Introduction
Human liver cancer, hepatoblastoma, is the most
common malignant liver tumor in pediatrics (1). Although surgical excision of the tumor
mass can be performed, challenges still remain for patients with
vascular invasion and metastatic disease (2). Inhibition of metastasis is hence a key
strategy for liver cancer treatment, and the discovery of potential
inhibitors of metastasis could lead to improvements in therapy.
Optional therapeutic schemes and auxiliary substances have been
widely explored; however, only few are effective (3). Thus, the development of new drugs is
required for optimal treatment with fewer complications.
All-trans retinoic acid (ATRA) has been
effectively used for inducing the differentiation of acute
promyelocytic leukemia cells (4);
however, it does not cure patients with liver cancer, as liver
cancer requires nearly a 10 times higher concentration than
leukemic cells (5). Such high
concentrations are not suitable for clinical use due to several
side effects, including retinoic acid syndrome, skin dryness, and
liver damage. N-(4-hydroxyphenyl)retinamide (4-HPR or fenretinide),
an artificial variant of ATRA, exhibits markedly different effects
to ATRA. Presently, 4-HPR is considered to be a drug with fewer
side effects (6). Vaccari et
al found that 4-HPR influences cell matrix interactions and
blocks tumor progression to locally invasive malignacy (7). 4-HPR was found to reduce the incidence
of breast cancer when used as a chemoprevention agent (8) and prevented secondary breast cancer in
a phase III trial (9). Furthermore,
6 years later Kang et al demonstrated that 4-HPR inhibited
the invasion of breast cancer cells (10). Similarly, Benelli et al
demonstrated that 4-HPR hindered the migration and invasion of
prostate cancer cells (11), and it
has now entered clinical phase II trials (12). In the present study, we compared the
antiproliferative effects of 4-HPR with ATRA on HepG2 cells, and
explored the functions and mechanisms of 4-HPR in modulating their
migration capacity.
Mitogen-activated protein kinases (MAPKs) are highly
conserved signaling pathway proteins, playing vital roles in
deciding cell fate (13,14). p38-MAPK is activated by different
stimuli, including chemical agents, cytokines and oxidative stress
(13–15). Sustained activation of p38-MAPK
induces cell death (14,16). 4-HPR-induced sustained activation of
p38-MAPK, accompanied by cell apoptosis, has been reported in
several types of tumors, including neuroblastoma, HeLa, T-cell
leukemia/lymphoma cells (17–19);
however, HepG2 cells were reported to be resistant to the apoptotic
effect of 4-HPR (20). Whether
4-HPR influences the p38-MAPK pathway in HepG2 cells remains
unclear.
Myosin light chain kinase (MLCK) is a crucial
Ca2+/calmodulin-dependent effector, and controls the
migration of smooth- and non-muscle cells through the
phosphorylation of Ser19 and Thr18 on myosin light chains (MLC)
(21). Previous studies have
reported that both MLCK and activated myosin II are abundant in the
lamellar protrusive structures of certain cell types during
migration (22,23). Several studies have revealed
p38-MAPK pathway links in the tumor cells treated with 4-HPR, and
also some researches have found that MLCK is involved in the
migration of tumor cells; however, the underlying mechanism that
explains how these factors influence liver cancer is still
unknown.
Therefore, we hypothesized that 4-HPR inhibits the
proliferation and migration of liver cancer cells via MLCK and
p38-MAPK signaling. This study was aimed to provide an experimental
basis for the further application of 4-HPR in liver cancer
therapy.
Materials and methods
Cell lines and major reagents
The human liver cancer cell line HepG2 (24) was obtained from the American Type
Culture Collection (ATCC; Manassas, VA, USA). HyClone Dulbecco's
modified Eagle's medium (DMEM; low glucose) was purchased from GE
Healthcare Life Sciences (Logan, UT, USA). Fetal bovine serum (FBS)
was purchased from Tianhang Biological Technology (Hanzhou,
Zhejiang, China). Primary antibodies: Rabbit antihuman monoclonal
MLCK (cat. no. ab92721) and E-cadherin (cat. no. ab40772), mouse
anti-human monoclonal F-actin (cat. no. ab205) were obtained from
Abcam (Cambridge, MA, USA); rabbit anti-human monoclonal
phosphpho-p38 MAPK (cat. no. 4511), mouse anti-human monoclonal
phospho-MLC (cat. no. 3675) were purchased from Cell Signaling
Technology (Danvers, MA, USA); rabbit anti-human polyclonal MLC
(cat. no. 15354-1-AP), mouse anti-human monoclonal GAPDH (cat. no.
60004-1-Ig) were obtained from Proteintech Group (Wuhan, Hubei,
China); rabbit anti-human polyclonal p38 MAPK (cat. no. sc-7149)
and mouse antihuman monoclonal β-actin (cat. no. sc-47778) were
obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). All
secondary antibodies (cat. nos. AP124P and AP132P) were obtained
from Millipore (Billerica, MA, USA). Western blot primary antibody
diluent was obtained from Beyotime Institute of Biotechnology
(Beijing, China). Enhanced chemiluminescence reagent Plus (ECL)
reagents were purchased from Thermo Fisher Scientific (Waltham, MA,
USA). 4-HPR was obtained from MedChem Express (Deer Park, NY, USA).
ATRA and ML-7 was purchased from DC Chemicals (Shanghai, China).
SB203580 was obtained from Selleck Chemicals (Houston, TX, USA).
4-HPR, ATRA, ML-7 and SB203580 were dissolved in a small amount of
dimethyl sulfoxide (DMSO) before addition to the complete cell
culture medium. MTS was purchased from Promega (Madison, WI,
USA).
Cell culture and morphologic
observation after drug treatment
Cells were seeded in 6-well plates, and cultured in
DMEM supplemented with 10% FBS, penicillin (100 U/ml) and
streptomycin (100 µg/ml). The plates were incubated at 37°C with 5%
CO2 in a humidified atmosphere. When the cell density
reached 40–50% confluency, the cells were treated with 4-HPR or
ATRA at 5 or 10 µM or with DMSO alone for 48 h. Cell morphology was
imaged using a microscope (Leica DMI3000B; Leica Microsystems,
Wetzlar, Germany).
Cell viability assay
Cells (5×103 cells/well) were plated in
96-well plates, and then treated with 4-HPR or ATRA (5 or 15 µM) at
different time intervals (24–72 h), or persistently treated with a
dose range of 1–25 µM of 4-HPR or ATRA for 48 h at 37°C. Following
treatment, MTS (20 µl/well) was added to each well and incubated
for 1 h at 37°C. Optical density (OD) values were measured at 490
nm using a Microplate reader (ELX800; BioTek, Winooski, VT, USA) at
37°C. Cell inhibition rate=(OD490 of the cell control group-OD490
of the experimental group)/OD490 of the cell control group.
Plate colony formation assay
When the cells were in logarithmic growth, they were
plated in 6-well plates. After culturing overnight, the cells were
treated with 4-HPR or ATRA (5 or 10 µM) for 48 h, and were then
collected as single cell suspensions. Approximately, 2,000
cells/wells were plated into a fresh 6-well plate, and the plate
was incubated for approximately 10 days. When the colonies were
clearly visible with the naked eye, the medium was discarded and
the cells were washed twice with phosphate-buffered saline (PBS),
fixed with 4% paraformaldehyde, and then stained with 1% crystal
violet. In five random visual fields, the colonies containing ≥50
cells were counted by an inverted microscope (Leica DMI3000B; Leica
Microsystems) for each well.
Wound healing assay
The migratory ability of the HepG2 cells was
determined by the wound healing assay. Cells were seeded in 12-well
plates. When the growth reached 95% confluency, the cell monolayer
was scratched with a sterilized 200-µl pipette tip, and then the
cells were washed thrice with PBS. Furthermore, two concentrations
(5 and 10 µM) of 4-HPR or ATRA were added to the cell culture
medium. The migration rate of cells was determined by observation
under a microscope at different time intervals (0, 24, and 48 h).
The nick distance of the wound was measured by Image-Pro Plus
software 6.0 (Media Cybernetics, Inc., Rockville, MD, USA).
Western blot analysis
Cells were treated with 4-HPR or ATRA (5 or 10 µM)
for 48 h. Total cellular proteins were extracted with lysis buffer
(Tris-HCl, pH 7.14, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1%
SDS, 1 mM leupeptin and 1 mM PMSF) on ice for 30 min. All the
samples were mixed with loading buffer, and then boiled for 5 min.
The proteins were separated by 8–12% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Furthermore,
the proteins were transferred to polyvinylidene difluoride (PVDF)
membranes, and blocked with 5% nonfat dry milk in Tris-buffered
saline Tween-20 (TBST) buffer (20 mM Tris-HCl, pH 7.6, 150 mM NaCl,
and 0.05% Tween-20) for 2 h at room temperature. The membranes were
then incubated in WB primary antibody diluent with the indicated
primary antibodies: MLCK (1:8,000), phospho-MLC (1:1,000), MLC
(1:1,000), phospho-p38 (1:1,000), p38 (1:1,000), F-actin (1:500),
E-cadherin (1:4,000), GAPDH (1:50,000), β-actin (1:1,000),
respectively overnight at 4°C. Thereafter, the membranes were
incubated with the corresponding horseradish peroxidase
(HRP)-conjugated secondary antibodies (diluted with TBST containing
5% non-fat dry milk) for 2 h at room temperature, and detected by
chemiluminescence using an ECL kit. Specific complexes were
revealed by enhanced chemiluminescence (Clinx Science Instruments,
Shanghai, China). The image data were quantified using Quantity One
software 4.6.2 (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
HepG2 cell treatment with p38-MAPK and
MLCK inhibitors
The pharmacological inhibitors of the p38-MAPK
signaling pathway (SB203580, 25 µM) and MLCK (ML-7, 20 µM) were
used in combination with 4-HPR or ATRA (10 µM) to investigate the
role of p38-MAPK and MLCK in 4-HPR/ATRA-induced inhibition of HepG2
cell migration. Thereafter, wound healing assay and western blot
analysis were used to measure the migration distances and protein
levels in the cells.
Statistical analysis
Results are expressed as means ± standard deviation
(SD). The data were assessed by one-way ANOVA using SPSS 10.0
software (SPSS, Inc., Chicago, IL, USA). P-values <0.05 were
considered as statistically significant.
Results
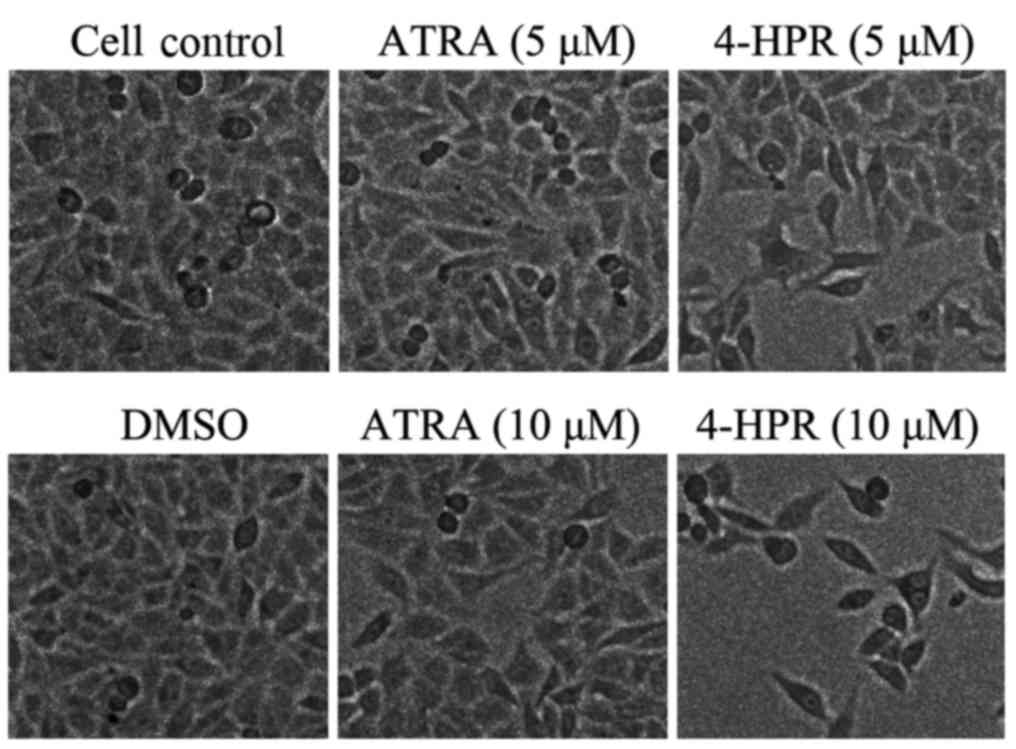
4-HPR alters the morphology of HepG2
cells
In a previous study, Yang et al (20) found that HepG2 cells were resistant
to the apoptotic effect of 4-HPR after 24 h treatment; however,
4-amino-2-trifluoromethyl-phenyl retinate (ATPR), another ATRA
derivative, has been observed to inhibit HepG2 cell proliferation
after 48 h of culture in our laboratory (25). Based on this report, we chose two
concentrations of 4-HPR (5 and 10 µM) to treat HepG2 cells for 48
h. Changes in the cell morphology and any inhibitory effects were
examined by microscopy. The cells congregated neatly and closely in
the solvent (DMSO) control (Fig.
1); however, after treatment with a low concentration of 4-HPR,
the cell density and cell-to-cell contact was reduced. In the high
concentration group, cell density was remarkably reduced, and the
cell morphology was altered into a slender shape containing more
filopodia when compared with the vehicle control (Fig. 1). These changes could be associated
with cell migration after 4-HPR treatment. Cell density was reduced
only slightly in the high concentration ATRA group, and was not
accompanied by any noticeable change in the cell morphology.
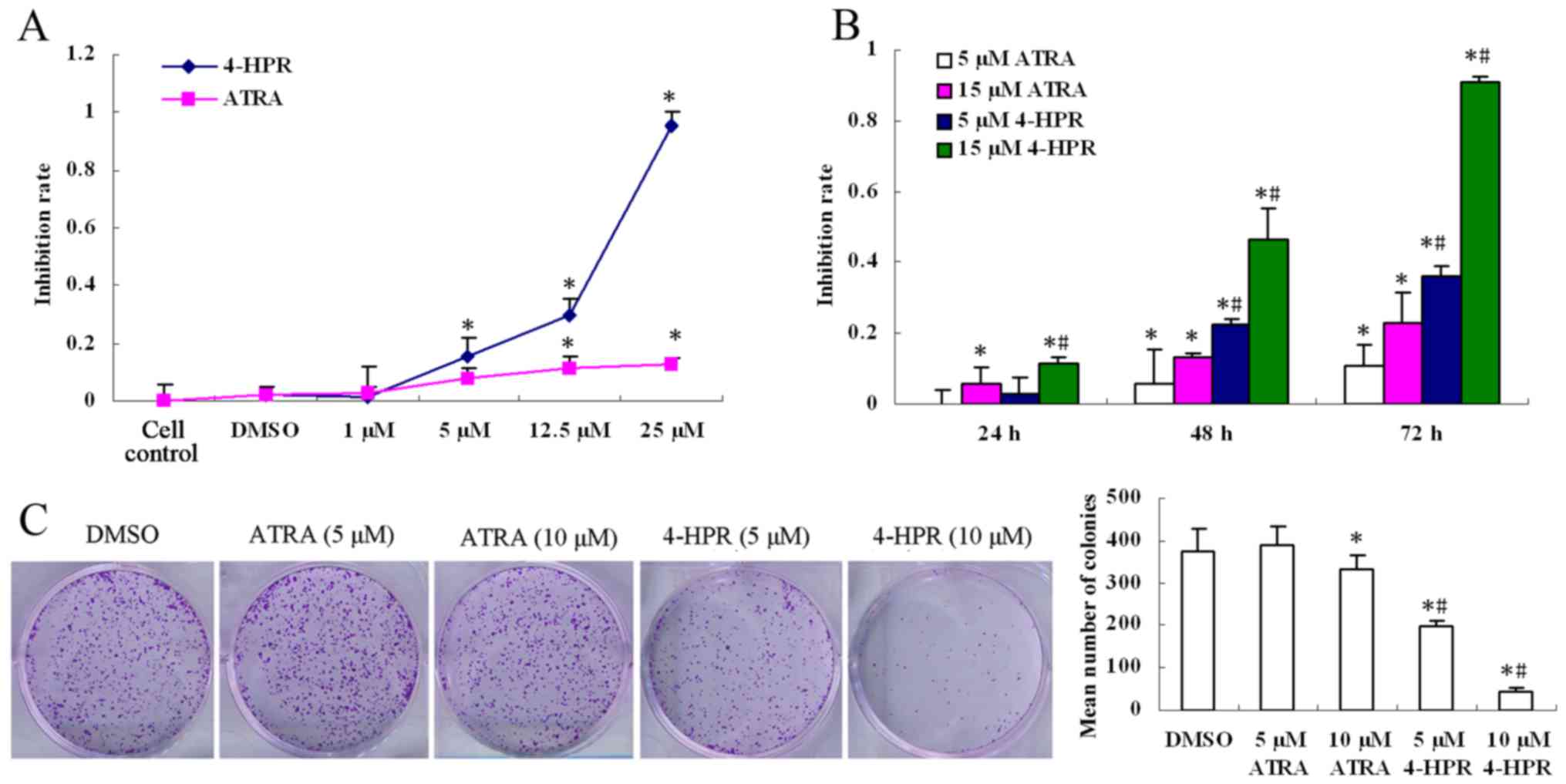
4-HPR inhibits the proliferation of
HepG2 cells
A cell viability assay was used to probe the effects
of 4-HPR and ATRA on the proliferation of HepG2 cells. Cell
viability was distinctly inhibited in a dose- and time-dependent
manner by 4-HPR (Fig. 2A and B). At
the same concentration, the inhibitory effect of 4-HPR was more
intense than that of ATRA (P<0.05). In addition, there was no
obvious difference between the cell control and the DMSO control
group. Based on the aforementioned data, the DMSO control, and an
incubation time of 48 h were chosen for further studies. A plate
colony formation assay was used to assess the colonizing ability of
HepG2 cells in vitro. After 10 days of culture, the density
and size of the colonies were both reduced in a dose-dependent
manner by 4-HPR (P<0.05); however, they were only changed
slightly at the high concerntration of ATRA compared with the
control group (P<0.05). The mean numbers of colonies in the
4-HPR group were lower than those in the ATRA group (P<0.05),
which was consistent with the morphological change and cell
viability analyses (Fig. 2C).
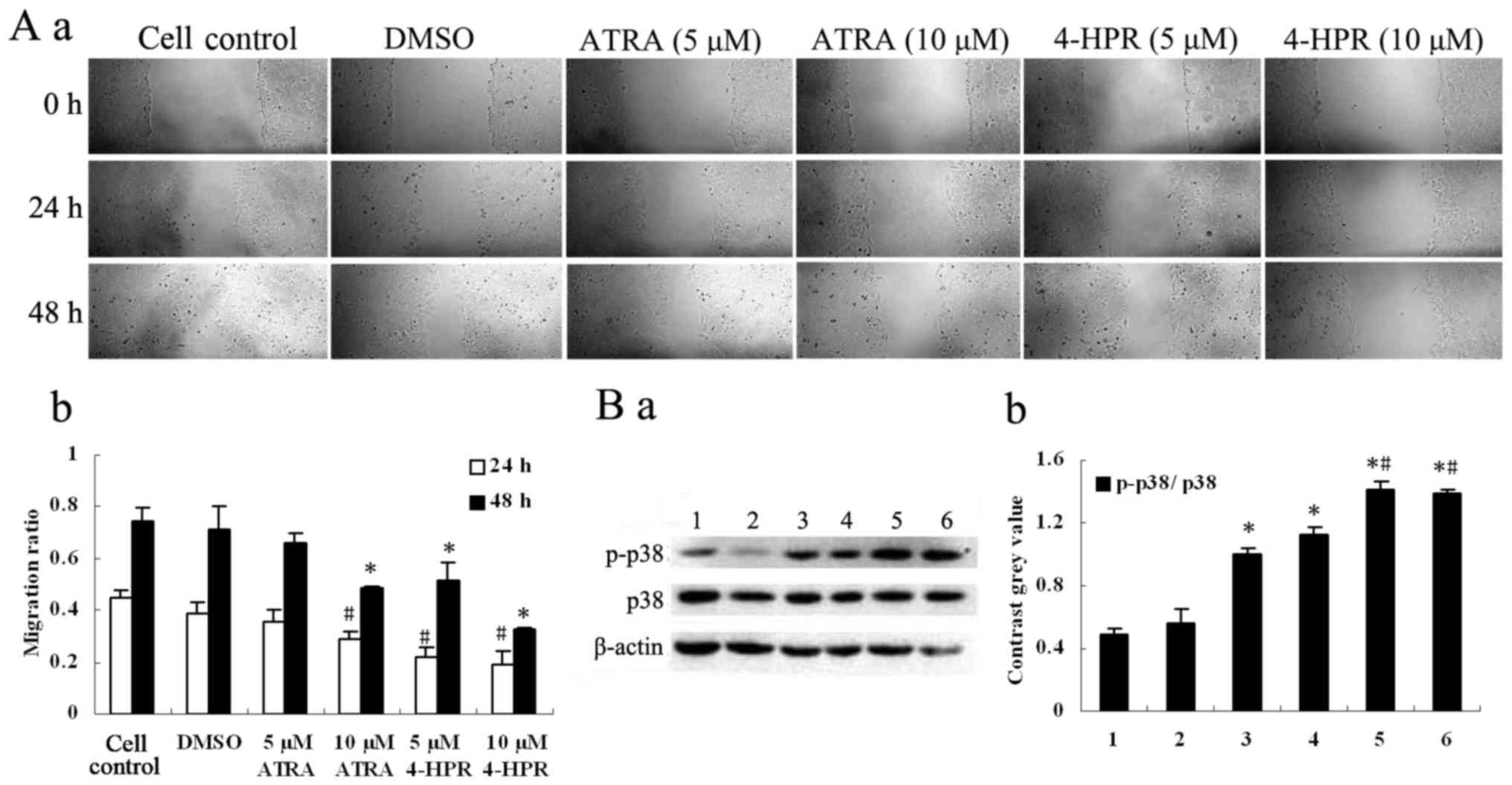
4-HPR hinders the migration of HepG2
cells
We investigated the effect of 4-HPR and ATRA on the
migration of HepG2 cells using a wound healing assay. 4-HPR
inhibited the migration of HepG2 cells (Fig. 3A) in a dose-dependent manner
(P<0.05, compared with the control), consistent with the data
from the cell proliferation assay. 4-HPR inhibited cell migration
at two concentrations of 5 and 10 µM (P<0.05, compared with the
control), whereas ATRA only produced inhibition at 10 µM
(P<0.05, compared with the control).
4-HPR increases the activation of
p38-MAPK in HepG2 cells
Treatment of cells with 4-HPR increased the
phosphorylation of p38 (p-p38) in a dose-dependent manner, and the
expression of p38 was also increased in the high concentration
group, as revealed by Western blot analysis (P<0.05, compared
with the control). ATRA also slightly increased the phosphorylation
of p38 (p-p38) (P<0.05, compared with the control); however, the
increased levels elicited by 4-HPR were much higher than those
produced by ATRA (P<0.05) (Fig.
3B). These results indicated that 4-HPR may inhibit the
migration of HepG2 cells through the p38-MAPK signaling
pathway.
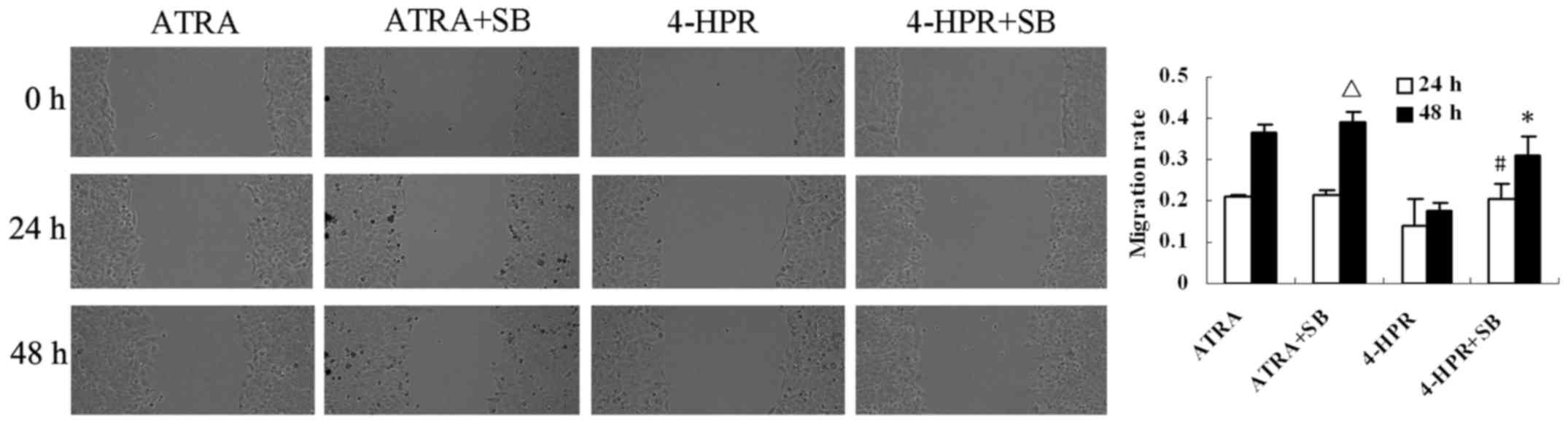
p38-MAPK inhibitor reverses the
inhibitory effect of 4-HPR on HepG2 cell migration
To determine whether 4-HPR inhibits HepG2 cell
migration via p38-MAPK signaling, the cells were pretreated with
the p38-MAPK inhibitor SB203580 for 1 h, and then exposed to 4-HPR
(10 µM) for 48 h. 4-HPR-induced inhibition of migration was
abrogated by the presence of the SB203580 at both 24 and 48 h
(P<0.05, compared with 4-HPR alone) (Fig. 4). These data indicate that 4-HPR
stimulates p38 activity leading to migration inhibition in the
liver cancer cells.
4-HPR decreases the expression of MLCK
and phosphorylation of MLC in HepG2 cells, and a p38-MAPK inhibitor
had an inverse effect
Since myosin light-chain kinase (MLCK) plays a
crucial role in cell migration and metastasis, we investigated the
expression of this protein, as well as its substrate (MLC) and
product (p-MLC). This revealed that 4-HPR markedly reduced the
expression of MLCK and the phosphorylation of MLC (p-MLC), and also
decreased the expression of MLC (P<0.05, compared with the
control) (Fig. 5A). Concomitantly,
4-HPR inhibited the expression of F-actin and increased the
expression of E-cadherin (P<0.05, compared with the control)
(Fig. 5B). ATRA reduced the
expression of p-MLC (P<0.05, compared with the control);
however, it caused no obvious change in the MLCK levels. 4-HPR
reduced the expression of MLCK and the phosphorylation of MLC to a
much greater extent than ATRA (P<0.05). However, when the cells
were pretreated with SB203580, p-MLC and F-actin were upregulated
and E-cadherin was downregulated when compared to 4-HPR or ATRA
treatment alone (P<0.05) (Fig. 5C
and D). These observations fit well with the wound healing
assay data presented above.
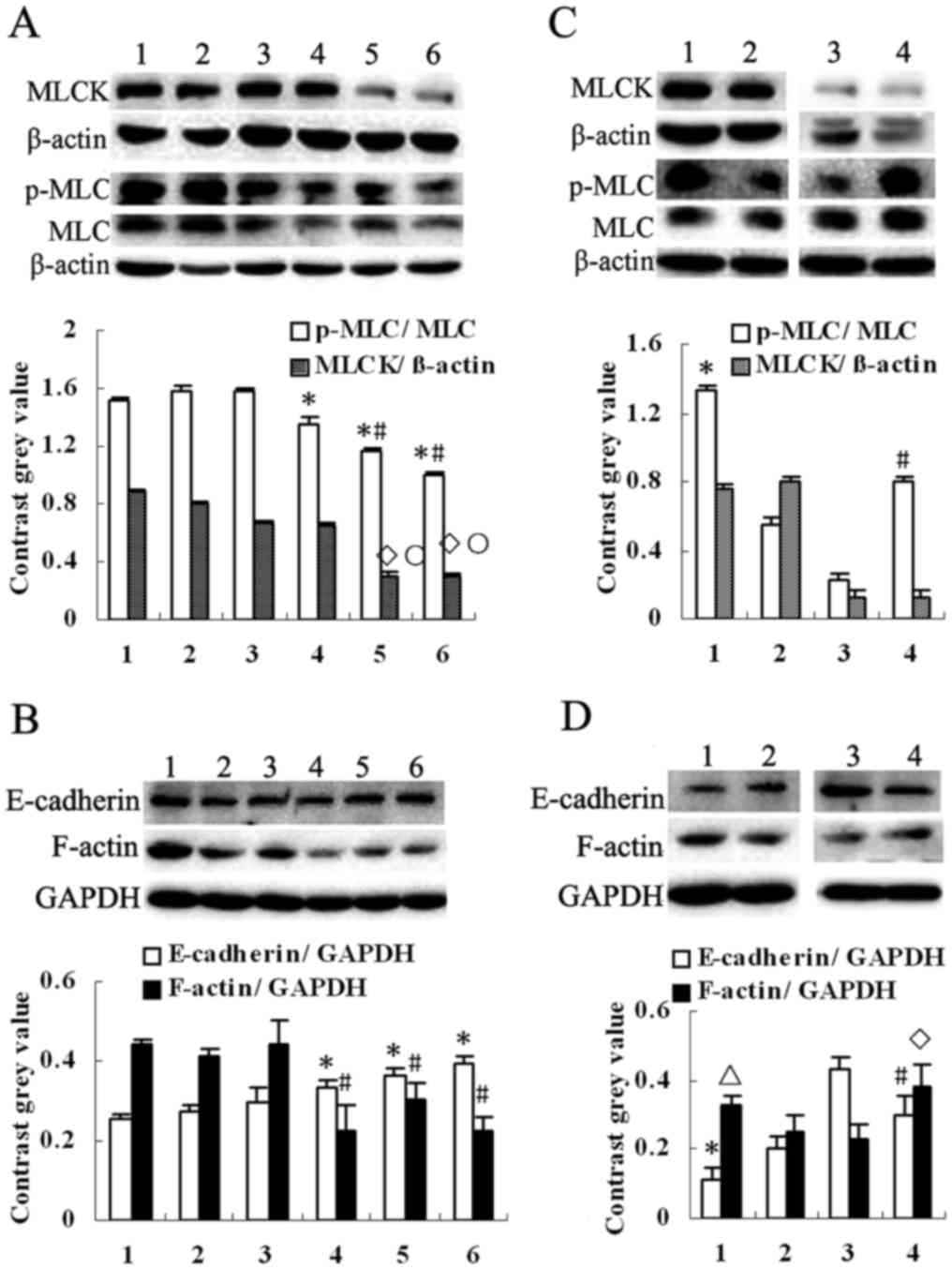 | Figure 5.Effect of 4-HPR and SB203580 on the
expression of MLCK, E-cadherin, F-actin and phosphorylation of MLC.
(A) After treatment of the HepG2 cells with 4-HPR or ATRA at
different concentrations for 48 h, the protein expression of MLCK
and phosphorylation of MLC in HepG2 cells were decreased. Moreover,
the protein expression of MLC was decreased by 4-HPR. Lane 1, cell
control; lane 2, DMSO; lane 3, 5 µM ATRA; lane 4, 10 µM ATRA; lane
5, 5 µM 4-HPR and lane 6, 10 µM 4-HPR. All values are presented as
mean ± SD. n=3, *P<0.05, ◊P<0.05 compared with
cell control; #P<0.05, ○P<0.05 compared
with ATRA group. (B) The protein expression of E-cadherin was
increased and F-actin was decreased in HepG2 cells. Lane 1–6, same
as in A. All values are presented as mean ± SD. n=3, *P<0.05,
#P<0.05 compared with cell control. (C) SB203580 was
used with 4-HPR or ATRA to treat HepG2 cells. Phosphorylation of
MLC were evidently increased. Lane 1, ATRA+SB; lane 2, ATRA; lane
3, 4-HPR; lane 4, 4-HPR+SB. All values are presented as mean ± SD.
n=3, *P<0.05 compared with the ATRA group; #P<0.05
compared with the 4-HPR group. (D) The protein expression of
E-cadherin was decreased and F-actin was increased. Lane 1–4, same
as in C. All values are presented as mean ± SD. n=3. *P<0.05,
ΔP<0.05 compared with the ATRA group;
#P<0.05, ◊P<0.05 compared with the
4-HPR group. 4-HPR, fenretinide; ATRA, all-trans retinoic
acid; SB203580, p38-MAPK inhibitor. |
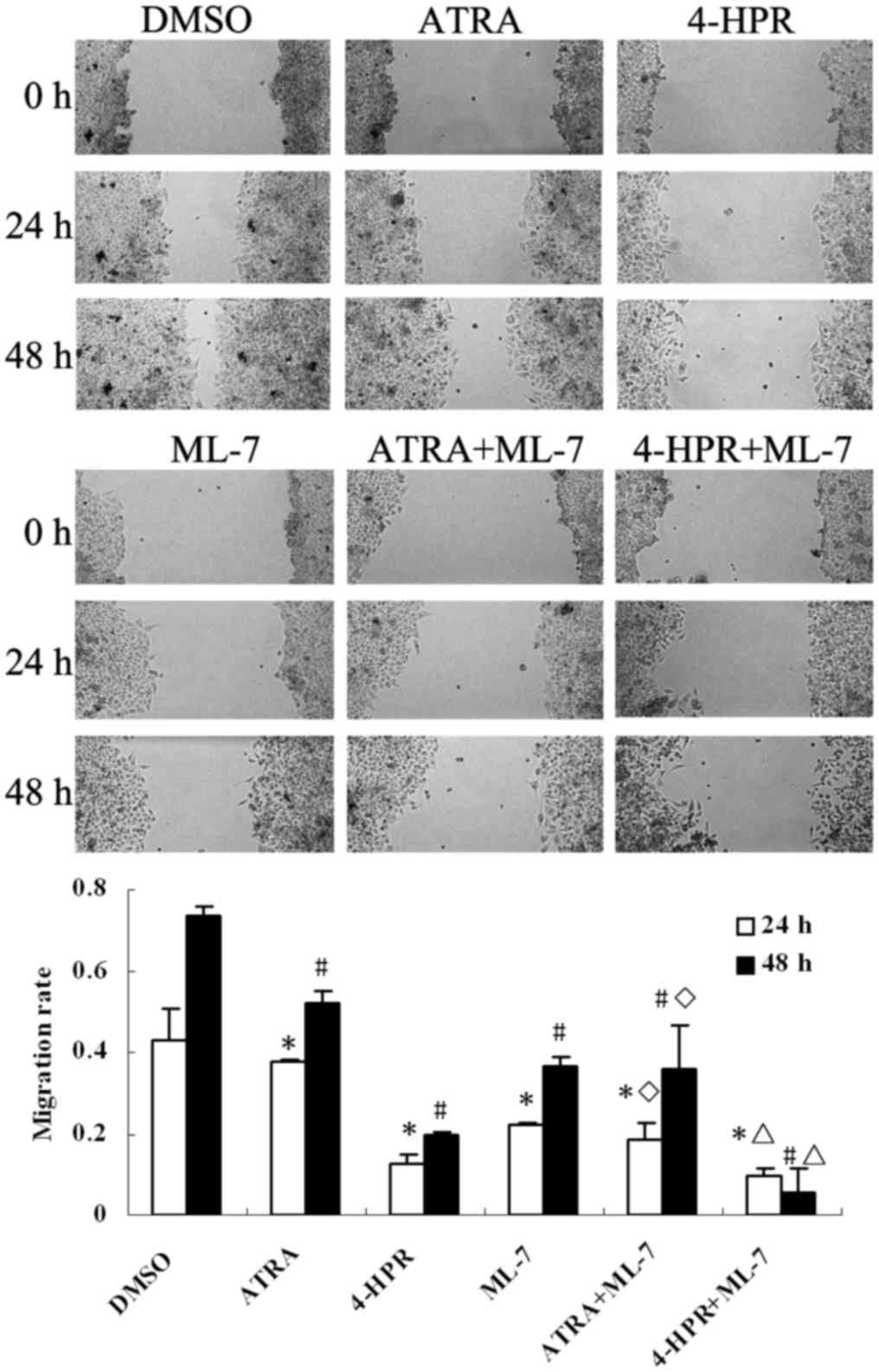
Reduction of MLCK activation inhibits
the migration of HepG2 cells
To verify the association between HepG2 migration
and the MLCK signaling pathway, a wound healing assay was performed
using ML-7 (a specific inhibitor of MLCK) in the culture medium.
After treatment with ML-7 for 24 or 48 h, the migration rate of
HepG2 cells was suppressed when compared with the control
(P<0.05) (Fig. 6). Moreover, the
group treated with both 4-HPR and ATRA combined with ML-7 exhibited
greater inhibition rates than the group treated with 4-HPR or ATRA
alone (P<0.05) (Fig. 6).
ML-7 increases the phosphorylation of
p38 and inhibits the activation of MLCK in HepG2 cells
The underlying mechanism, that is, the nature of the
signaling pathway tied to MLCK repression and p38-MAPK activation
after 4-HPR treatment, remained unclear. Therefore, we investigated
the protein expression after ML-7 treatment (Fig. 7A-C). ML-7 not only inhibited the
activity of MLCK by reducing the expression of p-MLC, but also
activated p38-MAPK by enhancing the expression of p-p38 in HepG2
cells (P<0.05, compared with the control). ML-7 also altered the
expression of F-actin and E-cadherin, and ML-7 combined with 4-HPR
or ATRA further increased the levels of p-p38 compared to 4-HPR or
ATRA alone (P<0.05).
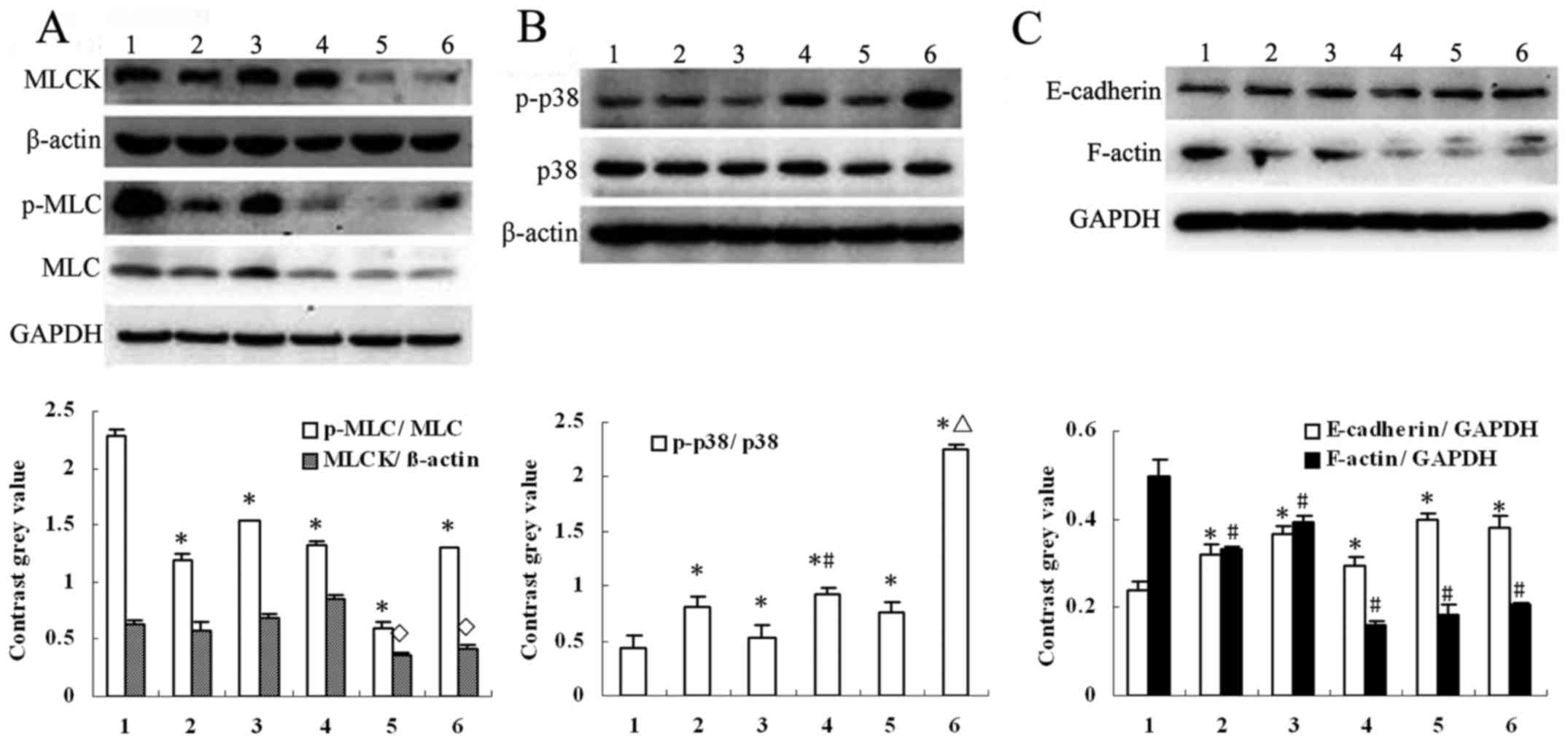 | Figure 7.Effect of 4-HPR and ML-7 on the
expression of MLCK, E-cadherin, F-actin and phosphoryled MLC and
p38. (A) ML-7 was used to treat HepG2 cells. The phosphorylation of
MLC was evidently decreased. In addition, the protein expression of
MLC was decreased by ML-7 combined with 4-HPR or ATRA. All values
are presented as mean ± SD. n=3, *P<0.05, ◊P<0.05
compared with DMSO group. (B) Phosphorylated (p)-p38 was increased
by ML-7. All values are presented as mean ± SD. n=3, *P<0.05
compared with DMSO group, #P<0.05 compared with ATRA
group and ΔP<0.05 compared with 4-HPR group. (C) The
protein expression of E-cadherin was increased and F-actin was
decreased by ML-7. All values are presented as mean ± SD. n=3,
*P<0.05, #P<0.05 compared with DMSO group. For
A-C: Lane 1, DMSO; lane 2, ML-7; lane 3, ATRA; lane 4, ATRA+ML-7;
lane 5, 4-HPR; lane 6, 4-HPR+ML-7. 4-HPR, fenretinide; ATRA,
all-trans retinoic acid; ML-7, a specific inhibitor of
MLCK. |
Discussion
Patients with liver cancer often have unfavorable
prognoses and short lifespans due to early metastasis (26). The process of metastasis involves
tumor cell escape, migration, invasion of the basement membrane,
and growth at new locations (27).
Knowledge concerning the circumstances that favor liver cancer cell
metastasis will aid in finding treatment options that can control
the growth and metastasis of liver cancer.
4-HPR is a known retinoid analog that is active
against several types of tumors that arise via different
ontological mechanisms (28). A
phase II clinical study in adults with prostate cancer revealed
good compatibility with 4-HPR (29), and similar results were obtained in
a neuroblastoma in a phase I study in children (30). Antitumor activity of 4-HPR was also
observed in medulloblastoma (31),
human pancreatic cancer (32),
chronic myeloid leukemia (33), and
in a lung cancer xenograft mouse model (34). Moreover, Sogno et al reported
that 4-HPR is effective in inhibiting angiogenesis (35).
In our study, we compared the effect of 4-HPR with
ATRA. 4-HPR potently inhibited the growth of and colony formation
of HepG2 cells, and suppressed cell migration. Compared with ATRA,
the inhibitory effect of 4-HPR on cell growth and colony formation
was achieved at a lower concentration (5 µM). We observed that the
IC50 of HepG2 cells was approximate 12.5 µM in the cell
viability assay. Notably, pediatric neuroblastoma patients who
received oral doses of 4-HPR achieved a blood serum concentration
of 12.9 µM (30). Thus, the
effective concentrations of 4-HPR in HepG2 cells implies that 4-HPR
may be a candidate for liver cancer therapy. When HepG2 cells were
treated with 4-HPR for only 24 h, a significantly slower migration
of the cells was observed; however, no effect could be detected
using ATRA for the same amount of time. ATRA required much higher
concentrations and longer incubation times to achieve the same
inhibition rates as 4-HPR. These results indicate that much lower
amounts of 4-HPR are required for growth suppression and migration
of liver cancer cells, and hence, the drug may be applied
clinically with less toxicity.
The main antitumor activity of 4-HPR is the
induction of apoptosis by retinoic acid receptor-dependent or
-independent mechanisms (8,36). 4-HPR also reduces the plasma
concentrations of retinol and retinol binding protein (37). To determine whether the cytotoxic
effect of 4-HPR in HepG2 cells is due to the induction of
apoptosis, we analyzed the expression levels of proteins involved
in this process; however, we found no marked changes in such
proteins upon 4-HPR treatment (data not shown). A previous report
is consistent with our results (20); hence, the mechanism of 4-HPR action
might be different in HepG2 cells. The p38-MAPK pathway has been
reported to mediate various cellular behaviors that are closely
related to tumor initiation and progression (38). Nevertheless, the regulation of
p38-MAPK in tumor development is complicated and controversial,
involving responses of various cells and cancer types (39). In this study, we found that 4-HPR
inhibited the migration of HepG2 cells by significantly inducing
the activation of p38-MAPK. When the p38-MAPK inhibitor SB203580
was added to the culture system preceding 4-HPR treatment, the
inhibitory effect on migration was ameliorated. This allows for the
preliminarily conclusion that 4-HPR inhibits the migration of HepG2
cells via stimulation of the p38-MAPK pathway.
MLCK confers the rat pituitary adenoma cells with a
slow and directional motility (40), and phosphorylation of MLC markedly
improves the invasion and migration ability of gastric cancer cells
(41). Leiomyosarcoma patients with
high expression of MLCK or p-MLC have shorter life spans than the
patients with low expression of these proteins (42). In our study, treatment of HepG2
cells with 4-HPR for 48 h resulted in the downregulated expression
of MLCK. Simultaneously, the expression of MLCK and p-MLC were
significantly suppressed. When the activation of MLCK was inhibited
by ML-7, cell migration was retarded and accompanied by reduced
p-MLC levels. In addition, ML-7 in combination with 4-HPR or ATRA
enhanced the inhibitory effect on the migration of HepG2 cells,
compared to 4-HPR or ATRA alone. E-cadherin and F-actin play
important roles in tumor cell migration (43,44).
E-cadherin protein levels often decrease and F-actin levels
increase in aggressive tumor cells (45,46).
4-HPR downregulated the expression of F-actin and upregulated the
expression of E-cadherin in HepG2 cells.
Based on our results, 4-HPR decreased the
proliferation and migration of HepG2 cells in association with
activation of p38-MAPK and inhibition of MLCK. We measured the
protein expression of MLCK and p-MLC after SB203580 pretreatment
and found that p-MLC increased compared to 4-HPR treatment alone.
Meanwhile, in cells treated with ML-7 and 4-HPR, p-p38 was
upregulated compared to treatment with 4-HPR alone. These results
provide evidence of a reciprocal cross-talk between MLCK and
p38-MAPK.
Erk-MAPK governs the cell movement via p-MLC
(47). We also found an altered
expression of p-p38 preceded that of p-MLC after 4-HPR treatment
(data not shown). Thus, we presently believe that 4-HPR generates
its effects on HepG2 cells via inhibiting MLCK activation through
the p38-MAPK signaling pathway. The exact mechanism of HepG2 cell
migration involving p38-MAPK via p-MLC, however, needs to be
further investigated.
Collectively, the present study using the HepG2 cell
line demonstrated a marked potential effects of 4-HPR on liver
cancer. 4-HPR potentially inhibits the biological behaviors
involved in liver cancer metastasis, and may be an alternative
therapeutic agent for its prevention. Despite these findings,
further studies on the specific targets of 4-HPR in these signaling
pathways are required, as well as therapeutic experiments using
in vivo models are warranted.
Acknowledgements
The authors thank Pro Zhilin Qi (Department of
Biochemistry, Wannan Medical College, Wuhu, China) for her
technical support.
Funding
The present study was financially supported by the
National Natural Science Foundation of China (nos. 81272399 and
81400695), the Natural Science Foundation of Anhui Province (no.
1508085QH185) and the Youth Projects of Wannan Medical College (no.
WK201403).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
YW, WW and LZ conceived and designed the study. LZ
performed the experiments. HL provided the cell line. QZ and HL
gave experimental guidance. LZ and DH analyzed the experimental
data and wrote the paper. YW reviewed and edited the manuscript.
All authors read and approved the manuscript and agree to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
All experimental protocols were approved by the
Institutional Review Board of Anhui Medical University (Hefei,
China).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Allan BJ, Parikh PP, Diaz S, Perez EA,
Neville HL and Sola JE: Predictors of survival and incidence of
hepatoblastoma in the paediatric population. HPB (Oxford).
15:741–746. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
McAteer JP, Goldin AB, Healey PJ and Gow
KW: Surgical treatment of primary liver tumors in children:
Outcomes analysis of resection and transplantation in the SEER
database. Pediatr Transplant. 17:744–750. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kremer N, Walther AE and Tiao GM:
Management of hepatoblastoma: An update. Curr Opin Pediatr.
26:362–369. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wu X, Wang X, Qien X, Liu H, Ying J, Yang
Z and Yao H: Four years' experience with the treatment of
all-trans retinoic acid in acute promyelocytic leukemia. Am
J Hematol. 43:183–189. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wu X, Lu H, Zhou L, Huang Y and Chen H:
Changes of phosphatidylcholine-specific phospholipase C in
hepatocarcinogenesis and in the proliferation and differentiation
of rat liver cancer cells. Cell Biol Int. 21:375–381. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cooper JP, Reynolds CP, Cho H and Kang MH:
Clinical development of fenretinide as an antineoplastic drug:
Pharmacology perspectives. Exp Biol Med (Maywood). 242:1178–1184.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vaccari M, Silingardi P, Argnani A, Horn
W, Giungi M, Mascolo MG, Grilli S and Colacci A: In vitro effects
of fenretinide on cell-matrix interactions. Anticancer Res.
20:3059–3066. 2000.PubMed/NCBI
|
|
8
|
Aoyama Y: Experimental studies on the
effects of the combined use of N-(4-hydroxyphenyl)retinamide
(4-HPR) and tamoxifen (TAM) for estrogen receptor (ER)-negative
breast cancer. Kurume Med J. 49:27–33. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Veronesi U, Mariani L, Decensi A, Formelli
F, Camerini T, Miceli R, Di Mauro MG, Costa A, Marubini E, Sporn MB
and De Palo G: Fifteen-year results of a randomized phase III trial
of fenretinide to prevent second breast cancer. Ann Oncol.
17:1065–1071. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kang H, Lee M, Choi KC, Shin DM, Ko J and
Jang SW: N-(4-hydroxyphenyl)retinamide inhibits breast cancer cell
invasion through suppressing NF-κB activation and inhibiting matrix
metalloproteinase-9 expression. J Cell Biochem. 113:2845–2855.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Benelli R, Monteghirfo S, Venè R, Tosetti
F and Ferrari N: The chemopreventive retinoid 4HPR impairs prostate
cancer cell migration and invasion by interfering with
FAK/AKT/GSK3beta pathway and beta-catenin stability. Mol Cancer.
9:1422010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Moore MM, Stockler M, Lim R, Mok TSK,
Millward M and Boyer MJ: A phase II study of fenretinide in
patients with hormone refractory prostate cancer: A trial of the
Cancer Therapeutics Research Group. Cancer Chemother Pharmacol.
66:845–850. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kyriakis JM and Avruch J: Mammalian
mitogen-activated protein kinase signal transduction pathways
activated by stress and inflammation. Physiol Rev. 81:807–869.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Matsuzawa A, Nishitoh H, Tobiume K, Takeda
K and Ichijo H: Physiological roles of ASK1-mediated signal
transduction in oxidative stress- and endoplasmic reticulum
stress-induced apoptosis: Advanced findings from ASK1 knockout
mice. Antioxid Redox Signal. 4:415–425. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Martindale JL and Holbrook NJ: Cellular
response to oxidative stress: Signaling for suicide and survival. J
Cell Physiol. 192:1–15. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xia Z, Dickens M, Raingeaud J, Davis RJ
and Greenberg ME: Opposing effects of ERK and JNK-p38 MAP kinases
on apoptosis. Science. 270:1326–1331. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ganeshan VR and Schor NF: p75 neurotrophin
receptor and fenretinide-induced signaling in neuroblastoma. Cancer
Chemother Pharmacol. 73:271–279. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cao J, Ying M, Xie N, Lin G, Dong R, Zhang
J, Yan H, Yang X, He Q and Yang B: The oxidation states of DJ-1
dictate the cell fate in response to oxidative stress triggered by
4-hpr: Autophagy or apoptosis? Antioxid Redox Signal. 21:1443–1459.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Makena MR, Koneru B, Nguyen TH, Kang MH
and Reynolds CP: Reactive oxygen species-mediated synergism of
fenretinide and romidepsin in preclinical models of T-cell lymphoid
malignancies. Mol Cancer Ther. 16:649–661. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang H, Nie Y, Li Y and Wan Y-JY: ERK1/2
deactivation enhances cytoplasmic Nur77 expression level and
improves the apoptotic effect of fenretinide in human liver cancer
cells. Biochem Pharmacol. 81:910–916. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhou X, Liu Y, You J, Zhang H, Zhang X and
Ye L: Myosin light-chain kinase contributes to the proliferation
and migration of breast cancer cells through cross-talk with
activated ERK1/2. Cancer Lett. 270:312–327. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chew TL, Wolf WA, Gallagher PJ, Matsumura
F and Chisholm RL: A fluorescent resonant energy transfer-based
biosensor reveals transient and regional myosin light chain kinase
activation in lamella and cleavage furrows. J Cell Biol.
156:543–553. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kolega J: Asymmetric distribution of
myosin IIB in migrating endothelial cells is regulated by a
rho-dependent kinase and contributes to tail retraction. Mol Biol
Cell. 14:4745–4757. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
López-Terrada D, Cheung SW, Finegold MJ
and Knowles BB: Hep G2 is a hepatoblastoma-derived cell line. Hum
Pathol. 40:1512–1515. 2009. View Article : Google Scholar
|
|
25
|
Liu H, Chen F, Zhang L, Zhou Q, Gui S and
Wang Y: A novel all-trans retinoic acid derivative
4-amino-2-trifluoromethyl-phenyl retinate inhibits the
proliferation of human hepatocellular carcinoma HepG2 cells by
inducing G0/G1 cell cycle arrest and apoptosis via upregulation of
p53 and ASPP1 and downregulation of iASPP. Oncol Rep. 36:333–341.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ryerson AB, Eheman CR, Altekruse SF, Ward
JW, Jemal A, Sherman RL, Henley SJ, Holtzman D, Lake A, Noone AM,
et al: Annual Report to the Nation on the Status of Cancer,
1975–2012, featuring the increasing incidence of liver cancer.
Cancer. 122:1312–1337. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mönig SP, Baldus SE, Hennecken JK,
Spiecker DB, Grass G, Schneider PM, Thiele J, Dienes HP and
Hölscher AH: Expression of MMP-2 is associated with progression and
lymph node metastasis of gastric carcinoma. Histopathology.
39:597–602. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kitareewan S, Spinella MJ, Allopenna J,
Reczek PR and Dmitrovsky E: 4HPR triggers apoptosis but not
differentiation in retinoid sensitive and resistant human embryonal
carcinoma cells through an RARgamma independent pathway. Oncogene.
18:5747–5755. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pienta KJ, Esper PS, Zwas F, Krzeminski R
and Flaherty LE: Phase II chemoprevention trial of oral fenretinide
in patients at risk for adenocarcinoma of the prostate. Am J Clin
Oncol. 20:36–39. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Garaventa A, Luksch R, Lo Piccolo MS,
Cavadini E, Montaldo PG, Pizzitola MR, Boni L, Ponzoni M, Decensi
A, De Bernardi B, et al: Phase I trial and pharmacokinetics of
fenretinide in children with neuroblastoma. Clin Cancer Res.
9:2032–2039. 2003.PubMed/NCBI
|
|
31
|
Reddy Damodar C, Guttapalli A, Adamson PC,
Vemuri MC, O'Rourke D, Sutton LN and Phillips PC: Anticancer
effects of fenretinide in human medulloblastoma. Cancer Lett.
231:262–269. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Messner MC and Cabot MC: Cytotoxic
responses to N-(4-hydroxyphenyl)retinamide in human pancreatic
cancer cells. Cancer Chemother Pharmacol. 68:477–487. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Du Y, Xia Y, Pan X, Chen Z, Wang A, Wang
K, Li J and Zhang J: Fenretinide targets chronic myeloid leukemia
stem/progenitor cells by regulation of redox signaling. Antioxid
Redox Signal. 20:1866–1880. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Durante S, Orienti I, Teti G, Salvatore V,
Focaroli S, Tesei A, Pignatta S and Falconi M: Anti-tumor activity
of fenretinide complexed with human serum albumin in lung cancer
xenograft mouse model. Oncotarget. 5:4811–4820. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sogno I, Venè R, Ferrari N, De Censi A,
Imperatori A, Noonan DM, Tosetti F and Albini A: Angioprevention
with fenretinide: Targeting angiogenesis in prevention and
therapeutic strategies. Crit Rev Oncol Hematol. 75:2–14. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sharp RM, Bello-DeOcampo D, Quader ST and
Webber MM: N-(4-hydroxyphenyl)retinamide (4-HPR) decreases
neoplastic properties of human prostate cells: An agent for
prevention. Mutat Res. 496:163–170. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Holven KB, Natarajan V, Gundersen TE,
Moskaug JO, Norum KR and Blomhoff R: Secretion of
N-(4-hydroxyphenyl) retinamide-retinol-binding protein from liver
parenchymal cells: Evidence for reduced affinity of the complex for
transthyretin. Int J Cancer. 71:654–659. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sui X, Kong N, Ye L, Han W, Zhou J, Zhang
Q, He C and Pan H: p38 and JNK MAPK pathways control the balance of
apoptosis and autophagy in response to chemotherapeutic agents.
Cancer Lett. 344:174–179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wagner EF and Nebreda AR: Signal
integration by JNK and p38 MAPK pathways in cancer development. Nat
Rev Cancer. 9:537–549. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ávila-Rodríguez D, Agama Solano C,
González-Pozos S, Méndez-Méndez Vicente J, Plata Ortiz A,
Arreola-Mendoza L and Mendoza-Garrido ME: The shift in GH3 cell
shape and cell motility is dependent on MLCK and ROCK. Exp Cell
Res. 354:1–17. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen Z, Liu S, Xia Y and Wu K: MiR-31
regulates Rho-associated kinase-myosin light chain (ROCK-MLC)
pathway and inhibits gastric cancer invasion: Roles of RhoA. Med
Sci Monit. 22:4679–4691. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li HS, Lin Q, Wu J, Jiang ZH, Zhao JB, Pan
J, He WQ and Zha JM: Myosin regulatory light chain phosphorylation
is associated with leiomyosarcoma development. Biomed Pharmacother.
92:810–818. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nürnberg A, Kitzing T and Grosse R:
Nucleating actin for invasion. Nat Rev Cancer. 11:177–187. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Shao JB, Gao ZM, Huang WY and Lu ZB: The
mechanism of epithelial-mesenchymal transition induced by TGF-β1 in
neuroblastoma cells. Int J Oncol. 50:1623–1633. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wu B, Yang S, Sun H, Sun T, Ji F, Wang Y,
Xu L and Zhou D: Keap1 inhibits metastatic properties of NSCLC
cells by stabilizing architectures of F-actin and focal adhesions.
Mol Cancer Res. 16:508–516. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Huang C, Jacobson K and Schaller MD: MAP
kinases and cell migration. J Cell Sci. 117:4619–4628. 2004.
View Article : Google Scholar : PubMed/NCBI
|