Introduction
Lung cancer is the leading cause of mortality
worldwide, being responsible for 1.59 million patient deaths every
year. Although early diagnosis increases the potential for curative
surgical resection, more than half of cases present with advanced
disease and require treatment with chemotherapy and radiotherapy
(1). A recent breakthrough, the
discovery of driver mutations and the development of corresponding
tyrosine kinase inhibitors (TKIs), has shown significant clinical
benefits to date (2,3). However, targeting KRAS, the
second major driver, is an emerging problem as its activation is
different from the usual kinase-based signaling. In addition,
overcoming an acquired resistance, including a mutation at exon 20
in ERBB2, (4,5) and activation of the HGF and MET
signaling pathway, (6,7) has yet been a problem to resolve
although the specific inhibitor targeting T790M in the EGFR
gene (8,9) has been developed.
The concept of synthetic lethality leading to cell
death in the presence of a combination of mutations in multiple
genes was first advanced in 1945 (10). This was later highlighted in 2005 by
reports indicating that breast cancer with the breast cancer
susceptibility gene mutation (BRCA1, DNA repair associated) was
very sensitive to inhibitors of poly(ADP-ribose) polymerase (PARP)
(11,12). Thus, it seemed that simultaneous
suppression of two major DNA repair genes could disrupt DNA
replication in cancer cells. Since this discovery, other
therapeutic approaches based on synthetic lethality have been
sought in various malignancies known to have specific gene
alterations (13). Notably, the
KRAS mutation has been a major target for synthetic
lethality, and the cosuppression of MEK with RAF1 or BCL-XL
was identified as being effective at inducing synthetic lethality
in cancer cells with this mutation (14,15).
To establish effective clinical treatments for lung cancer with
KRAS mutations, it is important to identify additional
combinations that cause synthetic lethality.
Rho GTPase-activating protein 35 (ARHGAP35) is a
RhoGAP protein (16,17) reported to be the principal substrate
of SRC and controller of RhoA activity (through the binding of
RhoA) (18,19) and also known with altered names such
as p190RhoGAP, GRF-1 and GRLF1. To date, however, the role of this
molecule in cancer has been uncertain, with both tumor suppressor
effects and oncogenic effects reported (20,21).
In proteome studies, the phosphorylation status of tyrosine Y1105
in ARHGAP35 has consistently been reported to change, being
dramatically suppressed by EGFR-TKI treatment for lung
adenocarcinoma in the presence of the EGFR mutation (22,23).
Therefore, we aimed to focus on ARHGAP35 as a possible key molecule
in the proliferation and metastasis of lung adenocarcinoma. To this
end, our group previously showed that ARHGAP35 messenger RNA (mRNA)
was overexpressed in lung cancer cell lines compared to normal
cells, and that positive protein expression was widely observed in
lung cancer cells in surgically resected specimens
immunohistochemically (24). It was
also observed that ARHGAP35 knockdown significantly
suppressed the viability, migration and invasion of lung
adenocarcinoma cells, including KRAS mutants (24).
In the present study, we investigated the effect of
long-term ARHGAP35 suppression in lung cancer cells. Our aim was to
identify a compensatory pathway that could be both a potential
mechanism of acquired resistance for SRC/ARHGAP35 inhibition and a
candidate for synthetic lethality when used in combination with
inhibition of the SRC/ARHGAP35 axis.
Materials and methods
Cell lines and culture
A549 and H1975 cell lines were obtained from the
American Type Culture Collection (ATCC, Manassas, VI, USA). PC9 was
obtained from DS Pharma Biomedical Co., Ltd. (Osaka, Japan). All
cells were maintained in RPMI-1640 medium (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) with 10% fetal bovine serum
(FBS) in a humidified 5% CO2 incubator at 37°C. These
cells are histologically adenocarcinomas, and A549 has the
wild-type (WT) EGFR and KRAS mutations (codon 12),
PC-9 has the EGFR mutation (exon 19 del), and H1975 has the
EGFR mutation (L858R and T790M). A549 and H1975 cells have
been reported to be EGFR-TKI resistant, whereas PC9 is EGFR-TKI
sensitive. To test whether there was concomitant inhibition of
STAT3 and SRC, cells were treated with combinations of different
concentrations of a STAT3 inhibitor (S3I-201; Santa Cruz
Biotechnology, Dallas, TX, USA) and SRC inhibitor (SKI-1; Abcam,
Cambridge, UK). Drug concentrations of S3I-201 were adjusted to 0,
100, 200 and 400 µM and those for SKI-1 were adjusted to 0, 2.5, 5
and 10 µM.
RNA interference (RNAi)
Lentiviral transfection with small hairpin RNA
(shRNA) was conducted using MISSION® shRNA
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). Cells were
transfected with shRNA directed to ARHGAP35 or negative control
shRNA using hexadimethrine bromide. Transfected cells were cultured
with media containing puromycin and selected clones were maintained
with puromycin over 1 month.
Western blot analysis
Cell lines were washed with ice-cold PBS and lysed
in ice-cold lysis buffer (pH 8.0 50 mM HEPES, 150 mM NaCl, 100 mM
NaF, 10 mM Na4P2O7H2O,
1.5 mM MgCl2, 1 mM EDTA, 10% glycerol and 1% Triton
X-100) which was added Complete Protease Inhibitor Cocktail (Roche,
Basel, Switzerland) and PhosSTOP phosphatase Inhibitor Cocktail
(Roche). The cell lysates were centrifuged at 15,000 × g for 20 min
at 4°C to collect the supernatant. We calculated protein
concentrations by Bradford method using Bio-Rad Protein assay (GE
Healthcare Life Sciences, Pittsburgh, PA, USA). Protein samples (10
µg) were separated by SDS-PAGE Mini-PROTEAN TGX Gel (GE Healthcare
Life Sciences) and transferred onto a polyvinylidene difluoride
membrane (PVDF; Hybond-P®; GE Healthcare Life Sciences).
The PVDF membranes with proteins were blocked with 5% non-fat dry
milk (NFDM) for 1 h at room temperature. The primary antibodies
dissolved in 5% bovine serum albumin (BSA) were used to detect the
target protein blots at 4°C overnight for incubation. Protein were
incubated on the membranes with primary antibodies dissolved in 5%
BSA (or 5% NFDM) at 4°C overnight and secondary antibodies labeled
with horseradish peroxidase dissolved in blocking buffer for 1 h at
room temperature. The proteins were visualized on ImageQuant LAS
4000 Mini (GE Healthcare Life Sciences), using the enhanced
chemiluminescence western blot detection system. The primary
antibodies used in the present study included: ARHGAP35 (1:1,000;
cat. no. 2860), SRC (1:1,000; cat. no. 2109), AKT (1:1,000; cat.
no. 4691), STAT3 (1:2,000; cat. no. 4904), MEK (1:1,000; cat. no.
9126), PRKCD (1:1,000, 5% NFDM; cat. no. 9616), PRKCZ (1:1,000;
cat. no. 9368), p-SRC (1:1,000; cat. no. 2101), p-AKT (1:2,000;
cat. no. 4060), p-STAT3 (1:2,000; cat. no. 9145), p-MEK (1:1,000;
cat. no. 9121) and β-actin (1:1,000; cat. no. 4967) all from Cell
Signaling Technology (Boston, MA, USA).
Quantitative assay for Rho kinase
activity
Measurement of Rho kinase (ROCK) activity was
performed using a ROCK Activity Immunoblot kit (Cell Biolabs, Inc.,
San Diego, CA, USA) according to the manufacturer's
instructions.
RT-qPCR
Total RNA isolation from cell lines and
complementary DNA synthesis was performed using TaqMan®
Gene Expression Cells-to-Ct™ Kits (Thermo Fisher Scientific, Inc.).
All mRNA was measured by qRT-PCR using an ABI PRISM 7000 Sequence
Detection System (Thermo Fisher Scientific) with primers (Thermo
Fisher Scientific; ARHGAP35: Hs00534180_m1, SRC: Hs01082246_m1,
STAT3: Hs00374280_m1, 18S rRNA: Hs99999901_s1). SRC, STAT3 and 18S
ribosomal RNA primer (Thermo Fisher Scientific). PCR reaction
conditions were performed as follows: 95°C for 10 min, and 40
cycles of 95°C for 15 sec and 60°C for 60 sec.
Cell viability assay
Measurement of cell viability was performed using a
CellTiter 96 AQueous One Solution Cell Proliferation Assay
(Promega, Madison, WI, USA). Cells were seeded onto a 96-well plate
at a concentration of 3,000 cells/well and were incubated at 37°C.
At 72 h, the optical density was measured at 490 nm using a
microtiter plate reader, and the rate of cell survival was
expressed as the absorbance. To assess the effect of an inhibitor,
cell viability was evaluated 48 h after treatment.
Cell migration assay
Cell migration was evaluated by scratch assay, as
described in a previous report (25). Cells were seeded onto a 24-well
plate at a concentration of 150,000 cells/well and were incubated
at 37°C. At 24 h, the cell monolayer was scratched in a straight
line to create a scratch with Cell Scratcher (AGC, Tokyo, Japan).
Twenty-four and 48 h later, we measured the width of the scratch
using an optical microscope without stain, and calculated the rate
of cell migration.
Statistical analysis
All experiments were performed in triplicate and
analyzed using JMP Pro 11 (SAS Institute, Inc., Cary, NC, USA).
Unpaired Student's t-tests were used for comparisons between two
groups. P<0.05 were considered to indicate a statistically
significant result. The Chou-Talalay method was used to evaluate
synergistic effect of drug combination as described in a previous
report (26). Combination index
(CI) was calculated based on the effect ratio of cell viability
under various drug concentrations using CompuSyn software
(ComboSyn, Inc., Paramus, NJ, USA). CI <0.9, 0.9–1.1 and >1.1
were regarded as synergism, additive effect and antagonism,
respectively (27).
Results
Suppressive effects of cell viability
are attenuated by long-term ARHGAP35 knockdown in lung
adenocarcinoma cell lines
We established lung adenocarcinoma cell clones by
continuous exposure to puromycin for a month after introducing
shRNA against ARHGAP35 into cells. qRT-PCR (Fig. 1A) and western blot (Fig. 1B) analyses showed that ARHGAP35 was
significantly suppressed in these cells. In the MTS assay, cell
viability with long-term ARHGAP35 knockdown was comparable
to the cell viability without knockdown in all cell lines (Fig. 1C). This suggested that the
suppressive effects of cell viability were attenuated by long-term
knockdown.
Molecular dynamics in EGFR signaling
pathway changes after long-term ARHGAP35 knockdown
We also evaluated the molecular dynamics of the
RAS/RAF1/MAPK, STAT, PI3K, MET and ERBB2 pathways (Figs. 2 and 3). Western blots showed increases in SRC
and STAT3 total protein, and increased levels of phosphorylated
STAT3 in A549 cells after long-term ARHGAP35 knockdown
(Fig. 2). PRKCZ, MET and ERBB2
levels were also increased with ARHGAP35 knockdown in the
A549 cells (Fig. 2 and data not
shown). By contrast, PRKCD, PRKCZ, MET and ERBB2 levels were
decreased after long-term ARHGAP35 knockdown in the H1975
cells (Fig. 2 and data not shown).
SRC, MEK, STAT3, AKT, PRKCD, PRKCZ and MET levels were decreased
with long-term ARHGAP35 knockdown in the PC9 cells (Fig. 2 and data not shown). qRT-PCR showed
increased SRC mRNA in A549 cells and decreased SRC (Fig. 3A) and STAT3 (Fig. 3B) mRNA in the H1975 cells. ROCK
activity, which is associated with cell migration, was within
normal limits in all cell lines with ARHGAP35 knockdown
(Fig. 4).
 | Figure 2.Dynamics of proteins in signaling
pathways associated with ARHGAP35. We evaluated the protein
dynamics of signaling pathways associated with ARHGAP35 by western
blot analysis when ARHGAP35 gene knockdown was continued in
the long term. Increases in SRC and STAT3, and consequently
phosphorylated STAT3, are shown following long-term ARHGAP35
knockdown in A549 cell line. PRKCZ, MET (data not shown) and ERBB2
(data not shown) were also increased in A549 cells with
ARHGAP35 knockdown. PRKCA (data not shown), PRKCZ, MET (data
not shown) and ERBB2 (data not shown) were decreased in H1975 cells
after long-term ARHGAP35 knockdown. SRC, MEK, STAT3, AKT,
PRKCD, PRKCZ and MET (data not shown) were decreased in PC9 cells
after long-term ARHGAP35 knockdown. |
STAT3 inhibitor is more effective
after long-term ARHGAP35 knockdown in A549 and PC9 cells
Using MTS assay, we measured cell viability after
long-term ARHGAP35 knockdown in cells treated with a STAT3
inhibitor (Fig. 5). A549 and PC9
cells were more sensitive to the STAT3 inhibitor after long-term
ARHGAP35 knockdown than when there was no knockdown, but the
opposite was noted in the H1975 cells.
Suppressive effect of ARHGAP35
knockdown on migration is maintained for a long time
Using scratch assays, we measured cell migration
after long-term ARHGAP35 knockdown in the cell lines
(Fig. 6A and B). This demonstrated
that cell migratory ability was reduced in cancers with
ARHGAP35 knockdown when compared with cancers without
knockdown. This was the case for all cell lines, and suggested that
the suppressive effect of ARHGAP35 knockdown on migration
was maintained for a long time.
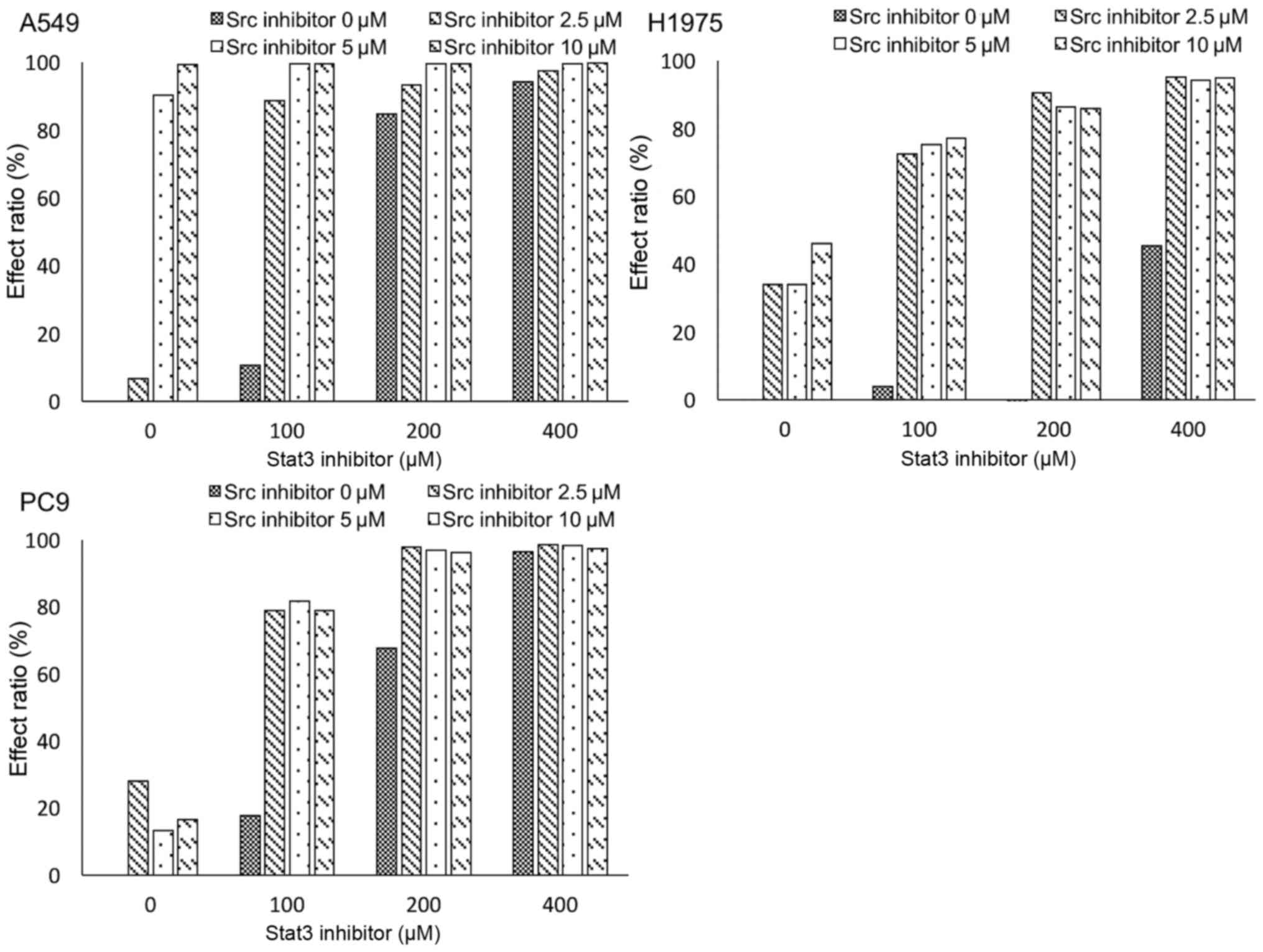
SRC inhibitor and STAT3 inhibitor
synergistically suppress cell growth in lung adenocarcinoma cell
lines
Finally, we measured cell viability under
combination treatment with an SRC inhibitor and a STAT3 inhibitor
in each cell line (Fig. 7). CIs of
SRC inhibitor and STAT3 inhibitor were 0.61, 0.08 and 0.17 for
A549, H1975 and PC9 cell lines, respectively; suggesting that the
two inhibitors synergistically suppressed cell growth.
Discussion
In our previous study, we showed that
ARHGAP35 knockdown by means of chemically modulated small
interfering RNA suppressed the proliferation of lung adenocarcinoma
cell lines with EGFR or KRAS mutations (24). In the present study, we obtained
clones in which ARHGAP35 was stably knocked down, and we
hypothesized that attenuated viability would be seen as a
compensatory mechanism for survival in those cells.
RAS activates the RAF1/MAPK pathway and EGFR
activates several downstream pathways, including the RAS/RAF1/MAPK,
PI3K/AKT and STAT pathways, and these play important roles when
regulating proliferation, invasion and migration (28). Among these pathways, ARHGAP35 has
been reported to be inactivated by the RAS/RAF1/MAPK pathway and to
regulate RhoA (24,29,30).
Importantly, ARHGAP35 is also activated by SRC, which is a
potential signal mediator of the EGFR pathway (19). We assumed that the RAS/RAF1/MAPK and
SRC pathways were compensatory mechanisms. In addition, based on
acquired resistance for EGFR-TKIs, we decided to screen MET, ERBB2,
PKC, AKT and STAT3 (31–33).
Western blots showed increased SRC, STAT3, PRKCZ,
MET and ERBB2 levels in A549 cells after long-term ARHGAP35
knockdown. A possible explanation for this is that increased SRC,
MET and ERBB2 might have accelerated STAT3 and PRKCZ downstream,
ultimately attenuating ARHGAP35 knockdown. This was
supported by the RT-qPCR findings. In contrast to this, the protein
and mRNA levels of the molecules associated with the EGFR pathway
were decreased in both the H1975 and PC9 cell lines after long-term
ARHGAP35 knockdown. We therefore assume the involvement of
another pathway.
Levels of STAT3 and phosphorylated STAT3 increased
in the A549 cell line after long-term ARHGAP35 knockdown.
STAT3 is a 92-kDa protein, encoded by STAT3 on 17q21, and is
a member of the STAT family of transcriptional activators (34–36).
This protein is associated with cell proliferation,
differentiation, invasion and apoptosis in the EGFR signaling
pathway (36–38). In lung cancer, STAT3 promotes cell
proliferation and invasion, while its inhibition leads to the
suppression of tumor proliferation in vitro and in
vivo (39–41).
After long-term ARHGAP35 knockdown, MTS assay
indicated that A549 cells were more sensitive to the STAT3
inhibitor. This was consistent with the hypothesis that increased
STAT3 levels in A549 cells after long-term ARHGAP35
knockdown would be a critical factor for cell survival. Similar but
modest results were obtained in the MTS assay for the PC9 cell
line, although STAT3 was not increased after long-term
ARHGAP35 knockdown. A possible explanation for the different
impact of the inhibitor in these two cell lines may be the
different dependency on the STAT3 pathway for proliferation. We
speculate that A549 cells depended heavily on the STAT3 pathway as
a compensatory mechanism for viability. This presented the
possibility of tumor suppression by combining STAT3 inhibition and
ARHGAP35 inhibition for lung cancer with the KRAS
mutation.
In contrast to A549 cells, however, opposite results
were obtained in H1975 cells after STAT3 inhibition. Although we
cannot explain these phenomena, we speculate that: i) Any specific
underlying molecular mechanisms may cause unexpected results after
STAT3 inhibition in lung cancer with acquired resistance for the
EGFR mutation or ii) sh-Neg treatment may become a stress
for H1975 since elevated protein levels of protein kinase C delta
(PRKCD) and zeta (PRKCZ) in sh-Neg treated H1975 cells were
observed (Fig. 2). The former could
be important because STAT3 inhibitors have been studied in clinical
trials for both hematological and solid malignancies (42,43).
For the latter, PKCs have been reported to be upregulated by
Toll-like receptors and positively regulate STAT3 in response to
stress (44,45). Accordingly, sh-Neg-treated H1975
cells may have relatively strong dependence to STAT3 and respond
strongly to STAT3 inhibitor. Our results that the ARHGAP35 protein
level was slightly decreased and the STAT3 protein level was
increased by sh-Neg treatment in H1975 cells (Figs. 1 and 2) and discrepancy between mRNA expression
and protein translation for SRC and STAT3 (Figs. 1 and 3) may be affected by same reason. However,
our important finding is that STAT3 inhibitor and SRC inhibitor
synergistically suppressed cell growth even in EGFR-mutant cells
(Fig. 7).
ARHGAP35 is poor therapeutic target because it lacks
a kinase domain. A natural alternative strategy is therefore to
target an upstream mediator, such as SRC, that can suppress
ARHGAP35 activity. Unfortunately, a phase II clinical trial of a
single-use SRC inhibitor for lung cancer has presented only modest
clinical benefit (46). Based on
the concept of synthetic lethality, we therefore, tested the
effects of cosuppression of the SRC/ARHGAP35 axis with both a STAT3
inhibitor and an SRC inhibitor in vitro. Concomitant
treatment caused a synergistic and strong effect on growth
inhibition in the KRAS- and EGFR-mutant cell lines,
especially in the KRAS-mutant cell line. Thus, cosuppression
of the STAT3 pathway and SRC/ARHGAP35 axis may be an effective
strategy for treating KRAS and EGFR mutant lung
adenocarcinoma. A further study is needed to confirm whether this
effect can be generalized to other cell lines.
Notably, a synergistic effect was observed for the
SRC and STAT3 inhibitors in both EGFR-mutant cell lines,
despite the fact that the H1975 cell line is usually resistant to
STAT3 inhibitors and the PC9 cell line is relatively resistant to
SRC inhibitors. Unfortunately, we cannot illuminate the mechanism
underlying this synergistic effect, but based on previous reports
of synthetic lethality, it is possible that mitosis regulation or
DNA duplication systems could be involved (11,12,14).
In a previous research, ARHGAP35 has been shown to regulate mitosis
by controlling RhoA (47). In
addition, a recent report showed that STAT3 upregulated TPX2, a
microtubule-associated protein known to be involved in mitosis, by
binding the 5′-flanking sequence of the TPX2 gene (48). Further study is warranted on this
topic as STAT3 and SRC have many potential targets.
In our previous study, short-term ARHGAP35
knockdown was also shown to suppress migration in lung cancer
(24). In the present study, cell
migration ability was reduced in all three cell lines after
ARHGAP35 knockdown, suggesting that the suppressive effect
on migration may be maintained in the long term. ROCK activity was
within normal limits, however, so this conflicting result will need
to be resolved in future research.
In conclusion, cosuppression of STAT3 and the
SRC/ARHGAP35 axis may be an effective strategy for treating
KRAS- and EGFR-mutant lung adenocarcinoma.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The analysed datasets generated during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
KO and HN performed the experiments. KO wrote the
manuscript. AS and CE made substantial contributions to conception,
design and intellectual content of the studies. TW, YM and MN made
key contributions to the analysis and interpretation of the data.
AS and YO reviewed and edited the manuscript. YO also contributed
to the planning of the research. All authors read and approved the
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Committee for Scientific Affairs, The
Japanese Association for Thoracic Surgery, ; Masuda M, Kuwano H,
Okumura M, Amano J, Arai H, Endo S, Doki Y, Kobayashi J, Motomura
N, Nishida H, et al: Thoracic and cardiovascular surgery in Japan
during 2012: Annual report by The Japanese Association for thoracic
surgery. Gen Thorac Cardiovasc Surg. 62:734–764. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Herbst RS and Shin DM: Monoclonal
antibodies to target epidermal growth factor receptor-positive
tumors: A new paradigm for cancer therapy. Cancer. 94:1593–1611.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Morita S, Okamoto I, Kobayashi K, Yamazaki
K, Asahina H, Inoue A, Hagiwara K, Sunaga N, Yanagitani N, Hida T,
et al: Combined survival analysis of prospective clinical trials of
gefitinib for non-small cell lung cancer with EGFR mutations. Clin
Cancer Res. 15:4493–4498. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mazières J, Peters S, Lepage B, Cortot AB,
Barlesi F, Beau-Faller M, Besse B, Blons H, Mansuet-Lupo A, Urban
T, et al: Lung cancer that harbors an HER2 mutation: Epidemiologic
characteristics and therapeutic perspectives. J Clin Oncol.
31:1997–2003. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang SE, Narasanna A, Perez-Torres M,
Xiang B, Wu FY, Yang S, Carpenter G, Gazdar AF, Muthuswamy SK and
Arteaga CL: HER2 kinase domain mutation results in constitutive
phosphorylation and activation of HER2 and EGFR and resistance to
EGFR tyrosine kinase inhibitors. Cancer Cell. 10:25–38. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Onitsuka T, Uramoto H, Nose N, Takenoyama
M, Hanagiri T, Sugio K and Yasumoto K: Acquired resistance to
gefitinib: The contribution of mechanisms other than the T790M,
MET, and HGF status. Lung Cancer. 68:198–203. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yano S, Wang W, Li Q, Matsumoto K,
Sakurama H, Nakamura T, Ogino H, Kakiuchi S, Hanibuchi M, Nishioka
Y, et al: Hepatocyte growth factor induces gefitinib resistance of
lung adenocarcinoma with epidermal growth factor
receptor-activating mutations. Cancer Res. 68:9479–9487. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kobayashi S, Boggon TJ, Dayaram T, Jänne
PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG and Halmos
B: EGFR mutation and resistance of non-small-cell lung cancer to
gefitinib. N Engl J Med. 352:786–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yun CH, Mengwasser KE, Toms AV, Woo MS,
Greulich H, Wong KK, Meyerson M and Eck MJ: The T790M mutation in
EGFR kinase causes drug resistance by increasing the affinity for
ATP. Proc Natl Acad Sci USA. 105:2070–2075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wright S and Ddbzhansky T: Genetics of
natural populations Xii. Experimental reproduction of some of the
changes caused by natural selection in certain populations of
drosophila-pseudoobscura. Genetics. 31:125–156. 1945.
|
|
11
|
Bryant HE, Schultz N, Thomas HD, Parker
KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ and Helleday T:
Specific killing of BRCA2-deficient tumours with inhibitors of
poly(ADP-ribose) polymerase. Nature. 434:913–917. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Farmer H, McCabe N, Lord CJ, Tutt ANJ,
Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I,
Knights C, et al: : Targeting the DNA repair defect in BRCA mutant
cells as a therapeutic strategy. Nature. 434:917–921. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
McLornan DP, List A and Mufti GJ: Applying
synthetic lethality for the selective targeting of cancer. N Engl J
Med. 371:1725–1735. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Corcoran RB, Cheng KA, Hata AN, Faber AC,
Ebi H, Coffee EM, Greninger P, Brown RD, Godfrey JT, Cohoon TJ, et
al: Synthetic lethal interaction of combined BCL-XL and MEK
inhibition promotes tumor regressions in KRAS mutant cancer models.
Cancer Cell. 23:121–128. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lamba S, Russo M, Sun C, Lazzari L,
Cancelliere C, Grernrum W, Lieftink C, Bernards R, Di Nicolantonio
F and Bardelli A: RAF suppression synergizes with MEK inhibition in
KRAS mutant cancer cells. Cell Rep. 8:1475–1483. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ellis C, Moran M, McCormick F and Pawson
T: Phosphorylation of GAP and GAP-associated proteins by
transforming and mitogenic tyrosine kinases. Nature. 343:377–381.
1990. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Settleman J, Narasimhan V, Foster LC and
Weinberg RA: Molecular cloning of cDNAs encoding the GAP-associated
protein p190: Implications for a signaling pathway from ras to the
nucleus. Cell. 69:539–549. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Brouns MR, Matheson SF and Settleman J:
p190 RhoGAP is the principal Src substrate in brain and regulates
axon outgrowth, guidance and fasciculation. Nat Cell Biol.
3:361–367. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chang JH, Gill S, Settleman J and Parsons
SJ: c-Src regulates the simultaneous rearrangement of actin
cytoskeleton, pl90RhoGAP, and pl20RasGAP following epidermal growth
factor stimulation. J Cell Biol. 130:355–368. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kusama T, Mukai M, Endo H, Ishikawa O,
Tatsuta M, Nakamura H and Inoue M: Inactivation of Rho GTPases by
p190 RhoGAP reduces human pancreatic cancer cell invasion and
metastasis. Cancer Sci. 97:848–853. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shen CH, Chen HY, Lin MS, Li FY, Chang CC,
Kuo ML, Settleman J and Chen RH: Breast tumor kinase phosphorylates
p190RhoGAP to regulate rho and ras and promote breast carcinoma
growth, migration, and invasion. Cancer Res. 68:7779–7787. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guo A, Villén J, Kornhauser J, Lee KA,
Stokes MP, Rikova K, Possemato A, Nardone J, Innocenti G, Wetzel R,
et al: Signaling networks assembled by oncogenic EGFR and c-Met.
Proc Natl Acad Sci USA. 105:692–697. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rikova K, Guo A, Zeng Q, Possemato A, Yu
J, Haack H, Nardone J, Lee K, Reeves C, Li Y, et al: Global survey
of phosphotyrosine signaling identifies oncogenic kinases in lung
cancer. Cell. 131:1190–1203. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Notsuda H, Sakurada A, Endo C, Okada Y,
Horii A, Shima H and Kondo T: p190A RhoGAP is involved in EGFR
pathways and promotes proliferation, invasion and migration in lung
adenocarcinoma cells. Int J Oncol. 43:1569–1577. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liang CC, Park AY and Guan JL: In vitro
scratch assay: A convenient and inexpensive method for analysis of
cell migration in vitro. Nat Protoc. 2:329–333. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: The combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhu H, Ding WJ, Wu R, Weng QJ, Lou JS, Jin
RJ, Lu W, Yang B and He QJ: Synergistic anti-cancer activity by the
combination of TRAIL/APO-2L and celastrol. Cancer Invest. 28:23–32.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Scaltriti M and Baselga J: The epidermal
growth factor receptor pathway: A model for targeted therapy. Clin.
Cancer Res. 12:5268–5272. 2006.
|
|
29
|
Chen JC, Zhuang S, Nguyen TH, Boss GR and
Pilz RB: Oncogenic Ras leads to Rho activation by activating the
mitogen-activated protein kinase pathway and decreasing
Rho-GTPase-activating protein activity. J Biol Chem. 278:2807–2818.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pullikuth AK and Catling AD: Extracellular
signal-regulated kinase promotes Rho-dependent focal adhesion
formation by suppressing p190A RhoGAP. Mol Cell Biol. 30:3233–3248.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Appleman LJ: MET signaling pathway: A
rational target for cancer therapy. J Clin Oncol. 29:4837–4838.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Moasser MM: The oncogene HER2: Its
signaling and transforming functions and its role in human cancer
pathogenesis. Oncogene. 26:6469–6487. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nakamura T, Sakai K, Nakamura T and
Matsumoto K: Hepatocyte growth factor twenty years on: Much more
than a growth factor. J Gastroenterol Hepatol. 26 Suppl
1:S188–S202. 2011. View Article : Google Scholar
|
|
34
|
Aggarwal BB, Kunnumakkara AB, Harikumar
KB, Gupta SR, Tharakan ST, Koca C, Dey S and Sung B: Signal
transducer and activator of transcription-3, inflammation, and
cancer: How intimate is the relationship? Ann NY Acad Sci.
1171:59–76. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Akira S, Nishio Y, Inoue M, Wang XJ, Wei
S, Matsusaka T, Yoshida K, Sudo T, Naruto M and Kishimoto T:
Molecular cloning of APRF, a novel IFN-stimulated gene factor 3
p91-related transcription factor involved in the gp130-mediated
signaling pathway. Cell. 77:63–71. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhong Z, Wen Z and Darnell JE Jr: Stat3: A
STAT family member activated by tyrosine phosphorylation in
response to epidermal growth factor and interleukin-6. Science.
264:95–98. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dauer DJ, Ferraro B, Song L, Yu B, Mora L,
Buettner R, Enkemann S, Jove R and Haura EB: Stat3 regulates genes
common to both wound healing and cancer. Oncogene. 24:3397–3408.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Turkson J and Jove R: STAT proteins: Novel
molecular targets for cancer drug discovery. Oncogene.
19:6613–6626. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li CJ, Li YC, Zhang DR and Pan JH: Signal
transducers and activators of transcription 3 function in lung
cancer. J Cancer Res Ther. 9 Suppl 2:S67–S73. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Siveen KS, Sikka S, Surana R, Dai X, Zhang
J, Kumar AP, Tan BK, Sethi G and Bishayee A: Targeting the STAT3
signaling pathway in cancer: Role of synthetic and natural
inhibitors. Biochim Biophys Acta. 1845:136–154. 2014.PubMed/NCBI
|
|
41
|
Song L, Rawal B, Nemeth JA and Haura EB:
JAK1 activates STAT3 activity in non-small-cell lung cancer cells
and IL-6 neutralizing antibodies can suppress JAK1-STAT3 signaling.
Mol Cancer Ther. 10:481–494. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bendell JC, Hong DS, Burris HA III, Naing
A, Jones SF, Falchook G, Bricmont P, Elekes A, Rock EP and Kurzrock
R: Phase 1, open-label, dose-escalation, and pharmacokinetic study
of STAT3 inhibitor OPB-31121 in subjects with advanced solid
tumors. Cancer Chemother Pharmacol. 74:125–130. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Verstovsek S, Mesa RA, Gotlib J, Levy RS,
Gupta V, DiPersio JF, Catalano JV, Deininger M, Miller C, Silver
RT, et al: A double-blind, placebo-controlled trial of ruxolitinib
for myelofibrosis. N Engl J Med. 366:799–807. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jain N, Zhang T, Kee WH, Li W and Cao X:
Protein kinase C δ associates with and phosphorylates Stat3 in an
interleukin-6-dependent manner. J Biol Chem. 274:24392–24400. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Olejniczak M, Galka P and Krzyzosiak WJ:
Sequence-non-specific effects of RNA interference triggers and
microRNA regulators. Nucleic Acids Res. 38:1–16. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Johnson FM, Bekele BN, Feng L, Wistuba I,
Tang XM, Tran HT, Erasmus JJ, Hwang LL, Takebe N, Blumenschein GR,
et al: Phase II study of dasatinib in patients with advanced
non-small-cell lung cancer. J Clin Oncol. 28:4609–4615. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chircop M: Rho GTPases as regulators of
mitosis and cytokinesis in mammalian cells. Small GTPases.
5:e297702014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cocchiola R, Grillo C, Altieri F,
Chichiarelli S, Turano C and Eufemi M: Upregulation of TPX2 by
STAT3: Identification of a novel STAT3 binding site. PLoS One.
9:e1130962014. View Article : Google Scholar : PubMed/NCBI
|