Introduction
Gastric cancer ranks as the fourth most common
cancer, and remains the second leading cause of cancer-related
death worldwide (1). High incidence
areas include Asia, Central and Eastern Europe and South America
(2). Despite the steadily declining
incidence, the clinical outcome of gastric cancer has improved
modestly in recent years. In addition, in 2013, more than 841,000
deaths were attributed to gastric cancer (3). The overall 5-year survival rate is
still less than 25% (4). One main
leading cause is the poor response of gastric cancer to currently
available treatments (5). Thus,
currently, targeted drugs are being explored extensively, which may
afford new therapeutic avenues for gastric cancer.
Poly(ADP-ribose) polymerase (PARP) inhibitors are a
type of promising targeted drugs, which have been approved by the
FDA to be used for the treatment of breast and ovarian cancer
patients with mutations of the BRCA1 or BRCA2 genes
(6). The PARP inhibitor is designed
to target cancers with impaired DNA damage repair abilities, such
as mutations of BRCA1/2, which is called ‘synthetic
lethality’ (7,8). However, several other molecular
biomarkers have also been revealed to predict the sensitivity to
PARP inhibitors, including CDK depletion (9), RAD51C deficiency (10), ATM deficiency (11) and AT-rich interactive domain
containing protein 1A (ARID1A) deficiency (12). Nonetheless, in gastric cancer, the
most recent clinical trial has reported that olaparib, an oral PARP
inhibitor, in combination with paclitaxel did not significantly
improve the overall survival in the overall or ATM-negative
population of Asian patients (13).
Thus, there is an urgent need to identify new drug combinations,
and new predictive biomarkers for the treatment of gastric
cancer.
Notably, phosphoinositide 3-kinase (PI3K) inhibition
has been revealed to impair BRCA1/2 expression, render tumor cells
more deficient in HR repair and sensitize breast cancer to PARP
inhibition (14). The PI3K
signaling pathway plays a critical role in regulating various
cellular processes, including proliferation, growth, apoptosis and
cell metabolism (15,16), and has been demonstrated to be an
attractive target for the treatment of various types of cancers.
BKM120, a selective pan-class I PI3K inhibitor, has been reported
to be effective in gastric cancer (17). In fact, the combined treatment with
PI3K inhibitor BKM120 and PARP inhibitor olaparib has been
demonstrated to be effective for breast (14,18),
prostate (19) and ovarian cancer
(20,21). However, the efficacy of the
combination in gastric cancer remains unclear, and potential
predictive biomarkers must be explored.
Recent evidence indicates that ARID1A deficiency
sensitizes tumor cells to PARP and PI3K inhibitors (12,22).
ARID1A is a novel chromatin remodeling gene, which has been
identified to be frequently mutated in a broad range of cancers
(23–25). The incidence of mutations of
ARID1A in gastric cancer varies from 8 to 27% (26–28).
ARID1A encodes the protein BRG1-associated factor 250a
(BAF250a), which is a crucial non-catalytic subunit of human
switch/sucrose non-fermentable (SWI/SNF) complex (29). The SWI/SNF complex plays an
important role in transcription, DNA replication and DNA damage
repair (12,30,31).
Mutations of ARID1A in tumors usually cause downregulated
protein expression (32) and ARID1A
is suggested as a bona fide tumor suppressor (33). ARID1A depletion has been reported to
promote gastric cancer cell growth in vitro, xenograft tumor
growth in vivo (22), and
cell migration and invasion (34),
which indicate that ARID1A is a potential target for gastric
cancer.
Therefore, in the present study, we investigated the
therapeutic role of the combined treatment of PI3K inhibitor BKM120
and PARP inhibitor olaparib on gastric cancer cells, and explored
ARID1A deficiency as a potential predictive biomarker of
therapeutic efficacy.
Materials and methods
Cell lines and reagents
Human gastric cancer cell lines AGS and SNU-1 were
purchased from the Cell Bank of the Shanghai Institutes for
Biological Sciences (Shanghai, China). Cells were grown in
RPMI-1640 medium (HyClone Laboratories; GE Healthcare, Chicago, IL,
USA) supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA). Cells were cultured in
a humidified incubator containing 5% carbon dioxide
(CO2) at 37°C. BKM120 and olaparib were obtained from
Selleck Chemicals (Houston, TX, USA) and dissolved in dimethyl
sulfoxide (DMSO).
Short hairpin RNA (shRNA)-mediated
ARID1A knockdown
Lentivirus-ARID1A-RNAi vector and the corresponding
empty vector were purchased from Shanghai GeneChem, Co., Ltd.
(Shanghai, China). We designed two independent shRNA constructs and
subcloned them into the lentivirus vectors (sequence: shARID1A #1,
5′-GCCTGATCTATCTGGTTCAAT-3′; shARID1A #2,
5′-CCTCTCTTATACACAGCAGAT-3′). The constructed plasmid was confirmed
by DNA sequencing. Then, the lentiviral vector and packaging mix
with Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific,
Inc.) were transfected into 293FT cells. The supernatant containing
the lentivirus was collected 48 h later, and was purified and
supplemented with 8 µg/ml Polybrene (Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, US). The virus solution was used to infect
the target cells. Another 72 h later, 2 µg/ml of puromycin (Sangon
Biotech, Shanghai, China) was added for 1 week to select the stably
transfected cells. Then, western blot analysis was used to evaluate
the protein expression level.
MTS assay
Cells were seeded into a 96-well culture plate. On
the next day, the indicated drugs were used to treat the cells.
Seventy-two hours later, the cells were washed and treated with 20
µl MTS reagent (Promega, Madison, WI, USA) per well for 2 h. Cell
viability was detected by a microplate reader at a wavelength of
490 nm. The half-maximal inhibitory concentration (IC50)
was analyzed using GraphPad Prism 6.0 (GraphPad Software, Inc., San
Diego, CA, USA). The combination index (CI) was calculated by
Chou-Talalay method (35). CI <1
indicates synergism, CI >1 indicates antagonism, and CI=1
indicates additive interactions.
Clonogenic assay
Cells were planted into 6-well culture plates. On
the next day, the cells were treated with the indicated drugs for 3
days and cultured for 2 weeks. Then, the clones were fixed with 4%
polymerised formaldehyde, and were stained with 0.01% crystal
violet (Sangon Biotech). The number of clones were counted.
Small interfering RNA (siRNA)-mediated
ARID1A knockdown
Cells were planted into a 6-cm cell culture dish, at
a density of 30%. On the next day, the cells were transfected using
ARID1A or non-target siRNAs (Shanghai GeneChem) with
Oligofectamine transfection reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) based on the manufacturer's protocol. The siRNA
sequences were as follows (34):
ARID1A, sense, 5′-GCCCUAACAUGGCCAAUAUTT-3′ and antisense,
5′-AUAUUGGCCAUGUUAGGGCTT-3′; non-target control: sense,
5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense,
5′-ACGUGACACGUUCGGAGAATT-3′.
Apoptosis assay
Cells were seeded into 6-well culture plates at an
~40% density. On the next day, the cells were exposed to the
indicating drugs. After 24 h, the cells were collected and
resuspended in binding buffer supplemented with Annexin V-PE. Then,
propidium iodide (PI) was added, and the Annexin V/PI apoptosis kit
(Invitrogen; Thermo Fisher Scientific, Inc.) was used. The rate of
apoptosis was detected by flow cytometric analysis (FACScan;
Beckman Coulter, Inc., Brea, CA, USA). In the figures demonstrating
the results, the percentage of cells in the upper right (including
necrotic or late apoptotic cells) and lower right corners
(including early apoptotic cells) were added to obtain the
percentage of apoptosis.
Western blot analysis
Cells were harvested and lysed in urine buffer
supplemented with 1% phosphorylation inhibitors and 1% protease
(Roche Diagnostics, Indianapolis, IN, USA). Total protein (30 µg)
for each sample was loaded to SDS-PAGE gel. After gel
electrophoresis, the protein was transferred to polyvinylidene
fluoride membranes (EMD Millipore, Billerica, MA, USA). Then, the
membranes were blocked using 5% non-fat dried milk for 1 h, and
washed in TBS with 0.1% Tween-20 for 3 times at room temperature.
Then, the membranes were incubated with primary antibodies
overnight at 4°C. The membranes were washed for three times, and
then incubated with horseradish peroxidise-conjugated goat
anti-mouse or goat anti-rabbit secondary antibodies (1:5,000; cat.
nos. 31430 and 31460; Invitrogen; Thermo Fisher Scientific, Inc.)
for 1 h. After washing for another 3 times, the expression of the
target proteins were visualized using an ECL detection kit (Thermo
Fisher Scientific, Inc.). The following were the antibodies used in
this study: ARID1A (1:500; cat. no. A301-040A; Bethyl Laboratories,
Montgomery, TX, USA); BRCA1, BRCA2 and cleaved-PARP (1:500; cat.
nos. 9010, 9012 and 9185; Cell Signaling Technology, Inc., Danvers,
MA, USA); AKT, p-AKT (Ser473) (1:500; cat. nos. ab28422 and
ab81283; Abcam, Cambridge, MA, USA); H2AX, γH2AX antibodies
(1:1,000; cat. nos. DR1016 and 05–636; EMD Millipore; Merck KGaA,
Darmstadt, Germany), N-cadherin, E-cadherin, β-catenin, ZEB1 and
vimentin (1:500; cat. no. 9782; Cell Signaling Technology, Inc.)
and β-actin antibody (1:2,000; cat. no. A5441; Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany).
Immunofluorescent staining
Cells on coverslips were fixed using 3%
paraformaldehyde for 20 min at room temperature, and then were
permeabilized with phosphate-buffered saline (PBS) containing 0.5%
Triton X-100 for 5 min. PBS containing 5% goat serum was used to
block the coverslips for 30 min. Then, the coverslips were
immunostained with anti-γH2AX (cat. no. 05-636; EMD Millipore)
antibody overnight at 4°C. On the next day, the coverslips were
washed for three times and incubated for 1 h using secondary
antibody Alexa Fluor 488-conjugated goat anti-mouse IgG (1:50; cat.
no. 4408; Cell Signaling Technology, Inc.) at room temperature. The
nuclei were counterstained using DAPI
(4′,6-diamidino-2-phenylindole; Sangon Biotech). Olympus laser
scanning confocal microscope (Olympus Optical Co., Tokyo, Japan)
was used to capture images. The γH2AX foci were counted from at
least 50 cells per sample.
Cell cycle analysis
Cells were treated with the indicated drugs, and
then were collected and fixed in 70% ethanol overnight at 4°C. On
the next day, the cells were washed, suspended and stained using
propidium iodide (PI) staining solution (50 µg/ml PI and 1 mg/ml
RNase in PBS). The cell cycle analysis was conducted using flow
cytometer FACScan (Beckman Coulter, Inc.).
Invasion and migration assays:
Polycarbonate membrane Transwell inserts (8- µm pore size) in a
24-well format (Corning, Inc., Corning, NY, USA) were used
For the invasion assay, the inserted membranes were
coated with Matrigel (BD Biosciences, San Jose, CA, USA). For the
migration assay, there was no Matrigel on the filter. The cells
treated with the indicated drugs were planted into the membranes of
the upper chambers in 200 µl of serum-free medium at a density of
1×105 cells/chamber, which was inserted into the lower
wells containing 500 µl of 10% FBS-supplemented medium. Twenty-four
hours later, the cells were fixed with 100% methanol and stained
with 0.1% crystal violet solution. Cells on the upper side of the
filter were removed with cotton swabs. Cells on the underside of
the filter were counted in three randomly chosen fields and
photographed using an inverted microscope (magnification, ×200;
Olympus Optical Co.).
Statistical analysis
Each experiment was repeated for at least three
times. The data are presented as mean ± standard deviation (SD) and
were analyzed using SPSS 17.0 software (SPSS, Inc., Chicago, IL,
USA). The Student's t-test or ANOVA was used. A probability level
of 0.05 was defined to indicate a statistically significant
difference.
Results
PI3K inhibitor BKM120 synergizes with
PARP inhibitor olaparib to inhibit the proliferation of gastric
cancer cells with ARID1A deficiency
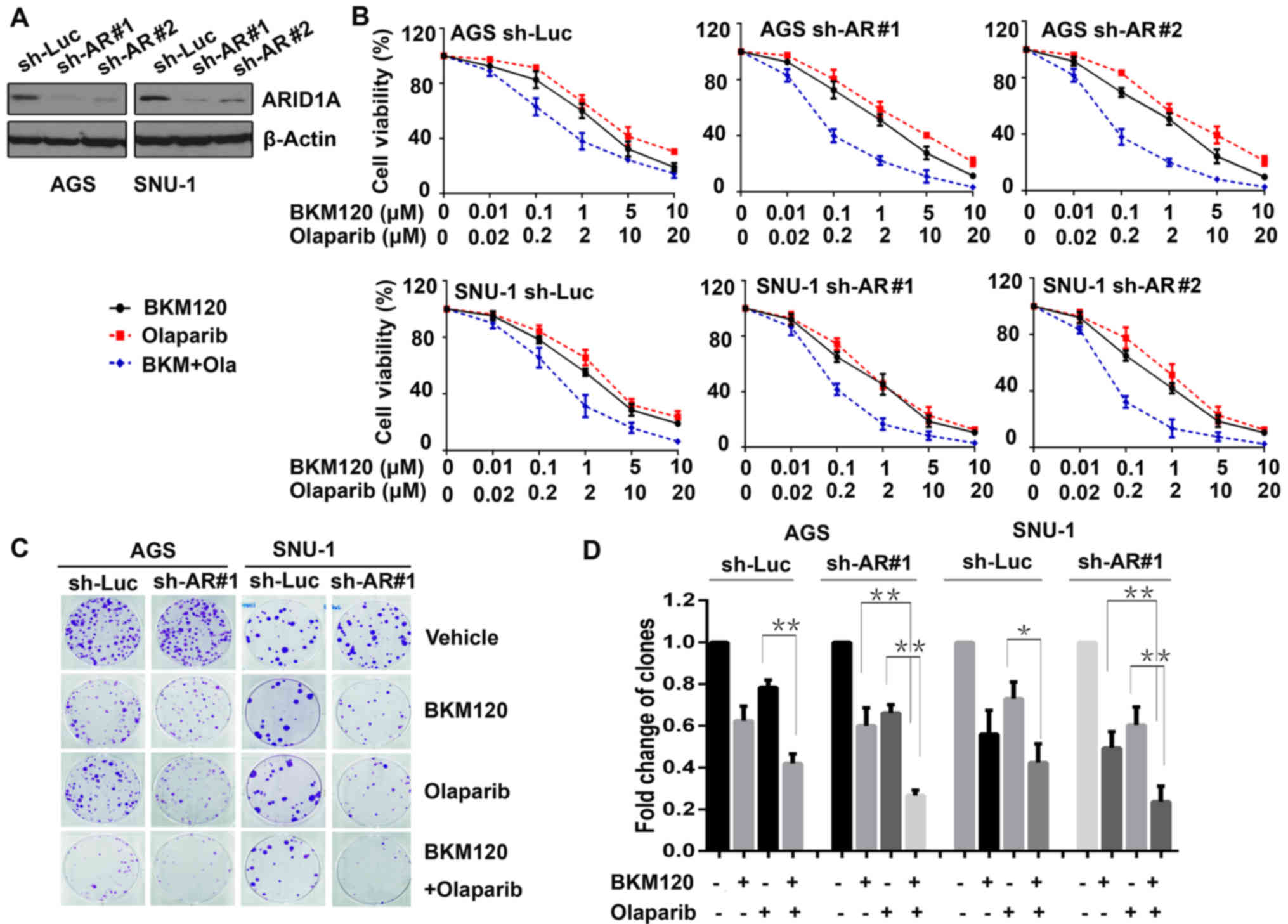
Human gastric cancer cell lines (AGS and SNU-1) were
transfected with ARID1A shRNAs (sh-AR#1 and sh-AR#2), and
the control cells were transfected with sh-Luc. The efficacy of
ARID1A knockdown was evaluated by western blot analysis
(Fig. 1A).
To explore the sensitivity of gastric cancer cells
to BKM120 and olaparib, the above cells were exposed to BKM120,
olaparib, or the combination at indicated concentrations. In
addition, the MTS assay was conducted. The results revealed that
BKM120 and olaparib inhibited the proliferation of both gastric
cancer cell lines in a dose-dependent manner (Fig. 1B). The IC50 values
calculated for BKM120 for the control (sh-Luc) of AGS and SNU-1
cells were higher (1.78±0.89 for AGS and 1.13±0.34 µmol/l for
SNU-1) than those for the ARID1A-knockdown cells (1.30±0.65 and
1.36±0.59 µmol/l for AGS, 0.86±0.43 and 0.78±0.32 µmol/l for SNU-1,
respectively). In addition, marked synergy was observed between
BKM120 and olaparib in the ARID1A-depleted cells (CI value was 0.74
and 0.70 in AGS, and 0.82 and 0.63 in SNU-1 cells), but was not
observed in the control cells (CI, 0.95 in AGS and 1.05 in
SNU-1).
Additionally, clonogenic assay was performed to
verify the synergistic role of BKM120 and olaparib (Fig. 1C). The results demonstrated that in
all AGS and SNU-1 cells, BKM120 as a single-agent markedly reduced
the number of clones, as well as olaparib. Compared with olaparib,
the combined treatment with BKM120 and olaparib led to
significantly additional inhibition in all gastric cancer cells
(P<0.05). Whereas, compared with BKM120, the combined treatment
resulted in significantly further inhibition only in
ARID1A-depleted cells, but not in the control cells (Fig. 1D), suggesting that the synergistic
role of the combination was notable in gastric cancer cells with
ARID1A depletion.
Combination with BKM120 and olaparib
induces a higher percentage of apoptosis in ARID1A-depleted gastric
cancer cells
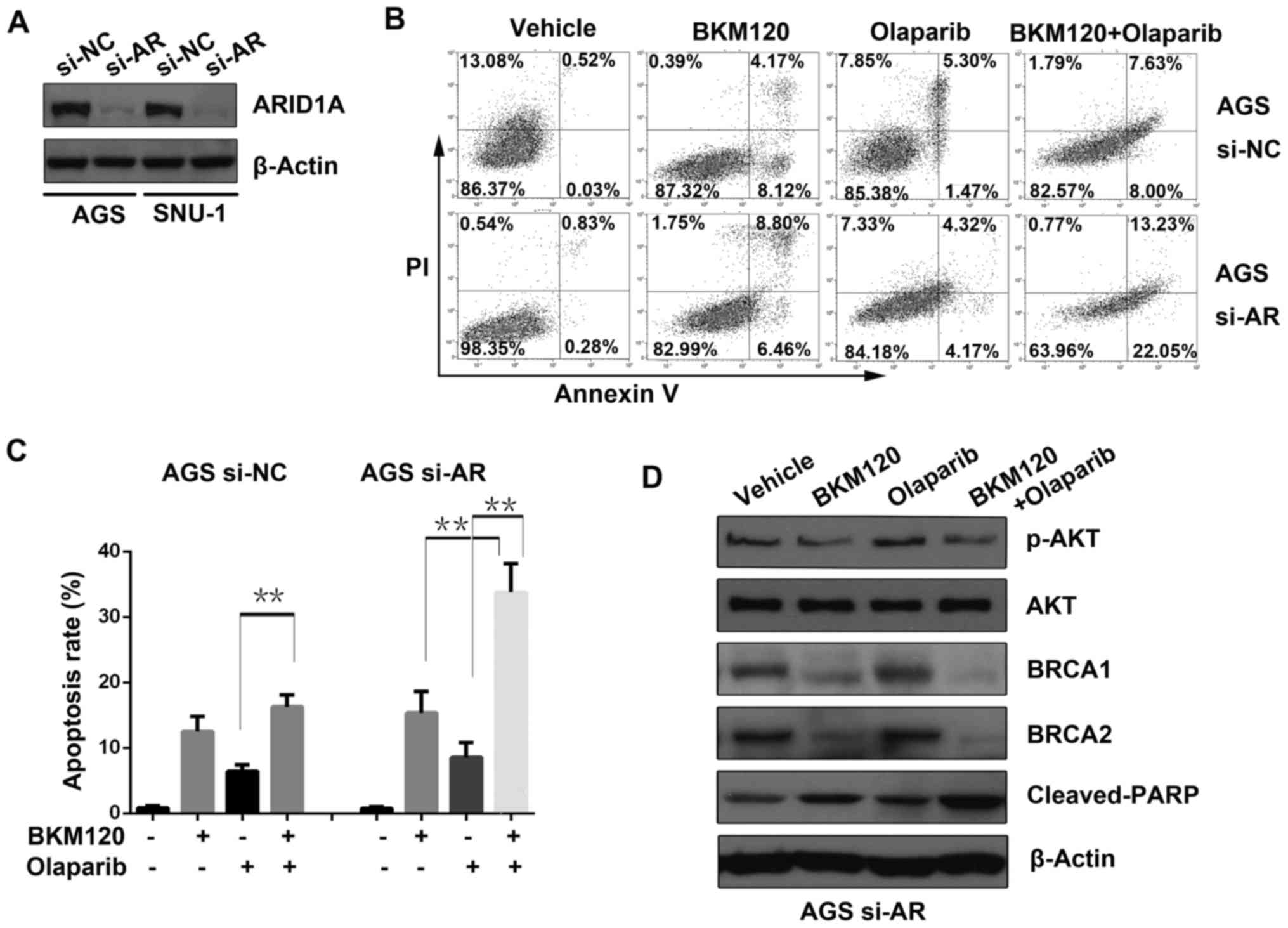
ARID1A was transiently knocked down in AGS
and SNU-1 cells by si-RNA (si-AR), and western blot analysis was
used to evaluate the knockdown efficacy (Fig. 2A).
To identify the possible mechanisms of the
synergistic effect between BKM120 and olaparib, an apoptosis assay
was performed (Fig. 2B). The
results demonstrated that BKM120 or olaparib induced apoptosis when
used alone. However, the combined treatment with BKM120 and
olaparib significantly increased apoptosis compared to that of
either single-agent in the ARID1A-depleted AGS cells, but not in
the control cells (si-NC) (Fig.
2C).
Furthermore, western blot analysis was applied to
detect the expression level of proteins including phosphorylated
AKT (p-AKT) at Ser473, AKT, BRCA1/2 and cleaved PARP in AGS cells
with ARID1A deficiency (si-AR). The results revealed that BKM120
either as a single-agent or in combination with olaparib
substantially reduced the abundance of p-AKT, a downstream effector
of PI3K, as well as BRCA1 and BRCA2. In accordance with the results
of the apoptosis assay, combined use of BKM120 and olaparib
significantly enhanced the abundance of cleaved PARP, a marker of
active apoptosis, compared to that of either BKM120 or olaparib
(Fig. 2D).
BKM120 combined with olaparib induces
significant DNA damage in gastric cancer cells with ARID1A
deficiency
To evaluate the effect of the drugs on DNA damage,
the DNA damage marker γH2AX was detected by immunofluorescence
assay (Fig 3A). The results
revealed that the γH2AX foci were markedly increased by BKM120 or
olaparib compared to that of the control (vehicle) in the AGS
cells. In the control (sh-Luc) cells, the dual inhibition of PI3K
and PARP significantly increased more γH2AX foci only when compared
with the cells treated with olaparib, but not to BKM120. However,
in the ARID1A-depleted (sh-AR#1) AGS cells, the combined treatment
significantly induced more γH2AX foci than both single BKM120 and
olaparib treatments (P<0.01) (Fig.
3B). These results suggested that the synergistic role of
BKM120 and olaparib in inducing DNA damage was more remarkable in
ARID1A-deficient gastric cancer cells.
Subsequently, western blot analysis was performed to
detect the abundance of γH2AX in ARID1A-depleted AGS cells treated
with the indicated drugs (Fig. 3C).
The results indicated that the combined treatment with BKM120 and
olaparib increased the abundance of γH2AX more significantly than
both the single-agent groups and the control group (vehicle), in
the ARID1A-knockdown AGS cells (AGS sh-AR#1) (Fig. 3D).
G2-M checkpoint arrest induced by the
combined treatment with BKM120 and olaparib is significantly
attenuated in ARID1A-deficient gastric cancer cells
Since G2-M checkpoint arrest is critical to DNA
damage repair, the cell cycle distribution was detected by FACS
analysis in AGS cells with or without ARID1A depletion (Fig. 4A). The results indicated that single
BKM120 or olaparib activated G2-M phase arrest compared with the
vehicle group, which was a response to the increased DNA damage
that they induced. The combination of BKM120 and olaparib enhanced
the G2-M phase arrest more significantly than single BKM120 or
olaparib. However, in the ARID1A-depleted AGS cells (AGS si-AR),
the G2-M phase arrest induced by the combination was weaker
compared to that of the control (si-NC) cells (Fig. 4B). The cell cycle analysis in SNU-1
cells revealed similar results (Fig. 4C
and D), suggesting that ARID1A depletion attenuated the G2-M
checkpoint arrest induced by the combined treatment.
BKM120 combined with olaparib inhibits
migration and invasion of gastric cancer cells with ARID1A
depletion
Invasion and migration are crucial to the growth and
metastasis of cancer. Thus, invasion and migration assays were
performed using ARID1A-depleted AGS (AGS sh-AR#1) cells treated
with vehicle, BKM120, olaparib or the combination (Fig. 5A). The results revealed that both
BKM120 and olaparib inhibited the migration and invasion of the
gastric cancer cells, compared to that of the control group.
However, the dual treatment with BKM120 and olaparib resulted in
the lowest degree of migration and invasion, compared to that of
either BKM120 or olaparib as a single-agent treatment (Fig. 5B).
Moreover, western blot analysis was conducted to
evaluate the abundance of proteins involving in invasion and
migration, including N-cadherin, E-cadherin, β-catenin, ZEB1 and
vimentin in the ARID1A-depleted AGS (AGS sh-AR#1) cells (Fig. 5C). The results showed that compared
to that of the control (vehicle) group, the abundance of E-cadherin
increased, while N-cadherin, ZEB1 and vimentin decreased in both
the BKM120 and olaparib groups. In the group treated with the
combination of BKM120 and olaparib, the increase in E-vadherin and
the decrease in ZEB1 were more significant than both of the
single-agent treatment groups (Fig.
5D).
Discussion
ARID1A has been demonstrated to regulate gastric
cancer cell proliferation and migration (22,34,36).
ARID1A-deficient cancer cells are reported to be sensitive to
inhibition of PI3K and PARP. In the present study, we revealed that
the combination of PI3K inhibitor BKM120 and PARP inhibitor
olaparib had synergistic antitumor effects on gastric cancer cells
with ARID1A deficiency, via inhibiting cell proliferation, inducing
apoptosis, increasing DNA damage, attenuating G2-M checkpoint
arrest and suppressing invasion and migration. To the best of our
knowledge, this is the first study to explore the effect of the
combination of BKM120 and olaparib on gastric cancer, suggesting
ARID1A deficiency as a potential predictive biomarker.
Recently, Zhang et al reported that ARID1A
deficiency increased the transcription of PI3K, and activated the
PI3K/AKT pathway in gastric cancer. Consequently, PI3K-inhibitor
LY294002 and AKT-inhibitor mk2206 were highly effective on gastric
cancer cells harboring deficient ARID1A in vitro and in
vivo (22). Similarly, several
other studies also revealed that the loss of ARID1A expression
upregulated the phosphorylation of AKT, which was a component of
the PI3K signaling pathway, in endometrial cancer (37), ovarian clear cell carcinoma
(38), and colon cancer (39). The synthetic lethal interaction
between loss of ARID1A expression and inhibition of the PI3K/AKT
pathway was also reported by Samartzis et al in breast and
ovarian cancer cells (40).
Meanwhile, Shen et al reported that ARID1A deficiency
impaired the DNA damage checkpoint and sensitized cells to PARP
inhibitors (12). In accordance
with the previous findings, herein, we demonstrated that BKM120 and
olaparib significantly suppressed the proliferation of gastric
cancer cells with ARID1A depletion.
PARP inhibitors show a synergistic effect with
several therapeutic reagents, such as EGFR inhibitors (41) and PI3K/mTOR inhibitors (42). The combination with olaparib and
BKM120 has been reported to be synergistically effective in breast
(14,18), prostate (19), and ovarian cancer with or without
PIK3CA mutations (20,21).
In the present study, the results demonstrated that olaparib
synergized with BKM120 to effectively inhibit the proliferation of
gastric cancer cells harboring ARID1A deficiency. Mechanistically,
BKM120 downregulated BRCA1/2 expression, which mimicked the
mutations of BRCA1/2, and might sensitize cells to PARP
inhibitors.
BKM120 has been reported to induce DNA damage in
breast (14,18), glioblastoma (43), ovarian and prostate cancer (19). We revealed that either BKM120 or
olaparib, when used as a single-agent treatment, induced DNA
damage. Nonetheless, the combined treatment with BKM120 and
olaparib potentiated the DNA damage, especially in ARID1A-deficient
gastric cancer cells, which may result in the synergistic
anticancer effect of BKM120 and olaparib.
Normally, after DNA damage, the cell cycle
checkpoint is activated, which leads to arrest injury, repair the
DNA damage and protect cells from apoptosis and unscheduled death
(44). The previous studies and our
results revealed that PI3K inhibitor and PARP inhibitor activated
the G2-M checkpoint, and caused cell cycle arrest, until the DNA
damage was repaired (44,45). We also revealed that the combination
with BKM120 and olaparib synergistically enhanced G2-M phase
arrest. However, the G2-M checkpoint arrest induced by the combined
treatment was significantly attenuated in ARID1A-deficient gastric
cancer cells, although the DNA damage in these cells was the most
serious. It has been reported that ARID1A is required for a proper
G2-M DNA damage checkpoint, and ARID1A deficiency impairs the G2-M
arrest and DNA damage repair (12).
Due to usual deficient function in G1-M checkpoint, most cancer
cells depend on the G2-M cell cycle checkpoint (44). Therefore, we propose that ARID1A
deficiency attenuates the G2-M checkpoint arrest induced by the
combined treatment, which may weaken the DNA damage repair, and
eventually lead to genomic instability, cell apoptosis and death.
Accordingly, another study in gastric cancer has reported that
ARID1A mutations are associated with increased microsatellite
instability (MSI), which is a form of genomic instability (26).
Invasion and migration are crucial to tumor growth
and metastasis. In addition, reduced expression of ARID1A has been
reported to enhance the migration and invasion of gastric cancer
cells via downregulation of E-cadherin transcription (34). Thus, for the drugs intended to treat
gastric cancer harboring ARID1A deficiency, it is important to gain
knowledge concerning the efficacy of these drugs on migration and
invasion. In addition, Wang et al reported that dual
treatment with BKM120 and olaparib significantly inhibited the
invasion and migration of ovarian cancer cells (20). Previous studies have reported that
ZEB1 plays an important role in suppressing E-cadherin expression
and in promoting tumor invasion and metastasis (46,47).
In accordance with these findings, our results revealed that the
dual blockade of PI3K and PARP notably suppressed the invasion and
migration in gastric cancer cells with ARID1A depletion through a
decrease in ZEB1 and an increase in E-cadherin, which is critical
for the utilization of such a drug combination in vivo and
in the clinic therapeutically.
In summary, we demonstrated that the PI3K inhibitor
BKM120 synergized with the PARP inhibitor olaparib to inhibit the
growth and migration of gastric cancer cells with ARID1A deficiency
in vitro. The present study provides a potential therapeutic
strategy for gastric cancer harboring ARID1A deficiency, which
warrants verification in vivo and in the clinic in future
research.
Acknowledgements
We would like to thank Dr Hai Zhang (Department of
Physiology and Research, Tongji Medical College, Huazhong
University of Science and Technology) for his contributions to the
immunofluorescent staining.
Funding
The present study was supported by the Natural
Science Foundation of Hubei Province (no. 2015CFB541) and the
Research Project of Hubei provincial health and Family Planning
Commission (no. WJ2017M114).
Availability of data and materials
The datasets used in the present study are available
from the corresponding author upon reasonable request.
Authors' contributions
GH, LY and YD designed and supervised the project.
LY, GY and YH performed the experiments. SL and LZ analyzed the
data and prepared the graphs. LY, WW and JW wrote the manuscript.
GH and YD edited the manuscript. All authors read and approved the
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
No animal or human experiments were included in the
present study.
Consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ARID1A
|
AT-rich interactive domain containing
protein 1A
|
|
PARP
|
poly(ADP-ribose) polymerase
|
|
PI3K
|
phosphoinositide 3-kinase
|
|
p-H3
|
phosphorylated histone H3 on Ser10
|
References
|
1
|
Venerito M, Link A, Rokkas T and
Malfertheiner P: Gastric cancer - clinical and epidemiological
aspects. Helicobacter. 21 Suppl 1:S39–S44. 2016. View Article : Google Scholar
|
|
2
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fitzmaurice C, Dicker D, Pain A, Hamavid
H, Moradi-Lakeh M, MacIntyre MF, Allen C, Hansen G, Woodbrook R,
Wolfe C, et al: Global Burden of Disease Cancer Collaboration: The
Global Burden of Cancer 2013. JAMA Oncol. 1:505–527. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ajani JA, D'Amico TA, Almhanna K, Bentrem
DJ, Chao J, Das P, Denlinger CS, Fanta P, Farjah F, Fuchs CS, et
al: Gastric Cancer, Version 3.2016, NCCN Clinical Practice
Guidelines in Oncology. J Natl Compr Canc Netw. 14:1286–1312. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Deeks ED: Olaparib: First global approval.
Drugs. 75:231–240. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Farmer H, McCabe N, Lord CJ, Tutt AN,
Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I,
Knights C, et al: Targeting the DNA repair defect in BRCA mutant
cells as a therapeutic strategy. Nature. 434:917–921. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bryant HE, Schultz N, Thomas HD, Parker
KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ and Helleday T:
Specific killing of BRCA2-deficient tumours with inhibitors of
poly(ADP-ribose) polymerase. Nature. 434:913–917. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Alagpulinsa DA, Ayyadevara S, Yaccoby S
and Reis Shmookler RJ: A cyclin-dependent kinase inhibitor,
dinaciclib, impairs homologous recombination and sensitizes
multiple myeloma cells to PARP inhibition. Mol Cancer Ther.
15:241–250. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Min A, Im SA, Yoon YK, Song SH, Nam HJ,
Hur HS, Kim HP, Lee KH, Han SW, Oh DY, et al: RAD51C-deficient
cancer cells are highly sensitive to the PARP inhibitor olaparib.
Mol Cancer Ther. 12:865–877. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kubota E, Williamson CT, Ye R, Elegbede A,
Peterson L, Lees-Miller SP and Bebb DG: Low ATM protein expression
and depletion of p53 correlates with olaparib sensitivity in
gastric cancer cell lines. Cell Cycle. 13:2129–2137. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shen J, Peng Y, Wei L, Zhang W, Yang L,
Lan L, Kapoor P, Ju Z, Mo Q, Shih IeM, et al: ARID1A deficiency
impairs the DNA damage checkpoint and sensitizes cells to PARP
inhibitors. Cancer Discov. 5:752–767. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bang YJ, Xu RH, Chin K, Lee KW, Park SH,
Rha SY, Shen L, Qin S, Xu N, Im SA, et al: Olaparib in combination
with paclitaxel in patients with advanced gastric cancer who have
progressed following first-line therapy (GOLD): A double-blind,
randomised, placebo-controlled, phase 3 trial. Lancet Oncol.
18:1637–1651. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ibrahim YH, García-García C, Serra V, He
L, Torres-Lockhart K, Prat A, Anton P, Cozar P, Guzmán M, Grueso J,
et al: PI3K inhibition impairs BRCA1/2 expression and sensitizes
BRCA-proficient triple-negative breast cancer to PARP inhibition.
Cancer Discov. 2:1036–1047. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Engelman JA: Targeting PI3K signalling in
cancer: Opportunities, challenges and limitations. Nat Rev Cancer.
9:550–562. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Park E, Park J, Han SW, Im SA, Kim TY, Oh
DY and Bang YJ: NVP-BKM120, a novel PI3K inhibitor, shows synergism
with a STAT3 inhibitor in human gastric cancer cells harboring KRAS
mutations. Int J Oncol. 40:1259–1266. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Juvekar A, Burga LN, Hu H, Lunsford EP,
Ibrahim YH, Balmañà J, Rajendran A, Papa A, Spencer K, Lyssiotis
CA, et al: Combining a PI3K inhibitor with a PARP inhibitor
provides an effective therapy for BRCA1-related breast cancer.
Cancer Discov. 2:1048–1063. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
González-Billalabeitia E, Seitzer N, Song
SJ, Song MS, Patnaik A, Liu XS, Epping MT, Papa A, Hobbs RM, Chen
M, et al: Vulnerabilities of PTEN-TP53-deficient prostate cancers
to compound PARP-PI3K inhibition. Cancer Discov. 4:896–904. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang D, Wang M, Jiang N, Zhang Y, Bian X,
Wang X, Roberts TM, Zhao JJ, Liu P and Cheng H: Effective use of
PI3K inhibitor BKM120 and PARP inhibitor Olaparib to treat PIK3CA
mutant ovarian cancer. Oncotarget. 7:13153–13166. 2016.PubMed/NCBI
|
|
21
|
Wang D, Li C, Zhang Y, Wang M, Jiang N,
Xiang L, Li T, Roberts TM, Zhao JJ, Cheng H, et al: Combined
inhibition of PI3K and PARP is effective in the treatment of
ovarian cancer cells with wild-type PIK3CA genes. Gynecol
Oncol. 142:548–556. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang Q, Yan HB, Wang J, Cui SJ, Wang XQ,
Jiang YH, Feng L, Yang PY and Liu F: Chromatin remodeling gene
AT-rich interactive domain-containing protein 1A suppresses gastric
cancer cell proliferation by targeting PIK3CA and
PDK1. Oncotarget. 7:46127–46141. 2016.PubMed/NCBI
|
|
23
|
Jones S, Wang TL, Shih IeM, Mao TL,
Nakayama K, Roden R, Glas R, Slamon D, Diaz LA Jr, Vogelstein B, et
al: Frequent mutations of chromatin remodeling gene ARID1A
in ovarian clear cell carcinoma. Science. 330:228–231. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kandoth C, Schultz N, Cherniack AD, Akbani
R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, Benz CC, et al:
Cancer Genome Atlas Research Network: Integrated genomic
characterization of endometrial carcinoma. Nature. 497:67–73. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Takeda T, Banno K, Okawa R, Yanokura M,
Iijima M, Irie-Kunitomi H, Nakamura K, Iida M, Adachi M, Umene K,
et al: ARID1A gene mutation in ovarian and endometrial
cancers (Review). Oncol Rep. 35:607–613. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang K, Kan J, Yuen ST, Shi ST, Chu KM,
Law S, Chan TL, Kan Z, Chan AS, Tsui WY, et al: Exome sequencing
identifies frequent mutation of ARID1A in molecular subtypes
of gastric cancer. Nat Genet. 43:1219–1223. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zang ZJ, Cutcutache I, Poon SL, Zhang SL,
McPherson JR, Tao J, Rajasegaran V, Heng HL, Deng N, Gan A, et al:
Exome sequencing of gastric adenocarcinoma identifies recurrent
somatic mutations in cell adhesion and chromatin remodeling genes.
Nat Genet. 44:570–574. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ali SM, Sanford EM, Klempner SJ, Rubinson
DA, Wang K, Palma NA, Chmielecki J, Yelensky R, Palmer GA, Morosini
D, et al: Prospective comprehensive genomic profiling of advanced
gastric carcinoma cases reveals frequent clinically relevant
genomic alterations and new routes for targeted therapies.
Oncologist. 20:499–507. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Roberts CW and Orkin SH: The SWI/SNF
complex - chromatin and cancer. Nat Rev Cancer. 4:133–142. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mao TL and Shih IeM: The roles of ARID1A
in gynecologic cancer. J Gynecol Oncol. 24:376–381. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nagl NG Jr, Patsialou A, Haines DS, Dallas
PB, Beck GR Jr and Moran E: The p270 (ARID1A/SMARCF1)
subunit of mammalian SWI/SNF-related complexes is essential for
normal cell cycle arrest. Cancer Res. 65:9236–9244. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wu JN and Roberts CW: ARID1A mutations in
cancer: Another epigenetic tumor suppressor? Cancer Discov.
3:35–43. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Guan B, Wang TL and Shih IeM:
ARID1A, a factor that promotes formation of SWI/SNF-mediated
chromatin remodeling, is a tumor suppressor in gynecologic cancers.
Cancer Res. 71:6718–6727. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yan HB, Wang XF, Zhang Q, Tang ZQ, Jiang
YH, Fan HZ, Sun YH, Yang PY and Liu F: Reduced expression of the
chromatin remodeling gene ARID1A enhances gastric cancer
cell migration and invasion via downregulation of E-cadherin
transcription. Carcinogenesis. 35:867–876. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chou TC: Drug combination studies and
their synergy quantification using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang DD, Chen YB, Pan K, Wang W, Chen SP,
Chen JG, Zhao JJ, Lv L, Pan QZ, Li YQ, et al: Decreased expression
of the ARID1A gene is associated with poor prognosis in
primary gastric cancer. PLoS One. 7:e403642012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liang H, Cheung LW, Li J, Ju Z, Yu S,
Stemke-Hale K, Dogruluk T, Lu Y, Liu X, Gu C, et al: Whole-exome
sequencing combined with functional genomics reveals novel
candidate driver cancer genes in endometrial cancer. Genome Res.
22:2120–2129. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chandler RL, Damrauer JS, Raab JR,
Schisler JC, Wilkerson MD, Didion JP, Starmer J, Serber D, Yee D,
Xiong J, et al: Coexistent ARID1A-PIK3CA mutations promote ovarian
clear-cell tumorigenesis through pro-tumorigenic inflammatory
cytokine signalling. Nat Commun. 6:61182015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xie C, Fu L, Han Y, Li Q and Wang E:
Decreased ARID1A expression facilitates cell proliferation and
inhibits 5-fluorouracil-induced apoptosis in colorectal carcinoma.
Tumour Biol. 35:7921–7927. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Samartzis EP, Gutsche K, Dedes KJ, Fink D,
Stucki M and Imesch P: Loss of ARID1A expression sensitizes cancer
cells to PI3K- and AKT-inhibition. Oncotarget. 5:5295–5303. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nowsheen S, Cooper T, Stanley JA and Yang
ES: Synthetic lethal interactions between EGFR and PARP inhibition
in human triple negative breast cancer cells. PLoS One.
7:e466142012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cardnell RJ, Feng Y, Mukherjee S, Diao L,
Tong P, Stewart CA, Masrorpour F, Fan Y, Nilsson M, Shen Y, et al:
Activation of the PI3K/mTOR pathway following PARP inhibition in
small cell lung cancer. PLoS One. 11:e01525842016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jane EP, Premkumar DR, Morales A, Foster
KA and Pollack IF: Inhibition of phosphatidylinositol 3-kinase/AKT
signaling by NVP-BKM120 promotes ABT-737-induced toxicity in a
caspase-dependent manner through mitochondrial dysfunction and DNA
damage response in established and primary cultured glioblastoma
cells. J Pharmacol Exp Ther. 350:22–35. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yin Y, Shen Q, Zhang P, Tao R, Chang W, Li
R, Xie G, Liu W, Zhang L, Kapoor P, et al: Chk1 inhibition
potentiates the therapeutic efficacy of PARP inhibitor BMN673 in
gastric cancer. Am J Cancer Res. 7:473–483. 2017.PubMed/NCBI
|
|
45
|
Jiang ZB, Huang J, Xie C, Li X, Liu L, He
J, Pan H, Huang L, Fan XX, Yao XJ, et al: Combined use of PI3K and
MEK inhibitors synergistically inhibits lung cancer with
EGFR and KRAS mutations. Oncol Rep. 36:365–375. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sánchez-Tilló E, de Barrios O, Siles L,
Cuatrecasas M, Castells A and Postigo A: β-catenin/TCF4 complex
induces the epithelial-to-mesenchymal transition (EMT)-activator
ZEB1 to regulate tumor invasiveness. Proc Natl Acad Sci USA.
108:19204–19209. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Schmalhofer O, Brabletz S and Brabletz T:
E-cadherin, beta-catenin, and ZEB1 in malignant progression of
cancer. Cancer Metastasis Rev. 28:151–166. 2009. View Article : Google Scholar : PubMed/NCBI
|