surgeries
(4,5).
Nevertheless, the exact underlying cellular and
molecular mechanisms of cholesteatoma are still unclear. Previous
studies indicated that, during cholesteatoma formation, epithelial
proliferation and keratinocyte differentiation are regulated by
high activity of growth factors (6). Cytokines are also supposed to play
important roles in the aggressive behavior of cholesteatoma
(6). In addition to those
inflammatory mediators and growth factors, aberrant expression of
regulatory microRNAs (miRNAs), the best-studied non-coding RNAs
(ncRNAs), play a role in cholesteatoma formation (7–10).
However, due to a complementarity of merely 6 base pairs (bp) nt
can be sufficient for target recognition, a miRNA can regulate
hundreds of mRNAs, and correspondingly, a mRNA can be regulated by
several miRNAs (11), leading to
the difficulties encountered when clarifying the molecular
mechanisms underlying cholesteatoma.
In 2011, Salmena et al introduced the
competitive endogenous RNA (ceRNA) hypothesis that all types of RNA
transcripts can communicate with and regulate each other by using
shared microRNA response elements (MREs) (12), which provides us with a new
perspective for research on the cholesteatoma formation process.
CeRNAs, also termed endogenous miRNA sponges, act as decoy
molecules that absorb active miRNAs and buffer regulation
activities of miRNAs on their targeted mRNAs (13).
Salmena et al proposed that the ceRNA
protagonists are miRNAs, the protein coding genes, pseudogenes, and
long non-coding RNAs (lncRNAs). lncRNAs, which are noncoding RNAs
longer than 200 nt, can regulate each other with miRNAs and
function in almost every aspect of human biology, including
chromatin modification, transcriptional regulation,
post-transcriptional processing, RNA editing, RNA trafficking, cell
cycle regulation, alternative splicing, and organelle biogenesis
(14,15). In the past decade, with the
emergence of microarray and high-throughput sequencing techniques,
lncRNAs have attracted the attention and have been involved in
epigenetic mechanisms of various diseases and display ceRNA
potential in many diseases (16–23).
Nevertheless, to date, no studies have addressed the expression
profiles of lncRNAs in cholesteatoma.
Therefore, in the present study, we performed
microarray analysis to identify the differentially expressed
patterns of lncRNAs and mRNAs between cholesteatoma and normal skin
tissues. Quantitative RT-PCR was applied to confirm the reliability
of microarray expression data. With specific bioinformatics
approaches, we constructed the lncRNA-miRNA-mRNA ceRNA network to
explore the ceRNA potential of lncRNAs in cholesteatoma.
Material and methods
Patients and specimens
Cholesteatoma tissue and matched post-auricular
normal skin tissue were sampled in each patient. A total of 7
patients aged 18–32 years who underwent unilateral middle ear
cholesteatoma surgeries between June 2016 and November 2016 at the
Department of Otorhinolaryngology, Peking Union Medical College
Hospital were enrolled in this study. All participants provided
written informed consent and were clinically and histologically
confirmed to present with cholesteatoma. All specimens were stored
at −80°C immediately after collection for later RNA extraction.
Ethical approval for the study was obtained from the Ethics
Committee of Peking Union Medical College Hospital (no.
S-K292).
RNA extraction and quality
control
Total RNA was extracted from cholesteatoma and
post-auricular skin tissues using TRIzol reagent (Invitrogen,
Carlsbad, CA, USA), according to the manufacturer's instructions.
RNA quantity and quality were assessed by NanoDrop ND-1000. RNA
integrity was assessed by standard denaturing agarose gel
electrophoresis or Agilent 2100 Bioanalyzer (Agilent Technologies,
Santa Clara, CA, USA).
Microarray analysis
Four pairs of cholesteatoma and post-auricular skin
tissues were used for microarray assay to determine differentially
expressed lncRNAs and mRNAs comparing cholesteatoma and
post-auricular skin specimens. Sample labeling and array
hybridization were performed according to the Agilent One-Color
Microarray-Based Gene Expression Analysis protocol (Agilent
Technologies, Englewood, CO, USA) with minor modifications. The
hybridized arrays were washed, fixed, and scanned using the Agilent
DNA Microarray Scanner (part no. G2505C). Agilent Feature
Extraction software (version 11.0.1.1) was used to analyze acquired
array images. Quantile normalization and subsequent data processing
were performed using the GeneSpring GX v12.1 software package
(Agilent Technologies, Englewood, CO, USA). After quantile
normalization of the raw data, lncRNAs and mRNAs in the 8 samples
flagged as ‘Present’ or ‘Marginal’ were chosen for further data
analysis. Differentially expressed lncRNAs and mRNAs with
statistical significance between the two groups were identified
through P-value/FDR filtering. Differentially expressed lncRNAs and
mRNAs between the two samples were identified through Fold Change
filtering. Pathway analysis and Gene ontology (GO) analysis were
applied to determine the roles played by these differentially
expressed mRNAs in these biological pathways or GO terms.
Hierarchical clustering and combined analysis were performed by
using in-house scripts.
Functional group analysis
GO provides a controlled vocabulary to describe gene
function and relationships between these concepts in any organism
(http://www.geneontology.org). GO covers
three aspects: Biological process, cellular component, and
molecular function. Fisher's exact test was applied to determine
whether the overlap between the differentially expressed (DE) list
and the GO annotation list is greater than that expected by chance.
The -log10 (P-value) was used to denote the significance
of the GO term enrichment in the DE genes. FDR represents the false
discovery rate. The lower the P-value is, the more significant the
GO term is (a P-value <0.05 is recommended). Pathway analysis
was performed to collect pathway clusters on the molecular
interaction and reaction networks by mapping genes to the Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathways (http://www.genome.jp/kegg/). The -log10
(P-value) denotes the significance of the pathway correlations. The
lower the P-value is, the more significant the correlation is (a
P-value <0.05 is recommended).
Quantitative real-time PCR
validation
The selected lncRNAs and primers used for qRT-PCR
were designed using Primer 5.0 and synthesized by Generay Biotech
(Shanghai, China). β-actin was used as an internal control for all
samples. Primer sequences were as follows: β-actin forward,
5′-GTGGCCGAGGACTTTGATTG-3′ and reverse,
5′-CCTGTAACAACGCATCTCATATT-3′; ENST00000415386 forward,
5′-TGGAGTAGGCACAGACGGAA-3′ and reverse,
5′-GACTGTGGTACAATCGCTTCG-3′; ENST00000420253 forward,
5′-CGATGTCAACCCTCGAACCT-3′ and reverse, 5′-TCAGATGTGGCCCACTGTCT-3′;
NR_024468 forward, 5′-GCTTACATTTTCTCTGCCATCTC-3′ and reverse,
5′-CCACTTTCCTTTCTCTCTCTTCT-3′; T044224 forward,
5′-TGCGAAAACACTGCGATTG-3′ and reverse, 5′-GGTTGGACCAGACCAGATGA-3′;
T347175 forward, 5′-GAAAGAGGCACTGCTGTTGA-3′ and reverse,
5′-GGCTGCTCCCAGAATAGATAG-3′; uc001kfc.1 forward,
5′-TTCCCAGAAGGCGTAGGTT-3′ and reverse,
5′-GACAGAGTCTCCCTCTATCATCC-3′; qRT-PCR was performed by using ViiA
7 Real-time PCR System (Applied Biosystems, San Jose, CA, USA) with
a SYBR expression assay system (Takara Biotechnology, Co., Ltd.,
Dalian, China). The PCR reaction conditions were as follows: An
initial denaturation at 95°C for 10 min, followed by 40 PCR cycles
at 95°C for 10 sec and 60°C for 60 sec. Then annealing and
extension at 95°C for 10 sec, 60°C for 60 sec and 95°C for 15 sec.
Each sample was assayed in triplicates. The 2−ΔΔCt
method was used to determine fold-change in gene expression in the
cholesteatoma samples relative to the normal skin samples. For
statistical analysis, we used unpaired t-test to compare the
expression of lncRNAs between cholesteatoma and normal skin
samples. P<0.05 was considered to indicate a statistically
significant difference.
Competing endogenous network
analysis
All potential miRNA response elements were searched
based on the sequences of lncRNAs and mRNAs. LncRNA/miRNA/mRNA
interactions were predicted by the overlapping of the miRNA seed
sequence binding site both on the chosen dysregulated lncRNAs and
the significantly dysregulated mRNA. miRBase V19 was selected to
interact with the lncRNAs and mRNAs (http://www.mirbase.org/). LncRNA/miRNA interactions
were predicted by miRcode (http://www.mircode.org/). miRNA/mRNA interactions were
predicted by miRanda (www.microrna.org/) and TargetScan (http://www.targetscan.org/).
Results
LncRNAs and mRNAs present
significantly different expression profiles in cholesteatoma
compared to matched normal skin specimens
A microarray analysis was performed to profile
differences in lncRNA and mRNA expression between 4 pairs of
cholesteatoma and matched normal skin samples. According to
microarray expression profiling data, 11,815 lncRNAs and 7,692
mRNAs were detected. The lncRNAs were carefully collected from the
most authoritative databases such as RefSeq, UCSC Known Genes,
Gencode, and other related sources. According to their relation
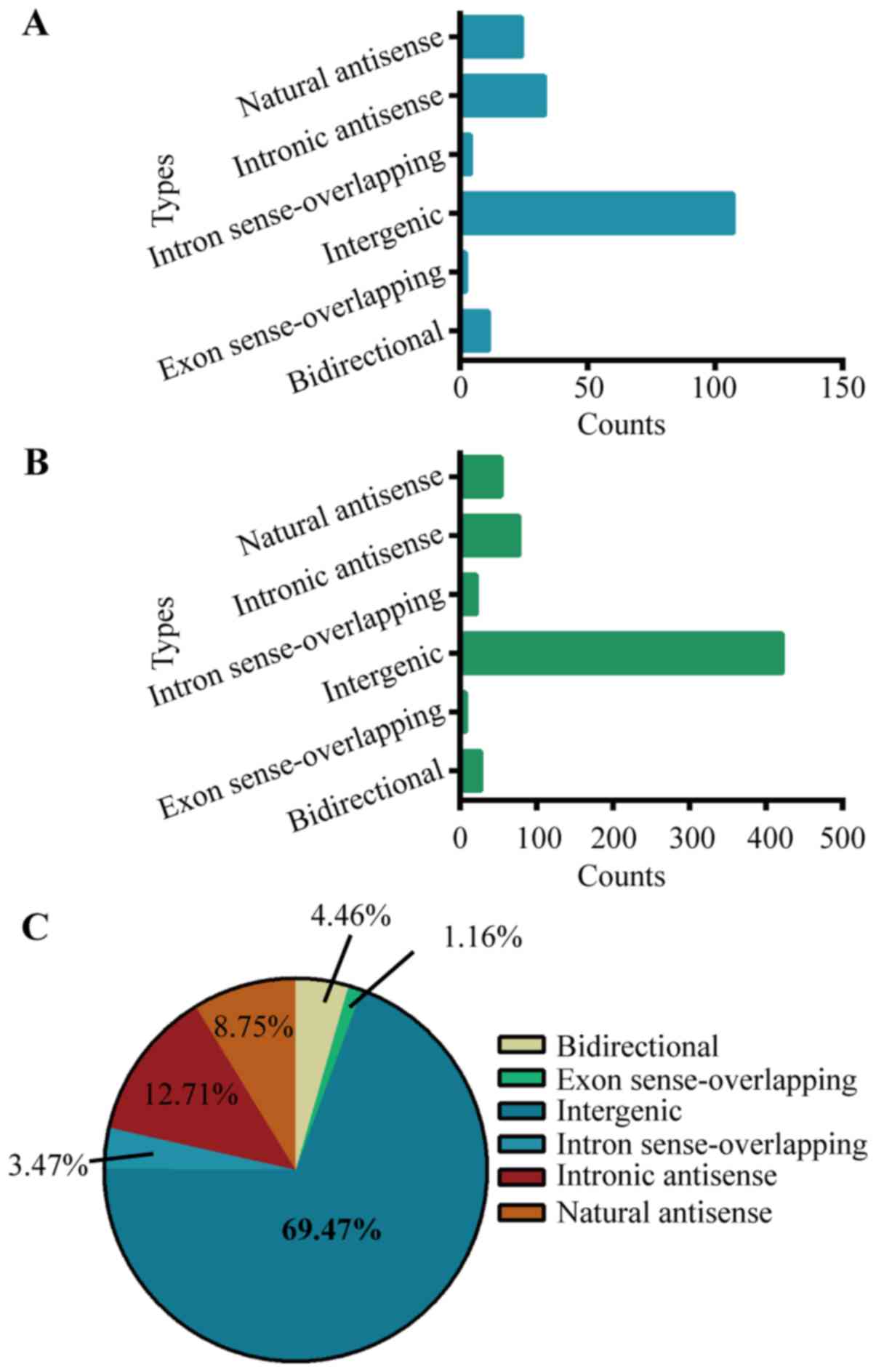
with protein-coding genes, all deregulated lncRNAs in cholesteatoma
were classified into six categories: Bidirectional (4.46%), exon
sense-overlapping (1.16%), intron sense-overlapping (3.47%),
natural antisense (8.75%), intronic antisense (12.71%) and
intergenic (69.47%) (Fig. 1).
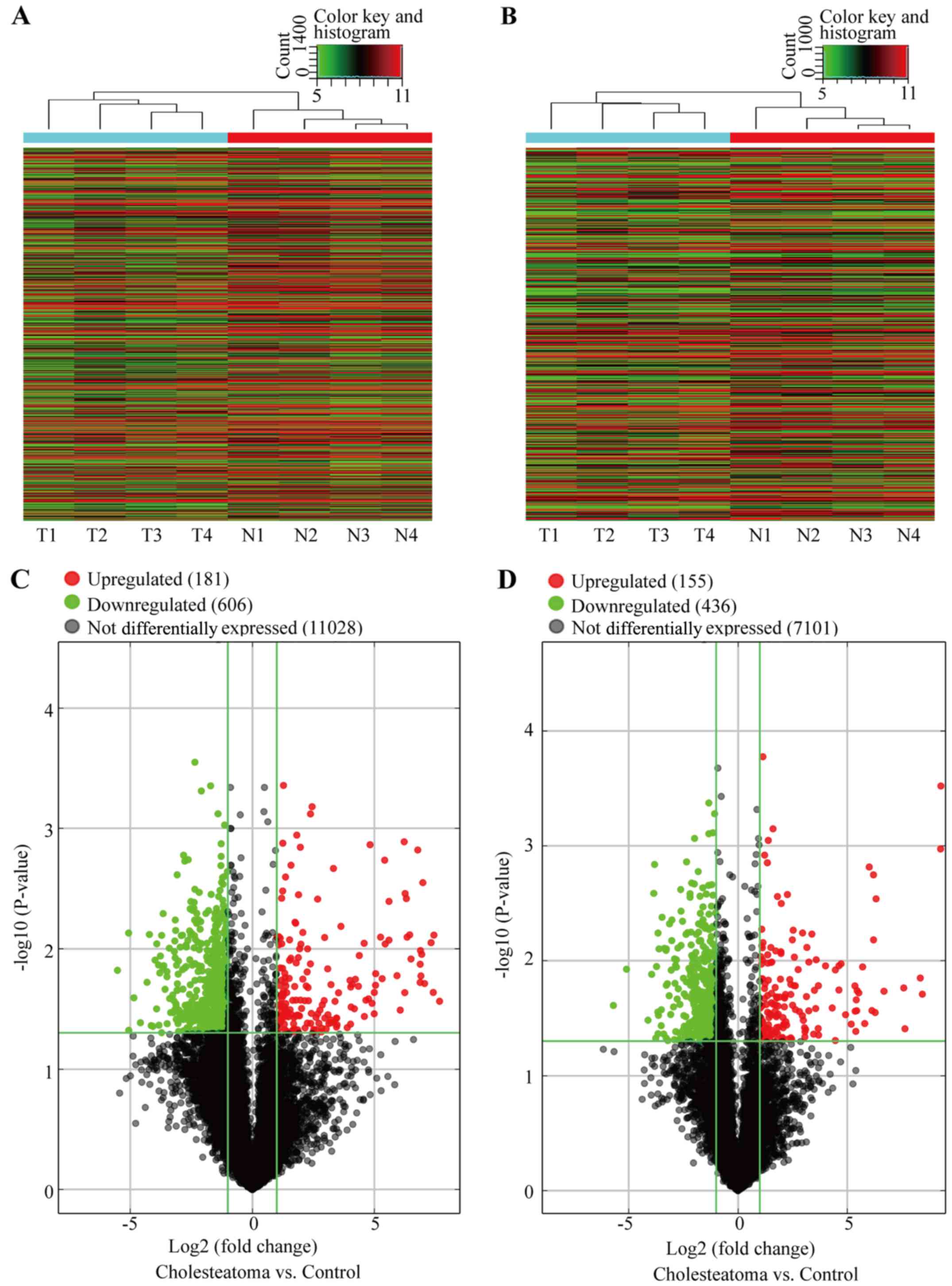
Hierarchical clustering revealed that lncRNA and mRNA expression
patterns between cholesteatoma and matched normal skin tissues were
distinguishable (Fig. 2A and B).
Volcano plots were used for visualization and assessment of the
variation (or reproducibility) of lncRNA and mRNA expression
between cholesteatoma and matched normal skin tissues (Fig. 2C and D).
Furthermore, by setting a threshold for differential
expression at changes ≥2.0-fold, we identified 787 lncRNAs and 591
mRNAs that were differentially expressed (P<0.05) between
cholesteatoma and matched normal skin tissues (Fig. 2C and D). Among them, in
cholesteatoma samples, 181 lncRNAs and 155 mRNAs were upregulated
(fold change ≥2.0, P<0.05) and 606 lncRNAs and 436 mRNAs were
downregulated (fold change ≥2.0, P<0.05) compared with normal
skin samples (Fig. 2C and D). The
microarray profile and RNAseq data sets have been deposited into
Gene Expression Omnibus (GEO) with accession number GSE102673
(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE102673).
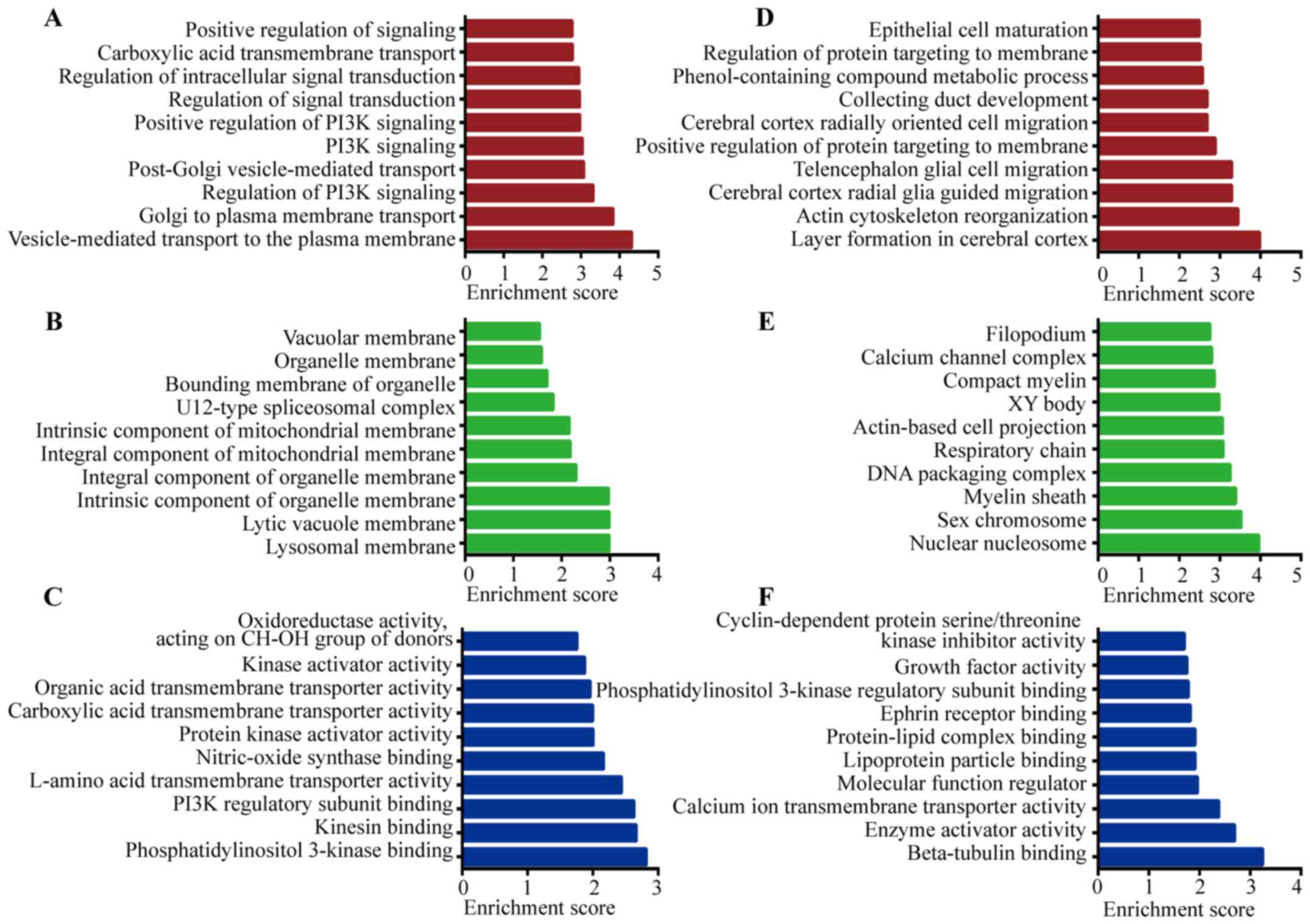
GO analyses display
cholesteatoma-related gene functions of differentially expressed
mRNAs
GO analysis was performed to determine the gene and
gene product enrichment in categories including biological process
(BP), molecular function (MF), and cellular component (CC). LncRNAs
can regulate the neighboring and overlapping coding gene
expression. Thus, GO enrichment analysis of differentially
expressed mRNAs may partially display the role of differentially
regulated lncRNAs. In the present study, the target genes of
deregulated mRNAs were analyzed and compared with those in
cholesteatoma. In the GO analysis, by setting P<0.05, the
upregulated and downregulated mRNAs were analyzed separately and
the top-10 enriched GO terms of BP, CC, MF were listed (Fig. 3). In BP analysis, regulation of
phosphatidylinositol 3-kinase signaling (GO:0014066),
phosphatidylinositol 3-kinase signaling (GO:0014065), and positive
regulation of phosphatidylinositol 3-kinase signaling (GO:0014068)
belonged to top-10 most enriched processes associated with the
upregulated mRNAs. Moreover, phosphatidylinositol 3-kinase binding
(GO:0043548) was the most significantly enriched function of the
upregulated mRNAs in MF analysis (Fig.
3C). It is notable, since phosphatidylinositol 3-kinase (PI3K)
has been reported to play a crucial role in the formation of
cholesteatoma (24). Furthermore,
growth factor activity (GO:0008083), one of the most significantly
enriched processes associated with the downregulated mRNAs in MF,
is also involved in cholesteatoma formation (25).
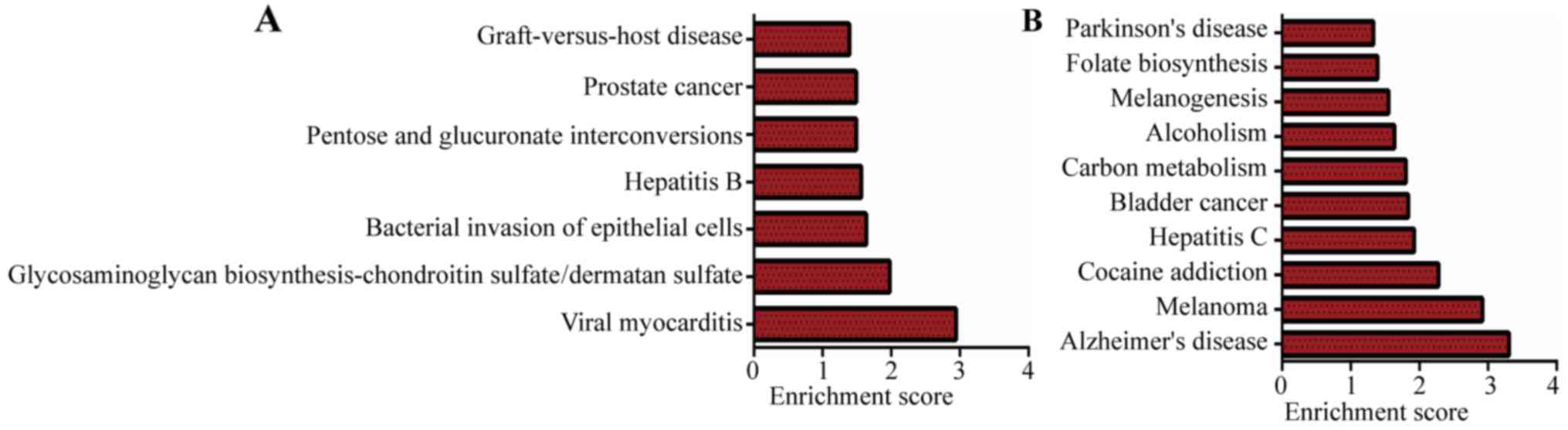
Pathway analyses reveal significant
enrichment of potential cholesteatoma-related pathways
By mapping genes to KEGG pathways, we performed
pathway analysis (http://www.genome.jp/kegg/pathway.html). KEGG pathway
enrichment analysis for differentially expressed mRNAs is devised
to comprehend pathways and molecular interactions related to genes.
Pathway analysis indicated that 10 enriched pathways corresponded
to downregulated mRNAs and 7 pathways corresponded to upregulated
mRNAs (P<0.05, Fig. 4). The
pathways enriched with upregulated lncRNAs were involved in a
category ‘bacterial invasion of epithelial cells (hsa05100)’ that
is involved in the molecular mechanisms of cholesteatoma (26). Furthermore, ‘viral myocarditis
(hsa05416)’ and ‘Hepatitis B (hsa05161)’ were also related to the
molecular biology of cholesteatoma (6).
Quantitative real-time PCR confirms
microarray expression results
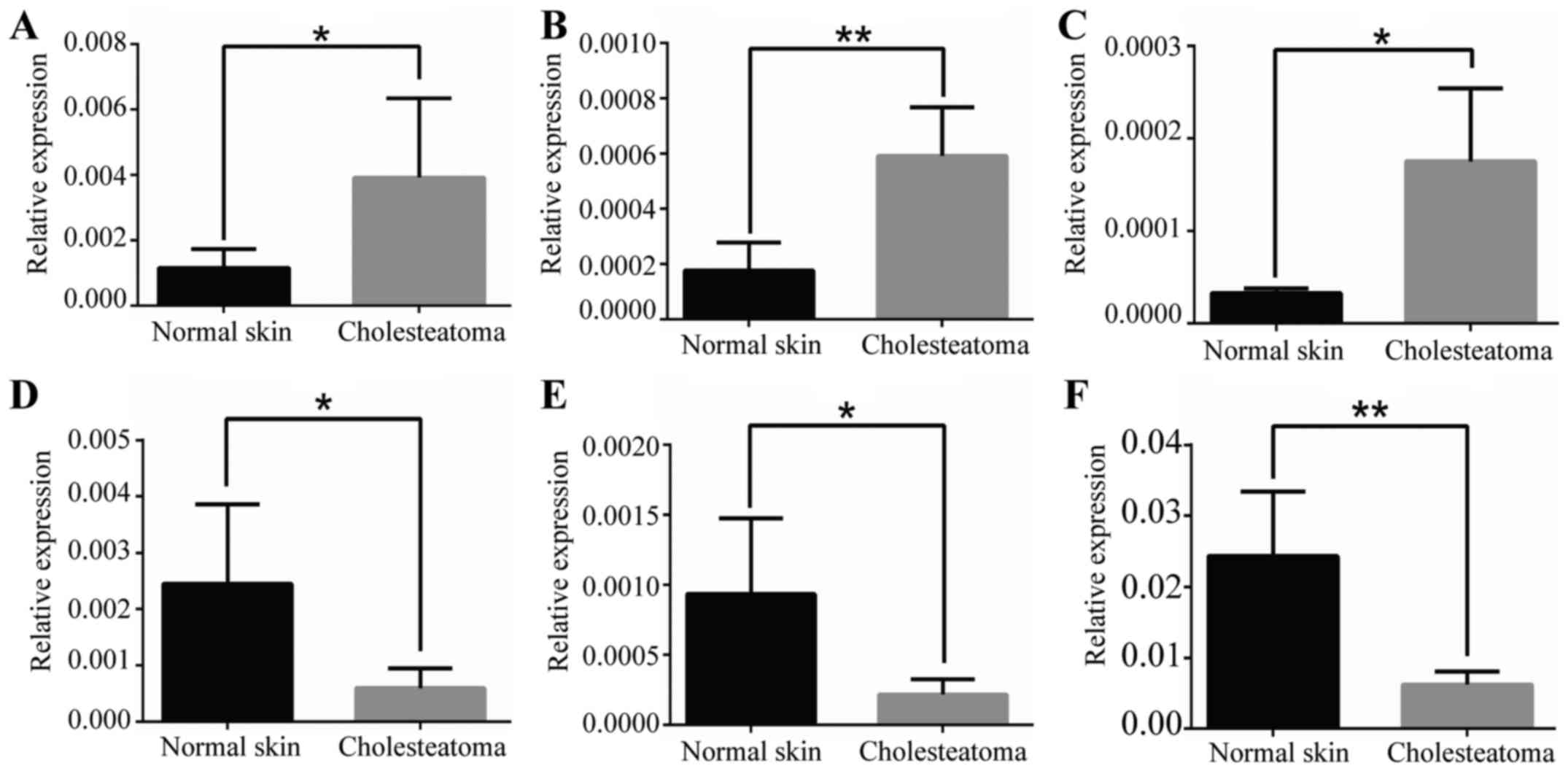
Using qRT-PCR, 3 upregulated and 3 downregulated
lncRNAs with fold-changes >2.0 were randomly selected to verify
the microarray data in 7 pairs of samples (4 pairs of original
tissues and 3 other pairs of cholesteatoma and normal skin
tissues). The qRT-PCR results and microarray data were consistent
(Fig. 5), demonstrating that the
microarray expression results are highly reliable.
CeRNA network analysis indicates that
lncRNAs have ceRNA potential in the pathogenesis of
cholesteatoma
CeRNAs are involved in a regulatory mechanism
between non-coding RNA and coding RNA based on shared MREs
(12). According to the ceRNA
hypothesis, ceRNA members can compete for the same MREs to regulate
each other. To explore whether lncRNAs have ceRNA potential in the
pathogenesis of cholesteatoma, in the present study, we constructed
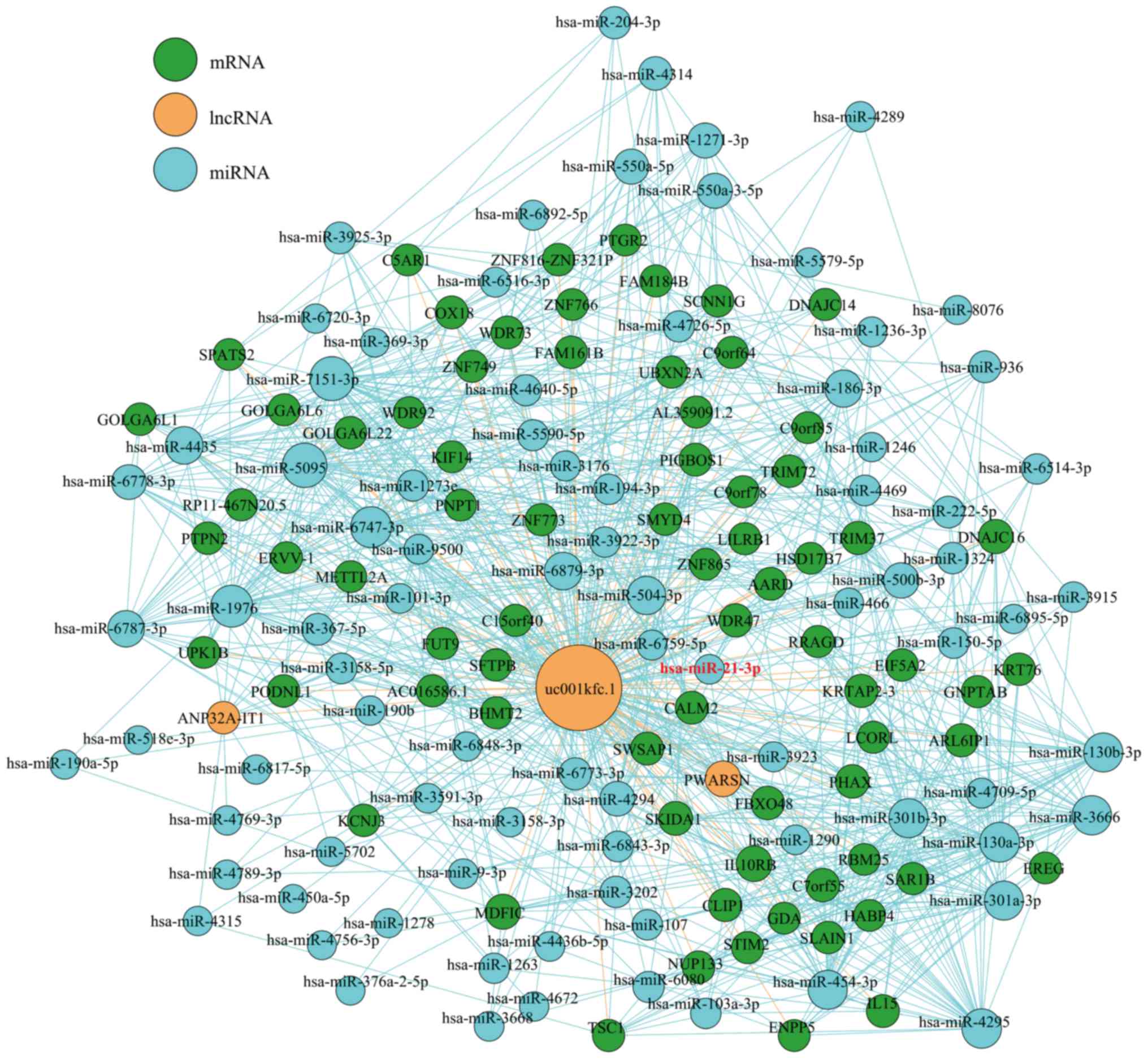
a ceRNA network in cholesteatoma based on the microarray data. We
selected 5 significantly differentially expressed lncRNAs (fold
change >2.0, P<0.05, Table
I), which shared common binding MREs with each other, to
constitute an lncRNA/miRNA/mRNA ceRNA network (available upon
request). The network was found to be composed of 20 lncRNA nodes,
399 miRNA nodes, and 137 mRNA nodes. In the network, we observed
that lncRNA-uc001kfc.1 interacted with miR-21-3p (Figs. 6 and 7A), a microRNA belonging to the miR-21
family, which promotes the formation and invasion of cholesteatoma
(7,8). (To make the lncRNA-uc001kfc.1-mediated
ceRNA network easier to identify, we separated it from the original
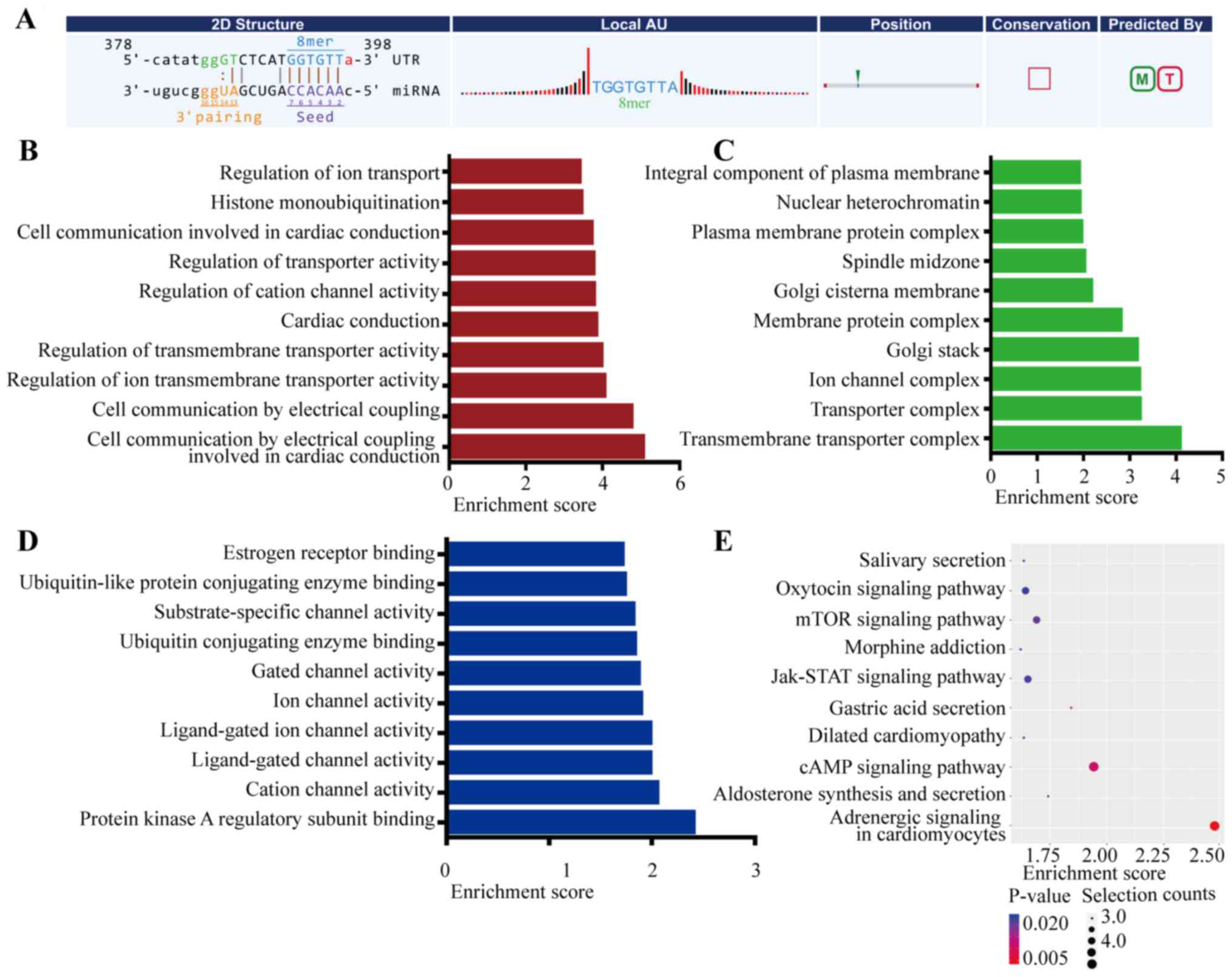
ceRNA network). The 2D structure of the miR-21-3p on uc001kfc.1
indicated that the binding site is 8 mer, perfectly matching
positions 2 to 8 of the mature miR-21-3p (Fig. 7A).
 | Table I.Significant differentially expressed
lncRNAs for ceRNA network construction (fold change >2.0,
P<0.05). |
Table I.
Significant differentially expressed
lncRNAs for ceRNA network construction (fold change >2.0,
P<0.05).
| lncRNAs | GeneSymbol | P-value | Fold change | Regulation |
|---|
| T162623 | G037602 | 0.0002817 |
5.1090401 | Down |
| T347175 | G081784 | 0.0013410 |
2.4223804 | Down |
| uc001kfc.1 | AK130076 | 0.0077363 | 13.0961017 | Down |
| T044224 | G010228 | 0.0101259 |
2.5568534 | Down |
|
ENST00000420253 | AC093495.4 | 0.0006598 |
5.4432596 | Up |
Furthermore, to better explore the ceRNA potential
of lncRNAs in cholesteatoma, we performed GO and KEGG pathway
analysis for the lncRNA/miRNA/mRNA network. In GO analysis, gene
product enrichment in BP, MF, and CC were analyzed (P<0.05) and
the top-10 enriched GO terms of the three aspects are listed in
Fig. 7B-D. GO analysis revealed
that protein kinase activator activity (GO:0030295) as well as
protein kinase A binding (GO:0051018) and protein kinase A
regulatory subunit binding (GO:0034237) were the most significant
molecular functions displayed in the ceRNA network. Protein kinase
can regulate protein phosphorylation, which is involved in many
aspects of cell biology and contributes to cholesteatoma formation
(27). In KEGG pathway analysis, as
depicted in Fig. 7E, the cAMP
signaling pathway, the JAK/STAT signaling pathway, and the mTOR
signaling pathway were related to cell migration, cell cycle, and
survival (http://www.genome.jp/kegg/).
Moreover, JAK/STAT signaling was also involved in the molecular
mechanisms underlying cholesteatoma (28).
Discussion
The present study provides a comprehensive analysis
of lncRNA and mRNA expression profiles in cholesteatoma and matched
normal skin tissues by microarray analysis. Microarray data
indicated that both lncRNA and mRNA profiles were distinctly
different between cholesteatoma and normal skin specimens. In
cholesteatoma tissues, a total of 181 upregulated lncRNAs and 606
downregulated lncRNAs were differentially expressed compared with
normal skin tissues, which indicated that lncRNAs may play
important roles in cholesteatoma. Among them, intergenic lncRNAs
account for the largest category (69.47%). This is meaningful,
since intergenic lncRNAs are a type of lncRNA that are transcribed
and act as cis-regulators when close to protein-coding genes
during gene translation, which makes them the best candidates for
in-depth study of transcriptional regulation of neighboring genes
(14). To verify the reliability of
our microarray data, 3 upregulated and 3 downregulated lncRNAs were
selected to conduct qRT-PCR in a total of 7 pairs of samples. The
results of qRT-PCR were consistent with the microarray assays,
which demonstrated that the microarray data were highly
reliable.
Since lncRNAs can regulate the expression of
neighboring and overlapping coding genes, GO enrichment analysis of
differentially expressed mRNAs can display the functional roles of
lncRNAs. Based on differentially expressed mRNAs data from
cholesteatoma specimens, we identified the framework of the gene
functions and analyzed the gene product activities in terms of BP,
MF, and CC by GO enrichment analysis. In BP analysis of upregulated
genes, we observed that regulation of phosphatidylinositol 3-kinase
signaling (GO:0014066), phosphatidylinositol 3-kinase signaling
(GO:0014065), and positive regulation of phosphatidylinositol
3-kinase signaling (GO:0014068) were three of the most significant
processes. Furthermore, in MF analysis of upregulated genes,
phosphatidylinositol 3-kinase binding (GO:0043548) was the most
significant gene activity process. All the gene activity processes
aforementioned are all related to upregulation of
phosphatidylinositol 3-kinase (PI3K) signaling in cholesteatoma
tissues. PI3Ks are enzymes that catalyze the phosphorylation of
phosphatidylinositol (PtdIns). PI3K signaling has been reported to
function in metabolic control, immunity, angiogenesis, and
cardiovascular homeostasis (29).
Furthermore, the PI3K pathway is also a critical regulator of cell
survival and proliferation (30).
During the past years, PI3K signaling activity has been reported to
promote the genesis of cholesteatoma. A previous study reported
that epithelial keratinocytes in cholesteatoma are protected
against programmed cell death by activation of the PI3K/Akt
signaling pathway (31). Akt, also
known as serine kinase PKB, is a downstream effector of PI3K
(29). In addition, Yune and Byun
discovered that cellular survival mechanisms are related to
cholesteatoma epithelial hyper-proliferation via reduced PTEN and
increased PI3K/Akt signaling pathway activation (32). KEGG pathway analysis for
differentially expressed mRNAs revealed that 7 upregulated pathways
and 10 downregulated pathways could participate in the mechanisms
underlying cholesteatoma. Bacterial invasion of epithelial cells is
one of the upregulated pathways. Our results support a previous
study in which it was revealed that bacterial infection may promote
the enlargement of cholesteatoma and destruction of local
structures (33). Therefore,
functional analysis implied that lncRNAs may play important roles
in cholesteatoma pathogenesis by regulating the neighboring and
overlapping coding genes.
During the past decade, though many studies have
reported that lncRNAs are involved in the epigenetic mechanisms of
many diseases, the exact regulatory pathways of most lncRNAs with
other transcripts are still largely unknown. In the present study,
we constructed an lncRNA/miRNA/mRNA network to discover the
relationship between lncRNAs, miRNAs, and mRNAs. The ceRNA
hypothesis proposes that, by shared MREs, lncRNAs can sequester the
miRNA activity, thereby upregulating the targeted mRNAs (12). In the present study, in the
lncRNA/miRNA/mRNA network, we observed that lncRNA-uc001kfc.1
shared common MREs with hsa-miR-21-3p. Compared with normal skin
tissues, lncRNA-uc001kfc.1 was confirmed to have low expression in
cholesteatoma by both microarray analysis and qRT-PCR. Previous
studies reported that hsa-miR-21 was overexpressed in
cholesteatoma; and when hsa-miR-21 is upregulated, the number of
proliferative as well as migrated cholesteatoma keratinocytes
increases significantly (7,8,34). In
the present study, lncRNA-uc001kfc.1 was significantly
downregulated. Therefore, we presume that lncRNA-uc001kfc.1 may
play a key role in cholesteatoma pathogenesis and function as an
‘endogenous sponge’ for miR-21-3p; and when lncRNA-uc001kfc.1 is
downregulated in cholesteatoma, hsa-miR-21-3p becomes
transcriptionally active, regulating relative genes, and thus
resulting in the hyper-proliferation and migration of
keratinocytes. This hypothesis gives us hope that lncRNA-uc001kfc.1
mimics may be a potential drug treatment for cholesteatoma. In
addition, more functional experiments are required to validate the
hypothesis.
To further explore the ceRNA potential of lncRNAs in
cholesteatoma, GO and KEGG pathway analyses were performed to
analyze the functions of lncRNA-related genes in the
lncRNA/miRNA/mRNA network. In MF fold enrichment of GO analysis,
protein kinase activator activity (GO:0030295) was one of the most
significantly enriched processes, which represents ‘Binds to and
increases the activity of a protein kinase, an enzyme that
phosphorylates a protein’ (http://www.geneontology.org). Protein phosphorylation
is important in the control of cell metabolism (35). Increased phosphorylated Akt (p-Akt)
expression can induce cell hyper-proliferation in cholesteatoma by
activating the PI3K/Akt pathway (32). In another study, phosphorylation of
HSP27, which is triggered by the Ras/Raf/ERK1/2 and MAPK pathways,
was involved in the activation of epithelial cell migration,
angiogenesis, and proliferation, subsequently resulting in the
growth of cholesteatoma (36).
Additionally, in the JAK/STAT signaling pathway, the
phosphorylation of STAT3 can promote cholesteatoma epithelial
hyper-proliferation by cell cycle acceleration, cellular
differentiation promotion, and inhibition of apoptosis (28). Among the most significant KEGG
pathways of the network, the JAK/STAT signaling pathway, just as
depicted above, has been previously reported to be involved in the
pathogenesis of cholesteatoma (28)
and our study further supported these findings. Moreover, the cAMP
signaling pathway, the JAK/STAT signaling pathway, and the mTOR
signaling pathway, which are associated with the PI3K/Akt signaling
pathway, may contribute to cholesteatoma formation through their
involvement in cell cycle, migration, and survival (http://www.genome.jp/kegg/). All functional analysis
of the lncRNA/miRNA/mRNA network further elucidated that lncRNAs
had ceRNA potential and may play key roles in cholesteatoma
pathogenesis.
In summary, this is the first study to examine
lncRNA expression profiles in cholesteatoma using a microarray
analysis and we discovered that lncRNA expression patterns were
significantly altered in cholesteatoma. In addition, by
constructing an lncRNA/miRNA/mRNA ceRNA network, we found that
lncRNAs may function as ceRNAs during cholesteatoma formation. Our
findings shed novel light on the pathogenesis of cholesteatoma and
provide potential therapeutic targets for the treatment of
cholesteatoma. In subsequent research, we will focus on the
functional studies of lncRNAs, such as gain-of-function or
loss-of-function of lncRNA-uc001kfc.1, to further explore the
precise molecular mechanisms of cholesteatoma.
Acknowledgments
Not applicable.
References
|
1
|
Louw L: Acquired cholesteatoma
pathogenesis: Stepwise explanations. J Laryngol Otol. 124:587–593.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kuo CL, Shiao AS, Yung M, Sakagami M,
Sudhoff H, Wang CH, Hsu CH and Lien CF: Updates and knowledge gaps
in cholesteatoma research. Biomed Res Int. 2015:8540242015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tono T, Sakagami M, Kojima H, Yamamoto Y,
Matsuda K, Komori M, Hato N, Morita Y and Hashimoto S: Staging and
classification criteria for middle ear cholesteatoma proposed by
the Japan Otological Society. Auris Nasus Larynx. 44:135–140. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Crowson MG, Ramprasad VH, Chapurin N,
Cunningham CD III and Kaylie DM: Cost analysis and outcomes of a
second-look tympanoplasty-mastoidectomy strategy for cholesteatoma.
Laryngoscope. 126:2574–2579. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Prasad SC, Piras G, Piccirillo E, Taibah
A, Russo A, He J and Sanna M: Surgical strategy and facial nerve
outcomes in petrous bone cholesteatoma. Audiol Neurootol.
21:275–285. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Maniu A, Harabagiu O, Schrepler Perde M,
Cătană A, Fănuţă B and Mogoantă CA: Molecular biology of
cholesteatoma. Rom J Morphol Embryol. 55:7–13. 2014.PubMed/NCBI
|
|
7
|
Friedland DR, Eernisse R, Erbe C, Gupta N
and Cioffi JA: Cholesteatoma growth and proliferation:
Posttranscriptional regulation by microRNA-21. Otol Neurotol.
30:998–1005. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen X and Qin Z: Post-transcriptional
regulation by microrna-21 and let-7a microRNA in paediatric
cholesteatoma. J Int Med Res. 39:2110–2118. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li N and Qin ZB: Inflammation-induced
miR-802 promotes cell proliferation in cholesteatoma. Biotechnol
Lett. 36:1753–1759. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang W, Chen X and Qin Z: MicroRNA let-7a
suppresses the growth and invasion of cholesteatoma keratinocytes.
Mol Med Rep. 11:2097–2103. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Salmena L, Poliseno L, Tay Y, Kats L and
Pandolfi PP: A ceRNA hypothesis: The Rosetta Stone of a
hidden RNA language? Cell. 146:353–358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bak RO and Mikkelsen JG: miRNA sponges:
Soaking up miRNAs for regulation of gene expression. Wiley
Interdiscip Rev RNA. 5:317–333. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ponting CP, Oliver PL and Reik W:
Evolution and functions of long noncoding RNAs. Cell. 136:629–641.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen LL and Carmichael GG: Decoding the
function of nuclear long non-coding RNAs. Curr Opin Cell Biol.
22:357–364. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rogoyski OM, Pueyo JI, Couso JP and
Newbury SF: Functions of long non-coding RNAs in human disease and
their conservation in Drosophila development. Biochem Soc Trans.
45:895–904. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Santoro M, Nociti V, Lucchini M, De Fino
C, Losavio FA and Mirabella M: Expression profile of long
non-coding RNAs in serum of patients with multiple sclerosis. J Mol
Neurosci. 59:18–23. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Elia L and Quintavalle M: Epigenetics and
vascular diseases: Influence of non-coding RNAs and their clinical
implications. Front Cardiovasc Med. 4:262017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang X, Liu G, Qiu J, Zhang N, Ding J and
Hua K: E2F1-regulated long non-coding RNA RAD51-AS1 promotes cell
cycle progression, inhibits apoptosis and predicts poor prognosis
in epithelial ovarian cancer. Sci Rep. 7:44692017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu J, Qian Y, Ye M, Fu Z, Jia X, Li W, Xu
P, Lv M, Huang L, Wang L, et al: Distinct expression profile of
lncRNA in endometrial carcinoma. Oncol Rep. 36:3405–3412. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pan B, Zhou HX, Liu Y, Yan JY, Wang Y, Yao
X, Deng YQ, Chen SY, Lu L, Wei ZJ, et al: Time-dependent
differential expression of long non-coding RNAs following
peripheral nerve injury. Int J Mol Med. 39:1381–1392. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jiao C, Song Z, Chen J, Zhong J, Cai W,
Tian S, Chen S, Yi Y and Xiao Y: lncRNA-UCA1 enhances cell
proliferation through functioning as a ceRNA of Sox4 in esophageal
cancer. Oncol Rep. 36:2960–2966. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sui J, Li YH, Zhang YQ, Li CY, Shen X, Yao
WZ, Peng H, Hong WW, Yin LH, Pu YP and Liang GY: Integrated
analysis of long non-coding RNAassociated ceRNA network reveals
potential lncRNA biomarkers in human lung adenocarcinoma. Int J
Oncol. 49:2023–2036. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu W, Ren H, Ren J, Yin T, Hu B, Xie S,
Dai Y, Wu W, Xiao Z, Yang X and Xie D: The role of
EGFR/PI3K/Akt/cyclinD1 signaling pathway in acquired middle ear
cholesteatoma. Mediators Inflamm. 2013:6512072013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ergun S, Zheng X and Carlsoo B: Expression
of transforming growth factor-alpha and epidermal growth factor
receptor in middle ear cholesteatoma. Am J Otol. 17:393–396.
1996.PubMed/NCBI
|
|
26
|
Likus W, Siemianowicz K, Markowski J,
Wiaderkiewicz J, Kostrząb-Zdebel A, Jura-Szołtys E, Dziubdziela W,
Wiaderkiewicz R and Łos MJ: Bacterial infections and
osteoclastogenesis regulators in men and women with cholesteatoma.
Arch Immunol Ther Exp. 64:241–247. 2016. View Article : Google Scholar
|
|
27
|
Huisman MA, De Heer E and Grote JJ:
Terminal differentiation and mitogen-activated protein kinase
signaling in human cholesteatoma epithelium. Otol Neurotol.
27:422–426. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu W, Xie S, Chen X, Rao X, Ren H, Hu B,
Yin T, Xiang Y and Ren J: Activation of the IL-6/JAK/STAT3
signaling pathway in human middle ear cholesteatoma epithelium. Int
J Clin Exp Pathol. 7:709–715. 2014.PubMed/NCBI
|
|
29
|
Hawkins PT, Anderson KE, Davidson K and
Stephens LR: Signalling through Class I PI3Ks in mammalian cells.
Biochem Soc Trans. 34:647–662. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Song G, Ouyang G and Bao S: The activation
of Akt/PKB signaling pathway and cell survival. J Cell Mol Med.
9:59–71. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huisman MA, De Heer E and Grote JJ:
Survival signaling and terminal differentiation in cholesteatoma
epithelium. Acta Otolaryngol. 127:424–429. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yune TY and Byun JY: Expression of PTEN
and phosphorylated Akt in human cholesteatoma epithelium. Acta
Otolaryngol. 129:501–506. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Brook I and Burke P: The management of
acute, serous and chronic otitis media: The role of anaerobic
bacteria. J Hosp Infect. 22 Suppl A:S75–S87. 1992. View Article : Google Scholar
|
|
34
|
Chen X, Li X and Qin Z: MicroRNA-21
promotes the proliferation and invasion of cholesteatoma
keratinocytes. Acta Otolaryngol. 136:1261–1266. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Humphrey SJ, James DE and Mann M: Protein
phosphorylation: A Major switch mechanism for metabolic regulation.
Trends Endocrinol Metab. 26:676–687. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ho KY, Yeh TS, Huang HH, Hung KF, Chai CY,
Chen WT, Tsai SM, Chang NC, Chien CY, Wang HM and Wu YJ:
Upregulation of phosphorylated HSP27, PRDX2, GRP75, GRP78 and GRP94
in acquired middle ear cholesteatoma growth. Int J Mol Sci.
14:14439–14459. 2013. View Article : Google Scholar : PubMed/NCBI
|