Introduction
HCC is the fifth most common type of cancer and the
second leading cause of cancer-related deaths worldwide (1). Notably, 10–20% of HCC cases are caused
by HBV infection (2). Although
long-term nucleoside analogue therapy produces efficient
suppression of HBV viremia, the incidence of HBV caused by HCC
remains very high (3). In addition,
it has been reported that patients with spontaneous HBV DNA
clearance, but residual HBsAg titers >1,000 IU/ml still showed
increased risk for HCC (4).
To prevent the development of HCC, it is important
to understand the molecular mechanism underlying its
carcinogenesis. Accumulation of HBV surface proteins, particularly
large HBV surface proteins, has been shown to be involved in HCC
carcinogenesis via activation of the NF-κB pathway (5). Mehdi et al reported that
recombinant hepatitis B surface antigen (rHBsAg) binds to
β2-glycoprotein I (β2GPI) (6).
β2GPI, also known as apolipoprotein H, is a human plasmatic protein
primarily synthesized in the liver. They found that the expression
of β2GPI was upregulated in HepG2.2.15 cells, and β2GPI enhanced
the ability of HBsAg to bind to cell surfaces of the liver
(7).
In our previous studies, we found that β2GPI
collocated with HBsAg in the cytosol of HepG2.2.15 cells (7). In addition, when we transfected β2GPI
and HBsAg into HCC cells, NF-κB was actived and transferred from
the cytoplasm into the nucleus (8).
All these findings indicate that β2GPI may be involved in the
development of HBsAg-caused HCC via the NF-κB pathway. However, the
mechanism through which the interaction between HBsAg and β2GPI
confer activation of the NF-κB pathway has not yet been
elucidated.
In the present study, we first identified the
functional domain of HBsAg which co-interacts with β2GPI for the
activation of NF-κB. Secondly, using a knockdown experiment with
siRNA, we identified the downstream molecules that are involved in
the β2GPI/HBsAg-induced NF-κB activation. Mechanically, we revealed
that HBsAg/β2GPI activated the NF-κB pathway through
phosphorylation of IκBα.
Materials and methods
Tissue specimens
HBV-associated HCC tissues (10 patients) and
non-cancerous tissues (8 patients) were obtained from HBV-infected
patients who underwent partial hepatectomy for HCC between July
2014 and July 2015, at the Department of Pathology, at the
Affiliated Hospital of Qingdao University, China. Controls (4
patients) were histologically normal tissues obtained from
positive-HBsAg carrier patients who underwent partial hepatectomy
for liver hemangiomas or biliary duct diseases. Their ages ranged
from 25 to 68 years and there were 14 male and 8 female cases. The
diagnosis was established based on commonly accepted clinical and
laboratory parameters and typical histologic features. These
patients had no other obvious underlying disease, tested positive
for HBV markers, and had normal levels of serum transaminase. All
tissue samples were collected with the written informed consent of
all patients and under the approval of the Ethics Committee of the
Affiliated Hospital of Qingdao University.
Cell culture and experimental
groups
HCC cells, SMMC-7721 were maintained as a monolayer
in Invitrogen™ RPMI-1640 medium (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS;
Gibco; Thermo Fisher Scientific, Inc.), 2 mM L-glutamine, 100 U/ml
penicillin and 100 µg/ml streptomycin at 37°C under 5%
CO2 and were passaged once every 2 days. A sTable cell
line β2GPI/SMMC-7721 was generated. pcDNA-3.1(−)-rLHBsAg
(recombinant large HBV surface antigen, rLHBs) and
pcDNA-3.1(−)-rSHBsAg (recombinant small HBV surface antigen, rSHBs)
plasmid were constructed (9).
SMMC-7721 cells were divided into nine groups as
follows: i) Group one, where β2GPI/SMMC-7721 cells were incubated
with 5 µg/ml of serum-derived HBsAg (donated by Professor Gao, The
First Hospital of Jilin University) for 48 h; ii) Group two, where
SMMC-7721 cells were incubated by 5 µg/ml of serum-derived HBsAg
for 48 h; iii) Group three, β2GPI/SMMC-7721 cells; iv) Group four,
where SMMC-7721 cells were transiently transfected with
pcDNA-3.1(−) vector; v) Group five, SMMC-7721 cells; vi) Group six,
β2GPI/SMMC-7721 cells were transiently transfected with
pcDNA-3.1(−)-rLHBs plasmid; vii) Group seven where β2GPI/SMMC-7721
cells were transiently transfected with pcDNA-3.1(−)-rSHBs plasmid;
viii) Group eight, where SMMC-7721 cells were transiently
transfected with pcDNA-3.1(−)-rLHBs plasmid; and ix) Group nine,
where SMMC-7721 cells were transiently transfected with
pcDNA-3.1(−)-rSHBs plasmid.
The transfections were performed using
FuGENE® HD transfection reagent according to the
manufacturer's instructions (Roche, Basel, Switzerland). Following
48 h of treatment, all cells were washed twice with PBS and
incubated for an additional 4–12 h in serum-free culture media to
collect secreted β2GPI, HBsAg and AFP. The procedure was performed
as described in a previous study (8).
Hematoxylin and eosin (H&E)
staining
The specimens were fixed in 10% formaldehyde,
dehydrated, embedded in paraffin and sliced into 4-µm sections.
Following hematoxylin and eosin (H&E) staining, pathological
changes in the liver tissues were observed under a light microscope
(Olympus BX-53; Olympus Corp., Tokyo, Japan).
Immunofluorescence detection
Detection of the expression of β2GPI and HBsAg in
the tissues and the activated NF-κB and the phosphorylation site of
IκBα in the cells were performed by immunofluorescence (8). Monoclonal rabbit anti-NF-κB p65
antibody (1:50 dilution; cat. no. bs-0465R; Beijing Biosynthesis
Biotechnology Co., Ltd., Beijing, China), p-IκBα-Ser32 mouse
monoclonal antibody (1:50 dilution; cat. no. SC-7977-R; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA), p-IκBα-Ser32/36 mouse
monoclonal antibody (1:50 dilution; cat. no. SC-101713; Santa Cruz
Biotechnology, Inc.), FITC-stained β2GPI protein and RBITC-stained
HBsAg protein were used.
The blue (Hoechst-stained DNA), green (FITC-stained
β2GPI protein) and red (RBITC-stained HBsAg protein or
p-IκBα-Ser32/36 or p-IκBα-Ser32) fluorescence was detected. The
activation of NF-κB was performed by assessments of fluorescence
intensity (FI) (8).
Cytokine measurements
The levels of β2GPI, HBsAg, TNF-α, IL-1β and AFP in
the supernatants of the cells were measured using commercially
available ELISA kits (Nanjing KeyGen Biotech. Co., Ltd., Nanjing,
China) according to the manufacturer's instructions.
Cell extract preparation and
non-radioactive electrophoretic mobility shift assay (EMSA)
Nuclear and cytoplasmic extracts from the HCC cell
groups were prepared from 1×107 cells, for analysis by
EMSA using a non-radioactive EMSA kit (Viagene Biotech Inc., Tampa,
FL, USA). The assay was performed according to the manufacturer's
instructions (8). The bands were
visualized using Viagene Cool Imager (Viagene Biotech Inc.). The
band intensities were quantified using Cool Imager.
Effectiveness of the inhibitors
For the in vitro experiments on the
inhibition of IκBα, the cells which showed the highest level of
activation of NF-κB were incubated. IκB phosphorylation by IκB
kinase (IκK) prepares IκB for ubiquitination and subsequent
degradation. Pharmacological inhibition of IκK by Bay 11–7082 or of
tyrosine kinase by piceatannol were studied. Bay 11–7082 (CAS
19542–67-7-Calbiochem; Merck Millipore, Darmstadt, Germany) or
piceatannol (CAS 10083-24-6-Calbiochem; Merck Millipore) were added
and incubated. The nuclear extracts of the cells that were treated
by the inhibitors at 15, 30 and 60 min, respectively were isolated.
The activation of NF-κB was analyzed by non-radioactive EMSA.
Co-immunoprecipitation
All cells in the above-mentioned nine groups were
prepared and then incubated with a p-Tyr-2C8 mouse monoclonal
antibody (1:50 dilution; cat. no. sc-81529; Santa Cruz
Biotechnology, Inc.) or p-Ser-4A4 mouse monoclonal antibody (1:25
dilution; cat. no. DAM1460187; EMD Millipore, Billerica, MA, USA)
overnight. The immunocomplex was precipitated with protein
A/G-Separate beads (Protein G Plus-Agarose Immunoprecipitation
Reagent SC-2002; Santa Cruz Biotechnology, Inc.) and then submitted
to 7.5% SDS-PAGE. Western blot (WB) analysis was performed using an
anti-IκBα mouse monoclonal antibody (H-4, 1:300 dilution; cat. no.
SC-1643; Santa Cruz Biotechnology, Inc.).
RNA interference assay
The cells were transfected with various siRNA
directed against TLR4, TLR2 and MyD88 proteins (cat. nos.:
si_TLR4-1, TLR4-homo-595; si_TLR4-2, TLR4-homo-1325; si_TLR4-3,
TLR4-homo-595; si_TLR2-1, TLR2-homo-950; si_TLR2-2, TLR2-homo-1375;
si_TLR2-3, TLR2-homo-1648; si_MyD88-1, MyD88-homo-316; si_MyD88-2,
MyD88-homo-760; si_MyD88-3, MyD88-homo-987), and negative siRNA
with a random sequence was used as a control (all from Shanghai
GenePharma Co., Ltd., Shanghai, China). The siRNA sequences are
shown in Table I. The cells were
seeded at a density of 5×105 cells/well into 6-well
dishes and cultured overnight at 37°C with 5% CO2 until
the cells reached 70% confluency. The cells were co-incubated with
the HBsAg protein (5 µg/ml; Ab73749; Abcam, Cambridge, MA, USA) and
β2GPI protein (10 µg/ml; 362225; Merck Millipore) for 24 h.
Subsequently, the transfections were performed using Invitrogen™
Lipofectamine 2000 reagent (Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. Gene and protein
knockdown efficiencies were examined by qPCR and western
blotting.
 | Table I.Sequences for real-time PCR and
si-RNA. |
Table I.
Sequences for real-time PCR and
si-RNA.
| Real-time PCR | Sequences |
|---|
| TLR2 | F:
5′-CGTTCTCTCAGGTGACTGCTC-3′ |
| TLR4 | R:
5′-CCTTTGGATCCTGCTTGC-3′ |
|
| F:
5′-AGCCATGGCCTTCCTCTC-3′ |
|
| R:
5′-TTCAGCTCCATGCATTGATAA-3′ |
| MyD88 | F:
5′-CTGCTCGAGCTGCTTACCA-3′ |
|
| R:
5′-CTTCAAGATATACTTTTGGCAATCC-3′ |
| GAPDH | F:
5′-TGTTGCCATCAATGACCCCTT-3′ |
|
| R:
5′-CTCCACGACGTACTCAGCG-3′ |
| si-RNA |
|
| MyD88-homo-316 | F:
5′-GCCUGUCUCUGUUCUUGAATT-3′ |
|
| R:
5′-UUCAAGAACAGAGACAGGCTT-3′ |
| MyD88-homo-760 | F:
5′-GGCAACUGGAACAGACAAATT-3′ |
|
| R:
5′-UUUGUCUGUUCCAGUUGCCTT-3′ |
| MyD88-homo-987 | F:
5′-CCCAUCAGAAGCGACUGAUTT-3′ |
|
| R:
5′-AUCAGUCGCUUCUGAUGGGTT-3′ |
| TLR2-homo-1375 | F:
5′-GCCCUCUCUACAAACUUUATT-3′ |
|
| R:
5′-UAAAGUUUGUAGAGAGGGCTT-3′ |
| TLR2-homo-1648 | F:
5′-GCAACUCAAAGAACUUUAUTT-3′ |
|
| R:
5′-AUAAAGUUCUUUGAGUUGCTT-3′ |
| TLR2-homo-950 | F:
5′-GGUGAAACAAAUUCAUUGATT-3′ |
|
| R:
5′-UCAAUGAAUUUGUUUCACCTT-3′ |
| TLR4-homo-1546 | F:
5′-GGGCUUAGAACAACUAGAATT-3′ |
|
| R:
5′-UUCUAGUUGUUCUAAGCCCTT-3′ |
| TLR4-homo-1325 | F:
5′-CCCACAUUGAAACUCAAAUTT-3′ |
|
| R:
5′-AUUUGAGUUUCAAUGUGGGTT-3′ |
| TLR4-homo-595 | F:
5′-CCACCUCUCUACCUUAAUATT-3′ |
|
| R:
5′-UAUUAAGGUAGAGAGGUGGTT-3′ |
| Negative
control | F:
5′-UUCUUCGAACGUGUCACGUTT-3′ |
|
| R:
5′-ACGUGACACGUUCGGAGAATT-3′ |
RNA extraction and quantitative
real-time PCR
Total RNAs of tissues and cells in different groups
were extracted with TRIzol® reagent (Invitrogen)
according to the manufacturer's instructions. RNA integrity was
electrophoretically verified by ethidium bromide staining and by
OD260/OD280 nm absorption ratio >1.95. Total RNA (2 µg) was used
for reverse transcription to synthesize cDNA (Applied Biosystems
High Capacity RNA-to-cDNA Kit (Thermo Fisher Scientific, Inc.). The
primers for β2GPI, TLR4, TLR2 and MyD88 were designed and
synthesized by Sangon Biological Engineering Technology and
Services Co. Ltd. (Shanghai, China). The PCR primers are listed in
Table I. Reaction system (25 µl)
was established and quantitative real-time PCR assays were
performed via the Applied Biosystems SYBR®-Green PCR
Master Mix (Thermo Fisher Scientific, Inc.) on the Applied
Biosystems 7500 Real-Time PCR system (Thermo Fisher Scientific,
Inc.). The expression data were normalized to housekeeping gene
GAPDH using the 2−∆CT/[2 −(CT of target genes - CT
of GAPDH)] method. All PCR experiments were performed in
triplicate.
Western blot analysis
The protein concentration of each sample was assayed
using the BCA protein assay kit (Beyotime Institute of
Biotechnology, Haimen, China) according to the manufacturer's
protocol as follows: Equal amounts (40 µl) of protein from each
sample were resolved by 10% SDS-PAGE and electrotransferred onto
polyvinylidene difluoride (PVDF) membranes. The membranes were
blocked in TBST containing 5% non-fat dried milk for 1 h at room
temperature followed by incubation with primary antibodies at 4°C
overnight and incubated with secondary antibodies in TBST at room
temperature for 1 h. The primary antibodies used were IκBα-H-4
mouse monoclonal antibody (1:300 dilution; cat. no. SC-1643; Santa
Cruz Biotechnology, Inc.), anti-TLR4 mouse monoclonal antibody
(1:200 dilution; cat. no. SC-293072; Santa Cruz Biotechnology,
Inc.), anti-TLR2 rabbit polyclonal antibody (1:200 dilution; cat.
no. SC-10739; Santa Cruz Biotechnology, Inc.), anti-MyD88 mouse
monoclonal antibody (1:200 dilution; cat. no. SC-11356; Santa Cruz
Biotechnology, Inc.). The immunoreacted proteins were detected
using an enhanced chemiluminescence (ECL) western blotting
detection kit (Thermo Fisher Scientific, Inc.). Image Lab v3.0
software (BioRad Laboratories, Inc., Hercules, CA, USA) was used to
acquire and analyze imaging signals.
Statistical analysis
Data are presented as the mean ± standard error of
the mean (SEM) or median. Differences between multiple groups were
analyzed using one way ANOVA test and the Bonferroni's post hoc
test for multiple comparisons. A P-value of <0.05 was considered
to indicate a statistically significant difference.
Results
β2GPI is highly expressed in
HBV-related HCC and co-localizes with HBsAg
To determine the expression level of β2GPI in
HBV-related HCC, we estimated the expression level of β2GPI using
HBV-related HCC, adjacent non-cancerous tissues and HBsAg-positive
liver tissues. We observed that the protein and mRNA levels of
β2GPI were significantly higher in HBV-related HCC tissues compared
with the levels noted in the adjacent non-cancerous and
HBsAg-positive liver tissues (Fig. 1A
and C). We hypothesized that β2GPI may co-interact with HBsAg.
To test our hypothesis, we conducted an immunohistochemical
analysis with double staining to analyze the localization of β2GPI
using HBV-related HCC, adjacent non-cancerous liver and
HBV-infected liver tissues. We observed that β2GPI was localized in
the cytoplasm and co-localized with HBsAg (Fig. 1B).
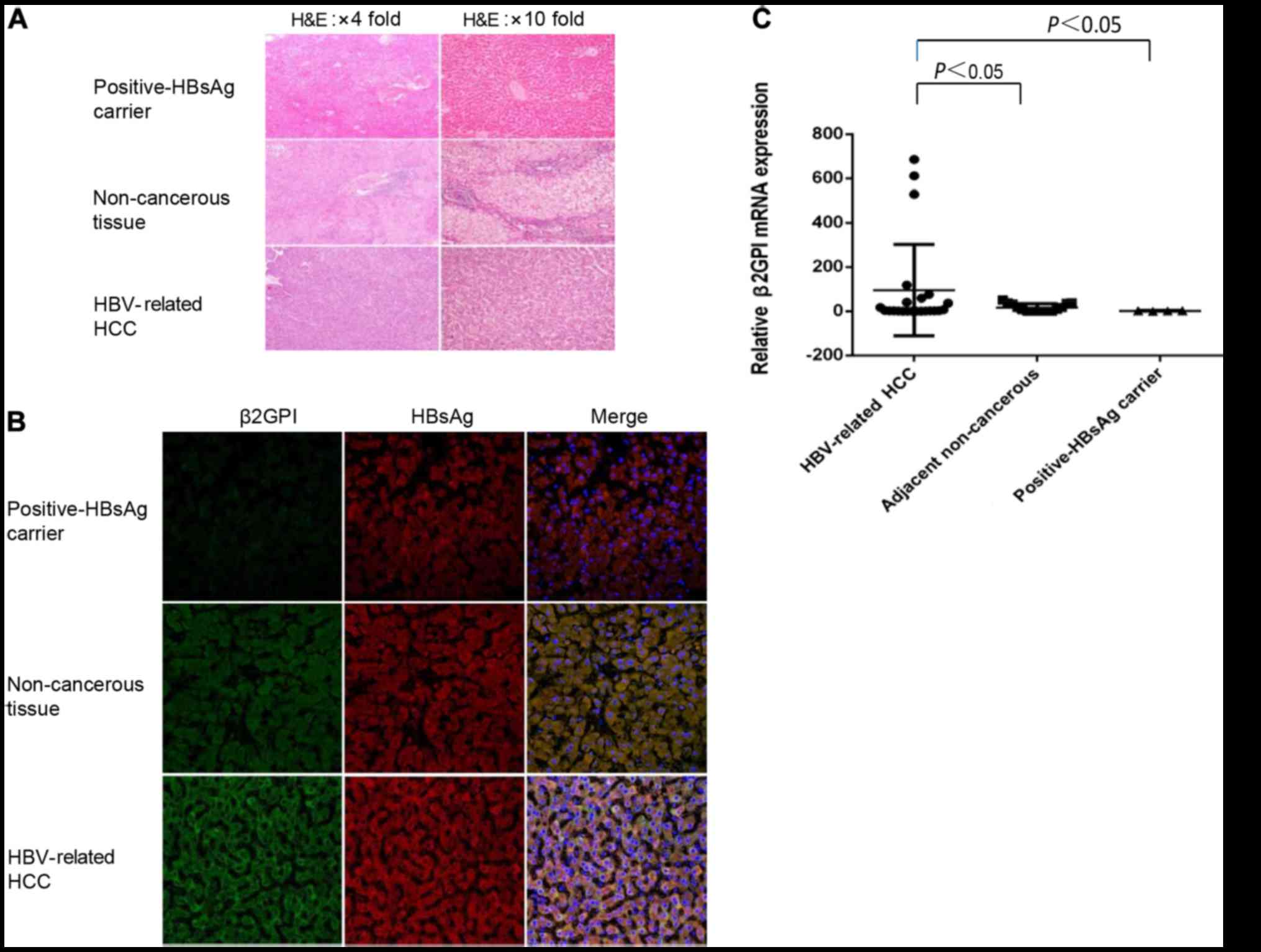 | Figure 1.β2GPI is highly expressed in
HBV-related HCC and co-localized with HBsAg. (A) Hematoxylin and
eosin (H&E) was used for staining of HBV-related HCC, adjacent
non-cancerous and HBsAg-positive liver tissues. Scale bars, 100 µm.
(B) Fluorescent immunohistochemical analysis was performed using
β2GPI pAb (FITG, green color) and HBsAg mAb (RO, red color) for
HBV-related HCC, adjacent non-cancerous and HBsAg positive liver
tissues. Double-stained tissues are shown (yellow color). Cell
nuclei were stained with Hoechst 33258 (blue color). The slides
were visualized using an Olympus FluoView FV1000 laser scanning
confocal microscope (LSCM). Original magnification: ×400, and scale
bars are 20 µm. The green fluorescence intensity was strongest in
the HBV-related tissue than in adjacent non-cancerous tissues.
β2GPI co-localized with HBsAg in the cytosol of HBV-related tissue
and adjacent non-cancerous (It showed white color when
double-stained was added to Hoechst stain). (C) Relative analysis
of β2GPI mRNA expression. The mRNA level of β2GPI of all tissues
was quantitated. P<0.05, compared to adjacent non-tumorous liver
and positive HBsAg carrier tissues. HBV, hepatitis B virus; HBsAg,
hepatitis B surface antigen; β2GPI, β2-glycoprotein I; HCC,
hepatocellular carcinoma. |
HBsAg interacts with β2GPI mostly via
the S domain to activate the NF-κB pathway
In our previous study, we found that HBsAg is able
to interact with β2GPI to activate the NF-κB signaling pathway. To
determine which domain of HBsAg co-interacts with β2GPI, we
designed three forms of HBsAg with a different composition:
Serum-derived HBsAg, rSHBs and rLHBs. Among them, we observed that
a combination of β2GPI and rSHBs significantly activated the NF-κB
signaling pathway more than the others (Fig. 2A). In addition, by
immunofluorescence assay, a combination treatment with β2GPI and
rSHBs induced a transfer of NF-κB from the cytoplasm to the nucleus
(Fig. 2B). To further validate our
findings, we analyzed the expression of AFP, known as a downstream
target gene of NF-κB. As displayed in Fig. 2C, the level of AFP was significantly
upregulated after a combination treatment with β2GPI and rSHBs.
These data indicate that HBsAg interacts with β2GPI through the S
domain to activate the NF-κB pathway.
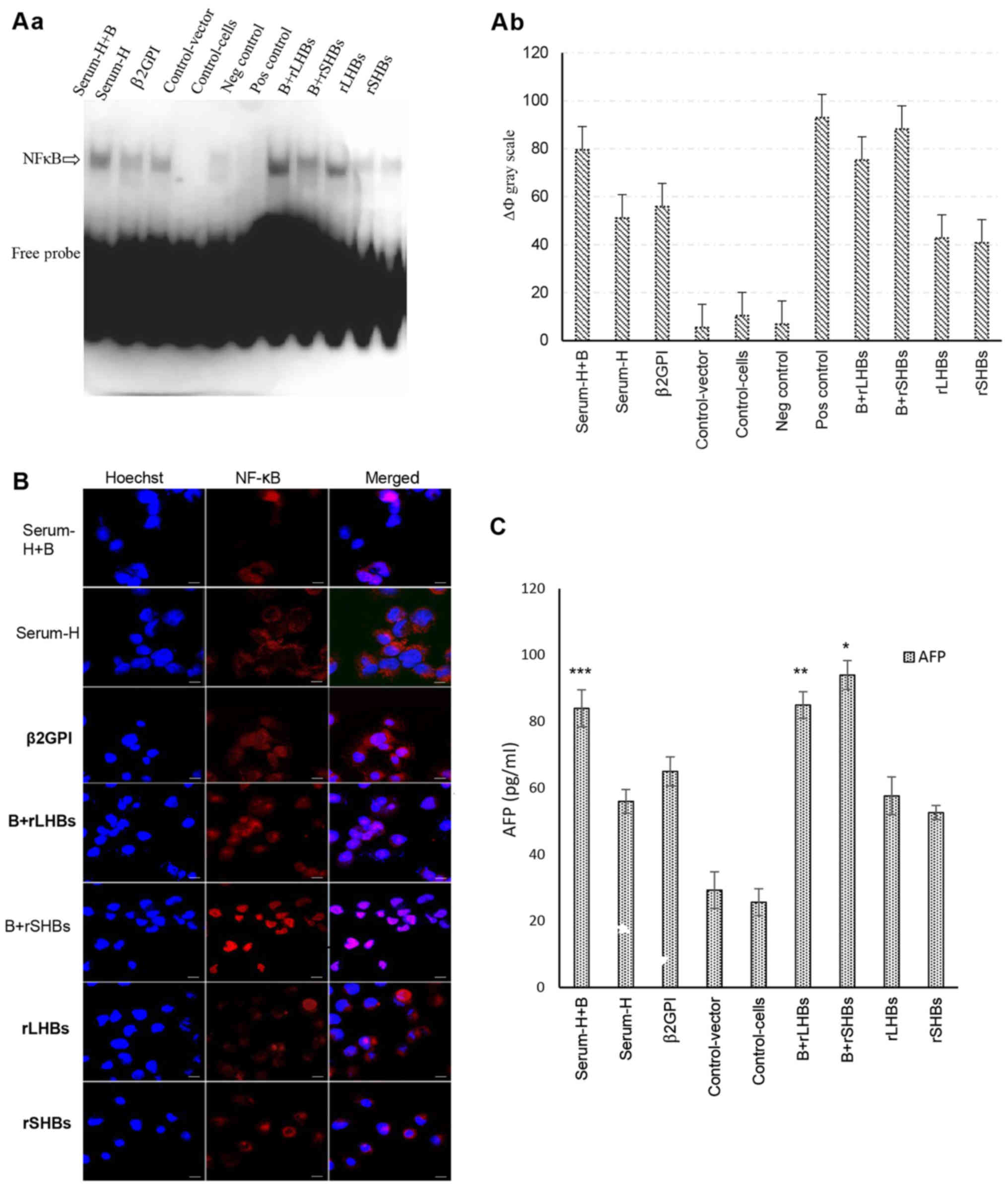 | Figure 2.HBsAg interacts with β2GPI mostly via
the S domain to activate the NF-κB pathway. (A-a) Activation of
NF-κB examined by non-radioactive EMSA. Cell nuclei from each of
the nine cell groups were assessed by non-radioactive EMSA. Lane 1:
Serum-H+B (group one), β2GPI/SMMC-7721 cells were incubated with 5
µg/ml of serum-derived HBsAg; lane 2: Serum-H (group two),
SMMC-7721 cells were incubated by 5 µg/ml of serum-derived HBsAg
for 48 h; lane 3: β2GPI (group 3), β2GPI/SMMC-7721 cells; lane 4:
Control-vector (group 4), SMMC-7721 cells were transiently
transfected with pcDNA-3.1(−) vector; lane 5: Control cells (group
5), SMMC-7721 cells; lane 6: Negative control,
cold-oligonucleotide; lane 7: Positive control, TNF-α; lane 8:
B+rLHBS (group 6), β2GPI/SMMC-7721 cells were transiently
transfected with pcDNA-3.1(−)-rLHBs plasmid; lane 9: B+rSHBS (group
7), β2GPI/SMMC-7721 cells were transiently transfected with
pcDNA-3.1(−)-rSHBs plasmid; lane 10: rLHBS (group 8), SMMC-7721
cells were transiently transfected with pcDNA-3.1(−)-rLHBs plasmid;
lane 11: rSHBs (group 9), SMMC-7721 cells were transiently
transfected with pcDNA-3.1(−)-rSHBs plasmid. (b) Gray scale of the
nine groups. ΔФ gray scale=total gray scale of each group/blank
group gray scale. The combination treatment groups
(β2GPI+serum-derived HBsAg, ΔФ gray scale=79.56, group one;
β2GPI+rLHBs, ΔФ gray scale=75.29, group six) exhibited stronger
activation of NF-κB compared with only β2GPI or rLHBsAg or rSHBsAg
or serum-derived-HBsAg treatment groups. The strongest activation
of NF-κB was observed in the β2GPI and rSHB treatment group, ΔФ
gray scale=88.13. The ΔФ gray scale of positive control was 92.9.
(B) Activation of NF-κB examined by immunofluorescence confocal
microscopy. The cells of the nine groups were plated onto glass
coverslips in a 24-well plate and were incubated for 36 h, then
fixed, permeabilized and stained with the indicated specific
antibodies against NF-κB (red color). Cell nuclei were stained with
Hoechst 33258 (blue color). It was suggested that NF-κB was
actived, when it was transferred from the cytoplasm into the
nucleus. Positive staining (pink color) in the nucleus was
determined in most of the cells, but the most significant positive
staining of NF-κB was shown in the β2GPI and rSHBs treatment group.
The slides were visualized using an Olympus FluoView FV1000 laser
scanning confocal microscope (LSCM). Original magnification ×600,
and scale bars are 20 µm. (C) The expression of downstream
cytokines of AFP was analyzed by ELISA. The expression of AFP was
significantly upregulated after a combination treatment with β2GPI
and rSHBsAg. Bars are the mean ± SEM (n=3). *P<0.05, compared
with other groups except Serum-H+B and rLHBs+B; **P<0.05,
compared with other groups except Serum-H+B and B+rSHBS;
***P<0.05, compared with other groups except B+rLHBS and
B+rSHBS. B+rSHBS: β2GPI+rSHBS; Serum-H+B: Serum-H+β2GPI; rLHBs+B:
rLHBs+β2GPI. HBsAg, hepatitis B surface antigen; β2GPI,
β2-glycoprotein I; NF-κB, nuclear factor κB. |
HBsAg/β2GPI contributes to the
activation of the NF-κB pathway via the TlR4/MyD88 axis
Generally, NF-κB activation was reported to be
mediated in the MyD88-dependent or -independent pathway. To test
whether the activation of NF-κB by HBsAg/β2GPI was mediated by
MyD88 dependence or not, we depleted TLR2, TLR4 or MyD88 with three
independent siRNAs using the cell line with co-incubated
HBsAg/β2GPI, and the siRNA with the highest knockdown efficiency
was selected (Fig. 3). Notably, we
found that both TLR4 and MyD88 depletion mutually downregulated the
expression of each other (Fig. 4A-a and
b). Both western blotting and real-time PCR revealed that when
TLR4 was knocked down, the expression of TLR2 and MyD88 was reduced
in addition to TLR4 itself. Based on the results of real-time PCR,
the level of TLR4 in the si_TLR4-1 group was significantly
different compared with the Negative, Positive and si_TLR2-3 group,
P<0.05; the level of TLR2 in the si_TLR4-1 group was significant
different compared with Positive and si_TLR2-3 group, P<0.05;
the level of MyD88 in the si_TLR4-1 group was significantly
different compared with Positive and si_MyD88-3 group, P<0.05).
In addition, when TLR2 was knocked down, the expression of TLR2,
TLR4 and MyD88 in the si_TLR2-3 group was significantly decreased
compared with the Positive groups (P<0.05). On the contrary,
these proteins were not significantly reduced, when compared with
the si_MyD88-3 group. Furthermore, when MyD88 was knocked out, the
expression of TLR2, TLR4 and MyD88 in the si_MyD88-3 group was
significantly reduced compared with the Positive and si_TLR2-3
groups, but, this was not statistically significant compared with
the si_TLR4-1 expected for the expression of MyD88, P<0.05.
Furthermore, TLR4 or MyD88 depletion reduced the activation of the
NF-κB pathway (Fig. 4B). In
addition, three downstream cytokines, IL-1β, TNF-α and AFP, were
obviously downregulated compared with the Positive and Negative
groups when TLR4, TLR2 or MyD88 was depleted. The level of AFP was
statistically different among the siRNA groups (P<0.05), which
was completely opposite of IL-1β. The expression of TNF-α among the
siRNA groups was statistically different except for the si_TLR4-1
group compared with si_MyD88-3 group (P<0.05) (Fig. 4C). These results indicated that
HBsAg/β2GPI primarily activated the NF-κB pathway via the
TLR4/MyD88 axis.
 | Figure 3.Selected siRNAs with the highest
knockdown efficiency. We constructed three independent siRNAs for
(A) TLR4, (B) TLR2 and (C) MyD88, named as follows: si_TLR4-1,
si_TLR4-2, si_TLR4-3; si_TLR2-1, si_TLR2-2, si_TLR2-3; si_MyD88-1,
si_MyD88-2, si_MyD88-3. The siRNA with the highest knockdown
efficiency was selected by WB (A-a, B-a and C-a) and real-time PCR
expression (A-b, B-b, C-b). si_TLR4-1, si_TLR2-3 and si_MyD88-3
were selected. *P<0.05, negative group compared with siRNA
treatment groups. TLR, Toll-like receptor; MyD88, myeloid
differentiation factor 88; WB, western blotting. |
 | Figure 4.HBsAg/β2GPI contributes to the
activation of the NF-κB pathway via the TlR4/MyD88 axis. (A-a)
Three independent siRNAs of TLR2, TLR4 or MyD88 using the cell line
with co-incubated HBsAg/β2GPI. The protein expression of TLR2, TLR4
or MyD88 was analyzed by western blotting with antibodies. (b) The
mRNA expression of TLR2, TLR4 or MyD88 in the cell line with
co-incubated HBsAg/β2GPI was determined by quantitative real-time
PCR. Both TLR4 and MyD88 depletion mutually downregulated the
expression of each other. *P<0.05, positive group compared with
other groups. (B-a) Activation of NF-κB as examined by
non-radioactive EMSA. TLR4 or MyD88 depletion reduced the
activation of the NF-κB pathway. (b) ΔФ gray scale of each
treatment group was as follows: The neg control=289.03, the
positive control=3,356.6, the siTLR4_1=1,081.8, the siTLR2_3=2,493
and siMyD88_3=1,403.8. (C) The expression of downstream cytokines
IL-1β, TNF-α and AFP was analyzed by ELISA. The result revealed
that expression of AFP, TNF-α and IL-1β in the si_TLR4-1, si_TLR2-3
and si_MyD88-3 groups was distinctly different compared with the
Positive and Negative groups (P<0.05). The expression of TNF-α
among the si_TLR4-1, si_TLR2-3 and si_MyD88-3 groups was
statistically different except the comparison between the si_TLR4-1
and si_MyD88-3 group (*P<0.05). The level of IL-1β was not
statistically different among the si_TLR4-1, si_TLR2-3 and
si_MyD88-3 groups. The level of AFP was statistically different
among any two groups, *P<0.05, positive group compared with
other groups. HBsAg, hepatitis B surface antigen; β2GPI,
β2-glycoprotein I; TLR, Toll-like receptor; MyD88, myeloid
differentiation factor 88; NF-κB, nuclear factor κB. |
HBsAg/β2GPI complex activates the
NF-κB pathway through the phosphorylation of IκBα
It has been reported that NF-κB activation is
induced by IκBα degradation. We also hypothesized that the
activation of NF-κB by HBsAg/β2GPI is induced by IκBα degradation.
To test this hypothesis and explore the exact mechanism, two
pharmacological substance inhibitors (Bay 11–7082 and piceatannol)
were used to treat the cells with co-incubated β2GPI and rSHBs
(Group 7). As displayed in Fig. 5A,
only Bay 11–7082 clearly abolished the activation of NF-κB in a
time-dependent manner, suggesting that IκBα degradation plays a key
role in the activation of NF-κB by HBsAg/β2GPI. To further
determine the mechanism of IκBα degradation, the phosphorylation of
IκBα in two well-known amino acid positions (p-Ser and p-Tyr) was
explored by co-immunoprecipitation using the purified cell extract
from the nine groups (Fig. 5B). We
observed that the serine phosphorylation of IκBα protein was
markedly increased in group six, seven and nine (β2GPI+rLHBs,
β2GPI+rSHBs and rSHBs treatment, respectively).
 | Figure 5.HBsAg/β2GPI complex activates the
NF-κB pathway through phosphorylation of IκBα. (A-a) The
pharmacological substance inhibitors (Bay 11–7082 and piceatannol)
were used to treat the cells with co-incubated β2GPI and rSHBs
(Group 7). (b) ΔФ Gray scale=total gray scale of each group/back
group gray scale. Activation of NF-κB examined by non-radioactive
EMSA. Bay 11–7082 clearly abolished the activation of NF-κB in a
time-dependent manner. (B-b) The p-Ser and p-Tyr were explored by
co-immunoprecipitation using the purified cell extract from the
nine groups). (a) The ΔФ Gray scale=total gray scale of each
group/back group gray scale. The serine phosphorylation of IκBα
protein was markedly increased in group six, seven and nine
(β2GPI+rLHBs, β2GPI+rSHBs, and rSHBs treatment). (C) The
phosphorylation of IκBα (Ser 32/36 and Ser 32) by western blotting
using the three groups with higher serine phosphorylation by WB
(β2GPI+rLHBs, Group six; β2GPI+rSHBs, Group seven; rSHBs, Group
nine). There was no staining in the p-IκBα-Ser32 group. We
confirmed that the phosphorylation of IκBα was observed in Ser
32/36 but not Ser 32. (D) Then we validated this finding by
immunofluorescence using the cells treated with β2GPI/rSHBs, which
were stained with the indicated specific antibodies (p-IκBα-Ser32
or p-IκBα-Ser32/36) (red color). Cell nuclei were stained with
Hoechst 33258 (blue color). All these data indicated that
HBsAg/β2GPI activated the NF-κB pathway through the phosphorylation
of IκBα in Ser 32/36. Original magnification, ×600, and scale bars
are 20 µm. B+rSHBS, β2GPI+rSHBS. HBsAg, hepatitis B surface
antigen; β2GPI, β2-glycoprotein I; NF-κB, nuclear factor κB; TLR,
Toll-like receptor; rHBsAg, recombinant hepatitis B surface
antigen; rLHBs, recombinant large HBV surface antigen; rSHBs,
recombinant small HBV surface antigen; EMSA, electrophoretic
mobility shift assay; IκK, IκB kinase. |
To further explore the site of serine
phosphorylation in IκBα, we analyzed the phosphorylation of IκBα in
two well-known amino acid site (Ser 32/36 and Ser 32) by western
blotting using the three groups with higher serine phosphorylation
(β2GPI+rLHBs, group six; β2GPI+rSHBs, group seven; rSHBs, group
nine). We confirmed that the phosphorylation of IκBα was observed
in Ser 32/36 but not Ser 32, and it was more obvious in group seven
(Fig. 5C). Subsequently we
validated this finding by immunofluorescence using the cells
treated with β2GPI/rSHBs (Fig. 5D).
All these data indicated that HBsAg/β2GPI activated the NF-kB
pathway through the phosphorylation of IκBα in Ser 32/36.
Discussion
In the present study, we demonstrated that β2GPI was
highly expressed in HBV-related HCC tissue compared with
non-cancerous tissue, and HBsAg-positive carrier normal liver
tissues. In our previous studies, we found that HBsAg interacting
with β2GPI conferred activation of NF-κB, which indicated the
probable action of β2GPI in HBV-induced HCC.
It was reported that HBV envelope proteins consist
of three isoforms: Small surface proteins (SHBs), middle surface
proteins (MHBs) and large surface proteins (LHBs) (10). SHBs consist of only the S domain,
MHBs contain the pre-S2 domain in addition to S, and LHBs contain
the pre-S1 domain in addition to pre-S2 and S (11). However, among these three isoforms,
it has been reported that β2GPI can only bind to the small S
proteins of rHBsAg (6). The preS1
can recognize the asialoglycoprotein receptor which is on the
surface of human hepatocytes or HCC cells (11), and preS1 not preS2 is essential for
virus infection (12). Therefore,
in the present study, we designed three different forms of HBsAg:
Serum-HBsAg, rSHBs and rLHBs. We found that rSHBs in combination
with β2GPI showed the highest activity for activation of NF-κB,
compared with serum-HBsAg, rLHBs, rSHBs, β2GPI alone or other
combination treatment, indicating that the S domain of HBsAg
sufficiently interacts with the β2GPI-activated NF-κB pathway. To
the best of our knowledge, this is probably due to β2GPI specially
binding to small S protein (6).
Approximately 80% of HCC patients experience chronic
liver inflammation, fibrosis or cirrhosis during the development
(1). It was reported that liver
exposure to various TLR ligands via the portal vein, leads to an
uncontrolled activation of innate immunity that may result in
inflammatory liver diseases (13).
TLRs, a family of transmembrane signaling receptors, contain
Toll/IL-1 receptor (TIR) motif to bridge receptors or corresponding
intracellular molecules for signal transportation (14). It has been reported that TLR2 or
TLR4 is upregulated in HCC (15),
and knockdown of either of them significantly decreases the
proliferation of HCC cells (16,17).
MyD88 was found to be an adaptor molecule to TIR, and it can
recruit IL-1R-associated kinase (IRAK) and TNF receptor-associated
factor-6 (TRAF6), leading to the activation of NF-κB (18). In addition, MyD88 was found to be
frequently upregulated in HCC, and overexpression of MyD88 was
found to promote HCC cell proliferation and invasion in
vitro (19). Therefore, it
appears that TLRs and MyD88 signaling are associated with
inflammation-associated liver cancer development (13).
In the present study, we found that knockdown of
TLR4 significantly reduced the activation of NF-κB, but not TLR2.
We also provided evidence that activation of NF-κB was prominently
inhibited by the small interfering (si)RNA-directed targeting of
MyD88. This finding indicated that the TLR4-associated
MyD88-dependent signaling pathway contributed to the activation of
NF-κB. Notably, we observed that the level of TLR2 protein was
decreased by knockdown of TLR4, and the expression of TLR2 and TLR4
proteins were downregulated by si-MyD88. This finding indicated
that there was a possible interaction between them. In agreement
with our findings, Sweeney et al (20) demonstrated that there exists a
TLR2/TLR4/TRAM native hepatic protein complex, which may have
important implications for the host response to sepsis. Yamamoto
et al (21) revealed that
TLR2/TLR4 shared the MyD88/TIRAP common signaling pathway.
The NF-κB signaling pathway plays a key role in HCC
carcinogenesis (22). NF-κB
transcription factors consist of five proteins, p65 (Rel A), RelB,
c-Rel, p105/p50 (NF-κB1) and p100/52 (NF-κB), which can form a
large number of homodimers and heterodimers (23). In unstimulated cells, NF-κB dimers
are sequestered in the cytoplasm by inhibitors of IκBs. IκBs are
degraded by upstream kinase complex IκB kinase (IKKs) upon
activation, thereby allowing nuclear translocation of NF-κB and the
subsequent induction of NF-κB response genes and release of
NF-κB-dependent interferons and cytokines, including tumor necrosis
factors (TNF-α) and IL-1β (5,23,24).
In the present study, we observed that the activation of NF-κB by
combination treatment of rSHBsAg and β2GPI was also evident on the
NF-κB canonical pathway, which depended on the inducible
degradation of inhibitor IκB. IκBs are degraded by upstream kinase
complex IκB kinases (IKKs), with the site 32/36 of serine of
phosphorylation.
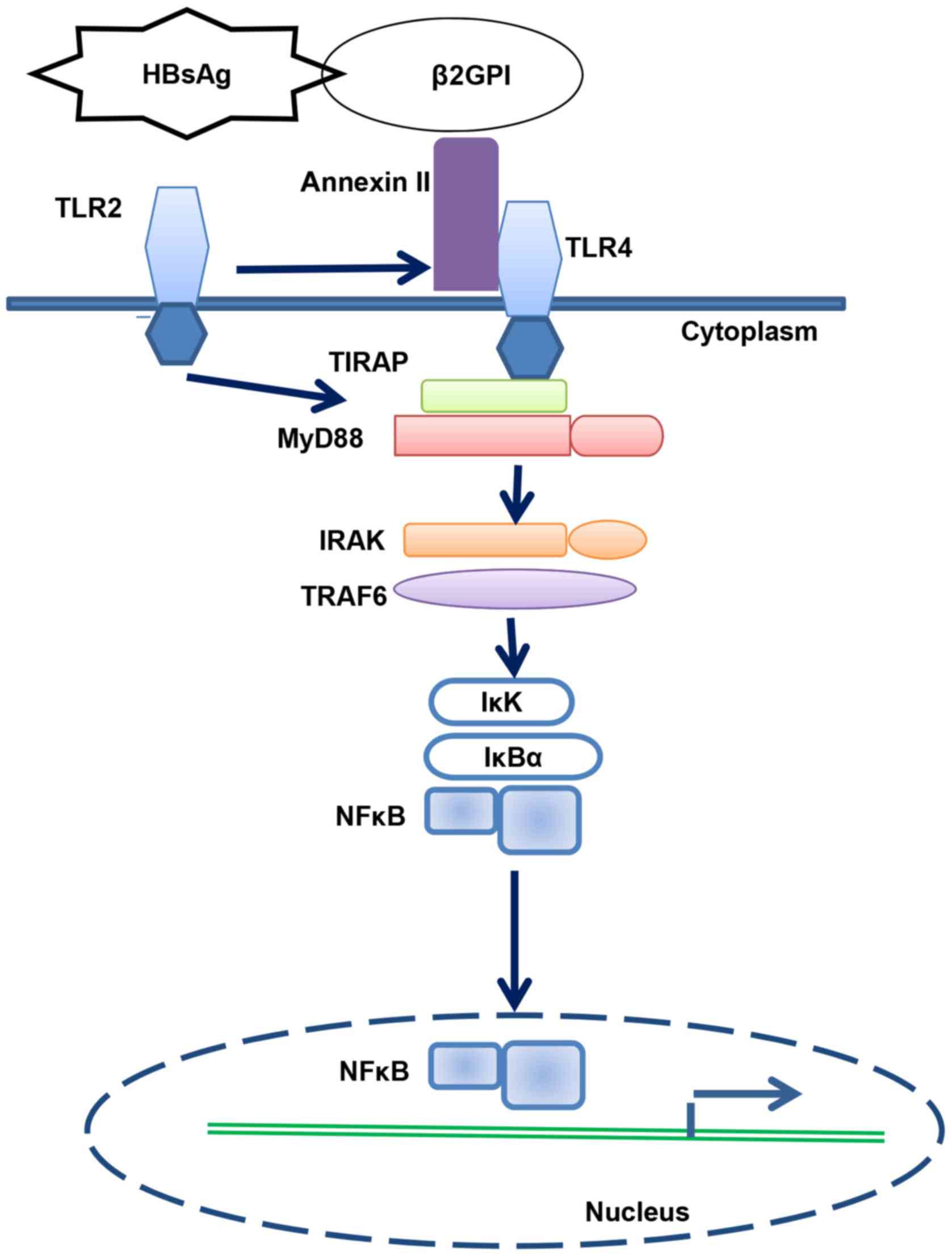
In conclusion, we revealed that HBsAg/β2GPI
activated the NF-κB pathway through the TLR4/MyD88/IκBα axis
(Fig. 6).
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Youth Foundation of China (no. 81101853) and the
Natural Science Youth Foundation of Shandong Province (no.
ZR2016HQ35).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
XJ acquired the data, obtained funding, conceived,
designed and drafted the manuscript; ZT and PG studied the
supervision; HX, XQ, YY, XD and LY collected the tissue samples and
analysed the data and LZ designed the study and drafted the
manuscript. All authors read and approved the manuscript and agree
to be accountable for all aspects of the research in ensuring that
the accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
All tissue samples were collected with the written
informed consent of all patients and under the approval of the
Ethics Committee of the Affiliated Hospital of Qingdao
University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
HBsAg
|
hepatitis B surface antigen
|
|
β2GPI
|
β2-glycoprotein I
|
|
HCC
|
hepatocellular carcinoma
|
|
NF-κB
|
nuclear factor κB
|
|
TLR
|
Toll-like receptor
|
|
MyD88
|
myeloid differentiation factor 88
|
|
rHBsAg
|
recombinant hepatitis B surface
antigen
|
|
rLHBs
|
recombinant large HBV surface
antigen
|
|
rSHBs
|
recombinant small HBV surface
antigen
|
|
EMSA
|
electrophoretic mobility shift
assay
|
|
IκK
|
IκB kinase
|
References
|
1
|
Sakamaki A, Kamimura K, Abe S, Tsuchiya A,
Takamura M, Kawai H, Yamagiwa S and Terai S: Spontaneous regression
of hepatocellular carcinoma: A mini-review. World J Gastroenterol.
23:3797–3804. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
McMahon BJ: The natural history of chronic
hepatitis B virus infection. Hepatology. 49 (5 Suppl):S45–S55.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tseng TC and Kao JH: Elimination of
hepatitis B: Is it a mission possible? BMC Med. 15:532017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu J, Yang HI, Lee MH, Lu SN, Jen CL,
Batrla-Utermann R, Wang LY, You SL, Hsiao CK, Chen PJ, et al:
Spontaneous seroclearance of hepatitis B seromarkers and subsequent
risk of hepatocellular carcinoma. Gut. 63:1648–1657. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Karin M: NF-kappaB as a critical link
between inflammation and cancer. Cold Spring Harbor Perspect Biol.
1:a0001412009. View Article : Google Scholar
|
|
6
|
Mehdi H, Kaplan MJ, Anlar FY, Yang X,
Bayer R, Sutherland K and Peeples ME: Hepatitis B virus surface
antigen binds to apolipoprotein H. J Virol. 68:2415–2424.
1994.PubMed/NCBI
|
|
7
|
Liu YM, Zhang WY, Wang ZF, Yan CY and Gao
PJ: High expression of beta2-glycoprotein I is associated
significantly with the earliest stages of hepatitis B virus
infection. J Med Virol. 86:1296–1306. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jing X, Piao YF, Liu Y and Gao PJ:
Beta2-GPI: A novel factor in the development of hepatocellular
carcinoma. J Cancer Res Clin Oncol. 136:1671–1680. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jing X: Positive effect of Beta2-GPI and
HBsAg in the pathogenesis of hepatocellular carcinoma. JiLin: JiLin
University; pp. 47–82. 2010
|
|
10
|
Hu J and Liu K: Complete and incomplete
hepatitis B virus particles: Formation, function, and application.
Viruses. 9:pii: E56. 2017. View
Article : Google Scholar
|
|
11
|
Li YW, Yang FC, Lu HQ and Zhang JS:
Hepatocellular carcinoma and hepatitis B surface protein. World J
Gastroenterol. 22:1943–1952. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Meier A, Mehrle S, Weiss TS, Mier W and
Urban S: Myristoylated PreS1-domain of the hepatitis B virus
L-protein mediates specific binding to differentiated hepatocytes.
Hepatology. 58:31–42. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Roh YS and Seki E: Toll-like receptors in
alcoholic liver disease, non-alcoholic steatohepatitis and
carcinogenesis. J Gastroenterol Hepatol. 28 (Suppl 1):S38–S42.
2013. View Article : Google Scholar
|
|
14
|
Li K, Qu S, Chen X, Wu Q and Shi M:
Promising targets for cancer immunotherapy: TLRs, RLRs, and
STING-mediated innate immune pathways. Int J Mol Sci. 18:pii: E404.
2017.
|
|
15
|
Zhe Y, Li Y, Liu D, Su DM, Liu JG and Li
HY: Extracellular HSP70-peptide complexes promote the proliferation
of hepatocellular carcinoma cells via TLR2/4/JNK1/2MAPK pathway.
Tumour Biol. 37:13951–13959. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shi W, Su L, Li Q, Sun L, Lv J, Li J and
Cheng B: Suppression of toll-like receptor 2 expression inhibits
the bioactivity of human hepatocellular carcinoma. Tumour Biol.
35:9627–9637. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang Y, Cai J, Zeng X, Chen Y, Yan W,
Ouyang Y, Xiao D, Zeng Z, Huang L and Liu A: Downregulation of
toll-like receptor 4 induces suppressive effects on hepatitis B
virus-related hepatocellular carcinoma via ERK1/2 signaling. BMC
Cancer. 15:8212015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liang B, Chen R, Wang T, Cao L, Liu Y, Yin
F, Zhu M, Fan X, Liang Y, Zhang L, et al: Myeloid differentiation
factor 88 promotes growth and metastasis of human hepatocellular
carcinoma. Clin Cancer Res. 19:2905–2916. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jia RJ, Cao L, Zhang L, Jing W, Chen R,
Zhu MH, Guo SW, Wu GB, Fan XY, Wang H, et al: Enhanced myeloid
differentiation factor 88 promotes tumor metastasis via induction
of epithelial-mesenchymal transition in human hepatocellular
carcinoma. Cell Death Dis. 5:e11032014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sweeney TE, Suliman HB, Hollingsworth JW,
Welty-Wolf KE and Piantadosi CA: A toll-like receptor 2 pathway
regulates the Ppargc1a/b metabolic co-activators in mice with
Staphylococcal aureus sepsis. PLoS One. 6:e252492011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yamamoto M, Sato S, Hemmi H, Sanjo H,
Uematsu S, Kaisho T, Hoshino K, Takeuchi O, Kobayashi M, Fujita T,
et al: Essential role for TIRAP in activation of the signalling
cascade shared by TLR2 and TLR4. Nature. 420:324–329. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Capece D, Fischietti M, Verzella D,
Gaggiano A, Cicciarelli G, Tessitore A, Zazzeroni F and Alesse E:
The inflammatory microenvironment in hepatocellular carcinoma: A
pivotal role for tumor-associated macrophages. Biomed Res Int.
2013:1872042013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Joe Y and Valerie C: The role of nuclear
factor-kappa B and endoplasmic reticulum stress in hepatitis B
viral-induced hepatocellular carcinoma. Translational Cancer Res. 5
(Suppl 1):S13–S17. 2016. View Article : Google Scholar
|
|
24
|
Sun B and Karin M: NF-kappaB signaling,
liver disease and hepatoprotective agents. Oncogene. 27:6228–6244.
2008. View Article : Google Scholar : PubMed/NCBI
|