Introduction
Liver cancer is the sixth most common cancer
worldwide and the second major tumor type in China (1,2).
Despite significant advances in the detection and treatment of
liver cancer, the survival of liver cancer patients remains poor
and the precise mechanisms underlying liver cancer remain unclear
(3). Therefore, novel diagnostic
markers are urgently needed to improve the survival rate of liver
cancer patients (4).
Previous investigations have demonstrated that
epithelial-to-mesenchymal transition (EMT) participates in the
progression and metastasis of various types of tumor, including
liver cancer (5,6). Since EMT is the initial step of tumor
metastasis, further understanding on the mechanisms of EMT will
shed new light on the use of targeted therapeutic strategies for
liver cancer. However, the molecular mechanism of EMT is yet
unknown. An increasing number of transcription factors that can
promote EMT have been identified, including zinc finger
E-box-binding homeobox 1 (ZEB1), ZEB2 and Twist (7).
C-C chemokine receptor type 2 (CCR2) is the specific
receptor of monocyte chemoattractant protein-1 (MCP-1), which is
also known as C-C motif chemokine ligand 2 (CCL2), and is also a
receptor for CCL7, CCL8, CCL11, CCL12 and CCL13. CCR2A and CCR2B
are the two isoforms of CCR2, both of which are derived from the
same gene, with the exception of a difference in the
carboxy-terminus, and CCR2B is the major functional form. CCR2 is
abnormally expressed in a variety of tumor cells, including
prostate cancer, renal cell carcinoma, non-small cell lung cancer,
myeloma and colorectal cancer (8–10).
Furthermore, it is associated with tumor invasion and metastasis
(11,12). However, the specific mechanism of
CCR2 in the regulation of liver cancer remains to be clarified.
Given the importance of CCR2 in tumor progression,
the aim of the present investigation was to elucidate the possible
mechanism of CCR2 in liver cancer cell invasion. Kaplan-Meier
survival analysis demonstrated that the survival time of patients
with high expression of CCR2 was lower in comparison with that of
patients with low expression of CCR2. In addition, wound healing
and the Transwell chamber invasion assays revealed that the number
of cells transfected with CCR2-small interfering RNA (siRNA) was
significantly reduced. Matrix metalloproteinase 2 (MMP2) expression
and activity were also significantly decreased following CCR2-siRNA
transfection. Furthermore, there was a positive correlation between
CCR2 and MMP2 in liver cancer tissues, and CCR2 interacted with
MMP2 in HepG2 cells. Taken together, the present study results
revealed that CCR2 promotes EMT through MMP2 in liver cancer, and
that CCR2 is an attractive, novel target for inhibiting the
invasion and metastasis of liver cancer cells.
Materials and methods
Clinical samples
Liver cancer tissues (n=39) and paired normal
tissues were obtained from the First Affiliated Hospital of the
Fourth Military Medical University (Xi'an, 710032, China) between
June 2004 and June 2007. All tissues were obtained by resection
surgery, and none of the patients had previously received
preoperative radiotherapy, chemotherapy or biotherapy. The present
study was approved by the First Affiliated Hospital of the Fourth
Military Medical University (approval no. KY20163226-1) and written
informed consent was obtained from all patients. The
clinicopathological data of the included cases are shown in
Table I. Tumor and paired tissues
were snap-frozen in liquid nitrogen and preserved at −80°C.
 | Table I.Association of CCR2 expression with
the clinicopathological characteristics of liver cancer
patients. |
Table I.
Association of CCR2 expression with
the clinicopathological characteristics of liver cancer
patients.
|
|
| CCR2 expression |
|
|---|
|
|
|
|
|
|---|
| Clinical
characteristics | Case no. | Low | High | P-value |
|---|
| Total | 39 | 18 | 21 |
|
| Sex |
|
|
| 0.573 |
| Male | 25 | 11 | 14 |
|
|
Female | 14 | 7 | 7 |
|
| Age (years) |
|
|
| 0.720 |
| ≤60 | 18 | 10 | 8 |
|
|
>60 | 21 | 9 | 12 |
|
| Tumor size (cm) |
|
|
| 0.0221a |
|
<5 | 21 | 14 | 7 |
|
| ≥5 | 18 | 4 | 14 |
|
| Histologic grade
(differentiation) |
|
|
| 0.215 |
|
Good/moderate | 20 | 10 | 10 |
|
| Poor | 19 | 8 | 11 |
|
| N status |
|
|
| 0.0187a |
| N0 | 15 | 10 | 5 |
|
|
N1/2 | 24 | 8 | 16 |
|
| Clinical stage |
|
|
| 0.0158a |
|
I–II | 17 | 10 | 7 |
|
|
III–IV | 22 | 8 | 14 |
|
Immunohistochemical staining
Liver cancer tissues samples were fixed in 10%
neutral formaldehyde for 24 h, dehydrated and embedded in paraffin.
Next, 5-µm sections were cut from each tissue. Subsequent to
dewaxing and blocking the endogenous peroxidase activity, specimens
were incubated with antibodies against CCR2 (dilution 1:100; cat.
no. LBP60766; Abcam, Cambridge, MA, USA) at 4°C overnight. Next,
specimens were incubated with the Biotin-labled goat anti-mouse
(1:1,000; ZSGB-BIO, Beijing, China) for 60 min at room temperature.
Following treatment with horseradish peroxidase-labeled
streptavidin solution (ZSGB-BIO) for 30 min at room temperature,
the samples were stained with 3,3′-diaminobenzidine. Finally, the
sections were counterstained with 0.02% hematoxylin and visualized
under a microscope.
Cell culture and treatment
Three human liver cancer cell lines (HepG2,
SMMC-7721 and MHCC97-H) and a normal liver cell line (HL-7702) were
purchased from the American Type Culture Collection (ATCC;
Manassas, VA, USA). All cell lines were cultured in Dulbecco's
modified Eagle's medium (DMEM)/high glucose supplemented with 10%
fetal bovine serum (FBS; both from Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) at 37°C in humidified chamber with 5%
CO2. Cells were transfected with CCR2-siRNA and negative
control siRNA (Shanghai GenePharma Co., Ltd., Shanghai, China) were
transfected using Lipofectamine® 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analyses
Total RNA was extracted from the cells or tissues
with Fast 200 kit (Fastagen, Shanghai, China), RNA concentration
was measured by NanoDrop. Next, 1–2 µg RNA was reverse transcribed
into cDNA using the RevertAid First Strand cDNA Synthesis kit
(Thermo Fisher Scientific, Inc.) and qPCR was performed on the
CFX96 Touch Real-Time PCR detection system (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). The conditions for the qPCR reaction were
as follows: Initial denaturation at 95°C for 10 sec, followed by 40
cycles of 95°C for 5 sec, 55°C for 15 sec and 72°C for 20 sec. The
primers used in PCR are listed in Table II. ΔCq values were normalized to
GAPDH, serving as the internal control, and comparative
quantification was performed with the 2−ΔΔCq method
(13).
| Table II.Primer and siRNA list. |
Table II.
Primer and siRNA list.
| mRNA | Sequence
(5′-3′) | Experimental
use |
|---|
| CCR2 | F:
GAGCGGTGAAGAAGTCACCA | qPCR |
|
| R:
CAGAAGCAAACACAGCCACC |
|
| GAPDH | F:
AAATCCCATCACCATCTTC | qPCR |
|
| R:
TCACACCCATGACGAACA |
|
| MMP2 | F:
GCATCCAGACTTCCTCAGGC | qPCR |
|
| R:
CCATTAGCGCCTCCATCGTAG |
|
| E-cadherin | F:
GCTGCTCTTGCTGTTTCTTCG | qPCR |
|
| R:
CCGCCTCCTTCTTCATCATAG |
|
| Vimentin | F:
AAGTTTGCTGACCTCTCTGAGGCT | qPCR |
|
| R:
CTTCCATTTCACGCATCTGGCGTT |
|
| CCR2-siRNA | F:
AAGCCAGGACGGTCACCTT | RNA
interference |
|
| R:
AAGGTGACCGTCCTGGCTT |
|
| Control-siRNA | F:
TTTTCGCATCGAGTCACGTCT | RNA
interference |
|
| R:
AGACGTGACTCGATGCGAAAA |
|
Western blot assay and antibodies
Cells were lysed in radioimmunoprecipitation assay
buffer containing 1 mM phenylmethane sulfonyl fluoride and cocktail
protease inhibitor (Roche Diagnostics, Nutley, NJ, USA). Protein
concentration was measured by BCA protein assay kit, then separated
by 12% SDS-PAGE gel and transferred to nitrocellulose membranes.
The nitrocellulose membrane was then blocked with 5% non-fat milk
in 0.01 M PBS buffer for 1 h at room temperature, followed by
incubation with primary antibodies against E-cadherin (1:1,000;
cat. no. ab76055; Abcam, Hong Kong, China), vimentin (1:500; cat.
no. 5741; Cell Signaling Technology, Danvers, MA, USA), CCR2
(1:1,000; cat. no. LBP60766; Abcam), MMP2 (1:1,000; cat. no. 40994;
Cell Signaling Technology, Inc.) and GAPDH (1:1,000; cat. no. 2118;
Cell Signaling Technology, Inc.). After washing with PBST, the
blots was reacted with horseradish peroxidase-conjugated anti-mouse
or anti-rabbit IgG (1:2,000; cat. nos. 7076 or 7074; Cell Signaling
Technology, Inc.). The immunoreactive bands were subsequently
detected with an enhanced chemiluminescence detection kit (Pierce;
Thermo Fisher Scientific, Inc.), and the signals were analyzed by
ChemiDoc MP Imaging System and (Bio-Rad Laboratories, Inc.).
Cell viability assay
Cells were seeded in 96-well plates
(3×103/well) and incubated for 24, 48 or 72 h.
Subsequently, cell growth was tested by Cell Counting Kit-8 (CCK-8;
7Sea, Shanghai, China), according to the protocol provided by the
manufacturer. At 2 h after addition of CCK-8, cell growth viability
was detected by measuring the optical density at 450 nm on an
EnSpire plate reader (PerkinElmer, Inc., Waltham, MA, USA).
Migration and invasion assays
The migration assay was conducted using a 24-well
Transwell chamber, while the invasion assay was performed in a
similar fashion using Matrigel-coated chambers. Briefly, cells
(5×105/ml in serum-free medium) were seeded in 100 µl in
the top chamber. The bottom wells were filled with complete medium
containing 20% FBS. After culturing for 24 h for the migration
assay and 48 h for the invasion assay, cells on the upper surface
was wiped off with cotton swabs. Cells in the lower membrane
surface were fixed with methanol and stained using 0.1% crystal
violet. Finally, cells were detected, counted and averaged in five
random fields under a phase contrast microscope (original
magnification, ×100).
Co-immunoprecipitation (Co-IP)
The Co-IP experiments were conducted utilizing the
Pierce Crosslink IP kit (Thermo Fisher Scientific, Inc.) following
the manufacturer's protocol. The Co-IP protocol was performed on
ice, unless otherwise indicated. Briefly, HepG2 cells (150
cm2 plate, 107 cell/plate) were treated with
1 ml extraction buffer, and the binding of the anti-CCR2 or
anti-MMP2 to Protein A/G Agarose was performed according to the
procedure described in the kit. Subsequently, the Protein A/G
Agarose was incubated with anti-CCR2 or anti-MMP2 antibody on a
mixer at 25°C for 1 h. The immunoprecipitated products were eluted
with Laemmli buffer. The eluting mixture was finally tested by
western blot assay.
Statistical analysis
Data are presented as the mean ± standard error of
the mean, and were analyzed by one-way analysis of variance using
the SPSS version 18.0 (SPSS, Inc., Chicago, IL, USA) and GraphPad
Prism version 5 (GraphPad Software, Inc., La Jolla, CA, USA)
statistical software. The patient survival rate was analyzed by the
Kaplan-Meier method, and the log-rank test was performed to
evaluate the presence of significant differences. Differences were
considered to be statistically significant at P<0.05.
Results
CCR2 is upregulated in liver cancer
tissues and associated with liver cancer progression
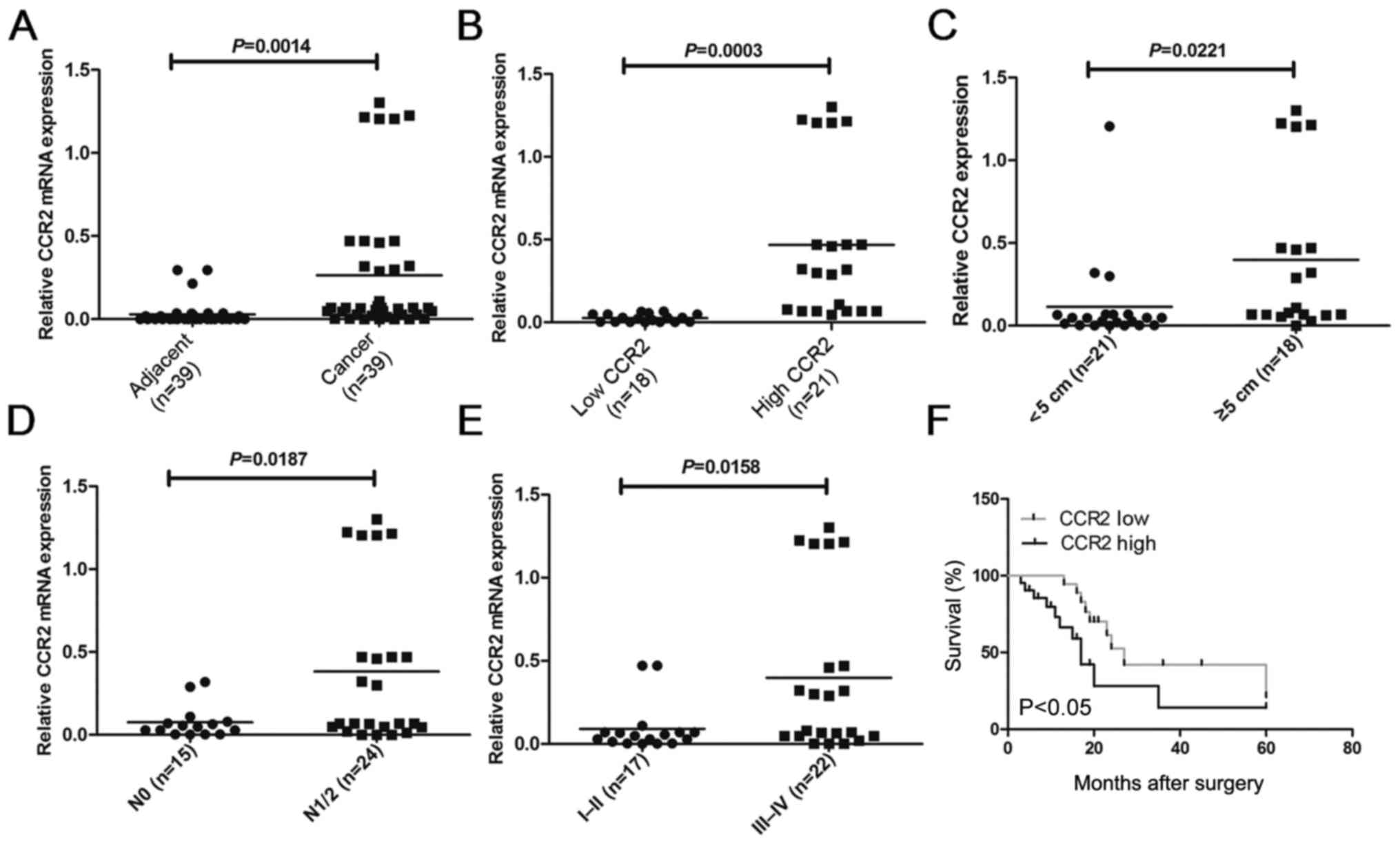
In order to compare the expression of CCR2 in
different liver cancer tissues, the mRNA levels of CCR2 were
detected in 39 liver cancer tissues and paired adjacent normal
liver tissues using RT-qPCR analysis. The results demonstrated that
the CCR2 level in liver cancer tissues was higher compared with
that in adjacent normal tissues (P=0.0014; Fig. 1A). To further investigate the
correlation of CCR2 expression with the clinicopathological
characteristics, the relative CCR2 expression in the liver cancer
tissues obtained from 39 patients was classified into two groups,
including patients with low (n=18; CCR2 expression ratio <
median ratio) and high (n=21; CCR2 expression ratio ≥ median ratio)
relative levels of CCR2 mRNA (P=0.0003; Fig. 1B). The results of the
clinicopathological analysis revealed that CCR2 was significantly
correlated with tumor size (P=0.0221; Fig. 1C), metastasis (P=0.0187; Fig. 1D) and clinical stage (P=0.0158;
Fig. 1E). However, no statistically
significant correlations were obtained between CCR2 expression and
other clinicopathological characteristics, including gender, age
and histologic grade (P>0.05; Table
I). Kaplan-Meier analysis and log-rank test were used to
evaluate the association between CCR2 expression in liver cancer
and patient survival. The median 5-year survival time was 27 months
in the low CCR2 expression group, whereas it was 17 months in the
high CCR2 expression group (P=0.0135; Fig. 1F). Taken together, these results
suggest that overexpression of CCR2 may be involved in liver cancer
development, progression and metastasis.
Knockdown of CCR2 inhibits tumor
development in liver cancer cells
The mRNA and protein expression levels of CCR2 were
also analyzed in three liver cancer cell lines (HepG2, SMMC-7721
and MHCC97-H) and the HL-7702 normal liver cell line. As shown in
Fig. 2A, the results indicated that
the CCR2 mRNA level in HepG2, SMMC-7721 and MHCC97-H cells was
markedly higher compared with that in the HL-7702 normal liver cell
line. Furthermore, the protein expression of CCR2 in cell lines
HepG2, SMMC-7721 and MHCC97-H was also higher than in HL-7702 cell
line (Fig. 2B). Due to the protein
level of CCR2 in SMMC-7721 and MHCC97-H cells which was roughly
similar, HepG2 and MHCC97-H cells were selected in subsequent
experiments. Subsequently, CCR2 expression in liver cancer cells
was manipulated to analyze its association with tumor progression.
The expression level of CCR2 was silenced by siRNA transfection,
and CCR2 expression level and liver cancer cell viability were
detected following CCR2 inhibition. Knockdown of CCR2 was observed
to successfully inhibit the CCR2 expression level (Fig. 2C and D) and viability (Fig. 2E and F) in liver cancer cells. Taken
together, these results suggest that CCR2 is required for tumor
development in liver cancer cells.
CCR2 regulates liver cancer cell
migration and invasion
In order to explore the role of CCR2 in the
regulation of liver cancer metastasis, the migration and invasion
of liver cancer cells (HepG2 and MHCC97H) were detected following
treatment with CCR2-siRNA. As shown in Fig. 3A and B, the silencing of CCR2 in
HepG2 and MHCC97H cells decreased cell mobility compared with that
in the control cells. In addition, downregulation of CCR2
significantly suppressed cell invasion subsequent to the treatment
of cells with CCR2-siRNA, in contrast to the control cells
(Fig. 3C and D). Therefore, these
data suggest that downregulation of CCR2 blocked EMT in liver
cancer cells.
CCR2 enhances EMT by MMP2 in liver
cancer cells
To determine whether CCR2 regulates the EMT in liver
cancer cells, the mRNA and protein levels of E-cadherin, vimentin
and MMP2 were initially evaluated in HepG2 and MHCC97H cells
transfected with CCR2-siRNA. As reported earlier, CCR2-siRNA
decreased CCR2 expression in liver cancer cells. This silencing
also enhanced the EMT in HepG2 and MHCC97H cells, as shown by the
decreased expression of E-cadherin and increased expression of
vimentin detected by RT-qPCR (Fig. 4A
and B) and western blotting (Fig.
4C and D). In order to further explore the molecular mechanisms
of CCR2 in liver cancer cells, the expression of MMP2 following the
treatment of cells with CCR2-siRNA was also analyzed. The results
demonstrated that the mRNA and protein expression levels of MMP2
were suppressed after silencing of CCR2 compared with the control
cells (Fig. 4A-D). Furthermore, the
activity of MMP2 was tested, and the results revealed that MMP2
activity was weakened by downregulation of CCR2 (Fig. 4E and F). The association between
CCR2 and MMP2 in 39 liver cancer tissues was also detected, and the
results suggest that the mRNA expression levels of CCR2 were
positively correlated with MMP2 (r=0.378, P=0.018; Fig. 4G). To further confirm the specific
binding between CCR2 and MMP2, the study also identified the
interaction between endogenous proteins independently in HepG2 cell
lines by Co-IP. The results indicated that endogenous CCR2
co-precipitated with MMP2 in HepG2 cells (Fig. 4F). Taken together, these findings
reveal that CCR2 is highly expressed in liver cancer tissues and
cells. In addition, CCR2 promoted EMT in liver cancer cells through
the regulation of MMP2 (Fig.
5).
 | Figure 4.CCR2 enhanced
epithelial-to-mesenchymal transition by MMP2 in liver cancer cells.
mRNA expression levels of E-cadherin, vimentin, CCR2 and MMP2 in
(A) HepG2 and (B) MHCC97H cells transfected with si-Ctrl or
si-CCR2. Protein expression level of E-cadherin, vimentin, CCR2 and
MMP2 in (C) HepG2 and (D) MHCC97H cells transfected with si-Ctrl or
si-CCR2. Relative MMP2 protein activity in (E) HepG2 and (F)
MHCC97H cells transfected with si-Ctrl or si-CCR2. (G) Correlation
between CCR2 and MMP2 mRNA expression in liver cancer tissues
(r=0.378, P=0.018). (H) Interaction between endogenous CCR2 and
MMP2 in HepG2 cell lines by Co-IP. *P<0.05 vs. control group.
CCR2, C-C chemokine receptor type 2; MMP2, matrix
metalloproteinase-2; IP, immunoprecipitation; NC, normal control;
si, small interfering RNA; Ctrl, control. |
Discussion
Mounting evidence has demonstrated that liver cancer
is one of the deadliest types of cancer due to its complexity,
heterogeneity, metastasis and reoccurrence (14). Tumor metastasis is one of the main
reasons for the poor prognosis of liver cancer patients. EMT, the
initial step of tumor metastasis that results in the loss of the
epithelial phenotype and leads to mesenchymal characteristics, has
become an important field in cancer research (15–18).
Further exploration of the molecular mechanisms of EMT will shed
new light on the development of liver cancer diagnostic and
therapeutic strategies (19–20).
Previous studies have demonstrated that CCR2 serves
an important role in tumor metastasis in breast, bladder, ovarian
and prostate cancer (21–25). CCR2 is the specific receptor of
MCP-1/CCL2. Hu et al (21)
reported that CCR2 acts as a competing endogenous RNA by inhibiting
the STARD13-RhoA-ROCK1-MLC-F-actin pathway in the regulation of
ovarian metastasis. In addition, Izumi et al (25) demonstrated that knockdown of the
androgen receptor in prostate cancer promoted PCa cell migration
and invasion through the CCL2/CCR2-STAT3 axis and EMT pathways. Rao
et al (23) also revealed
that upregulation of estrogen receptor β in mast cells and bladder
cancer cells resulted in an enhanced CCL2/CCR2/EMT/MMP9 axis. Thus,
exploration of the mechanisms of CCR2 in liver cancer may be useful
for developing novel diagnostic markers and therapeutic drugs for
liver cancer patients.
In the present study, the hypothesis that CCR2
serves an important role in liver cancer progression through the
regulation of EMT was proposed. To that end, CCR2 was detected in
39 liver cancer and paired adjacent tissues. The results revealed
that CCR2 levels in liver cancer tissues were higher when compared
with those in adjacent normal tissues. Further investigation into
the correlation of CCR2 expression with clinicopathological
characteristics indicated that CCR2 was significantly correlated
with tumor size, metastasis and clinical stage. The median 5-year
survival time was evaluated, and the results indicated that this
was higher in the low CCR2 expression group in comparison with that
in the high CCR2 expression group. These results suggest that the
overexpression of CCR2 may be involved in liver cancer development,
progression and metastasis. To further dissect the role of CCR2 in
the regulation of liver cancer EMT, CCR2 was silenced in liver
cancer cells by siRNA transfection. The results indicated that
knockdown of CCR2 inhibited liver cancer cell viability, migration
and invasion. Furthermore, the molecular mechanism of CCR2 in the
regulation of EMT in liver cancer cells was evaluated, and the
results revealed that CCR2-siRNA inhibited the expression levels of
E-cadherin and MMP2, while it increased the expression of vimentin.
In addition, the activity of MMP2 was weakened by downregulation of
CCR2. The mRNA expression levels of CCR2 were also found to be
positively correlated with MMP2 in liver cancer tissues, while
endogenous CCR2 co-precipitated with MMP2 in HepG2 cells.
In conclusion, CCR2 was aberrantly increased in
liver cancer tissues and significantly associated with the tumor
diameter, metastasis and stage. The survival of patients with high
CCR2 expression was lower than that of patients with low CCR2
expression. CCR2 can promote cell viability, cell migration and
invasion in liver cancer cells. Moreover, CCR2 was found to
participate in the regulation of EMT by measuring RNA and protein
expression levels of vimentin and E-cadherin, downregulating of
CCR2, which reduced the expression of E-cadherin and MMP2, activity
of MMP2, and enhanced the expression of vimentin. In addition, the
specific binding between CCR2 and MMP2 was confirmed by Co-IP in
HepG2 cells. These results revealed that CCR2 promotes EMT in liver
cancer. Thus, CCR2 is an attractive novel target for inhibiting
invasion and metastasis of liver cancer cells (Fig. 5).
Acknowledgements
The authors would like to thank Dr Haimin Li from
the First Affiliated Hospital of the Fourth Military Medical
University for his assistance with the design and technical
support.
Funding
The present study was supported by the Nature
Science Basic Research Program of Shanxi Province (grant no.
201701D221177) and the Youth Tuoju Project of Shaanxi Association
for Science and Technology (grant no. 20170408).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HJL and HL conceived and designed the study. HJZ,
LTL, HJL, XPL, WQL and TD performed the experiments. HJL and XPL
wrote the paper. HJL, HL, HJZ and TD reviewed and edited the
manuscript. All authors read and approved the manuscript and agree
to be accountable for all aspects of the research in ensuring that
the accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
The present study was approved by the First
Affiliated Hospital of the Fourth Military Medical University
(approval no. KY20163226-1; Xi'an, China), and written informed
consent has been provided by all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in china,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jayachandran A, Dhungel B and Steel JC:
Epithelial-to-mesenchymal plasticity of cancer stem cells:
Therapeutic targets in hepatocellular carcinoma. J Hematol Oncol.
9:742016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Etik Ozer D, Suna N and Boyacioglu AS:
Management of hepatocellular carcinoma: Prevention, surveillance,
diagnosis, and staging. Exp Clin Transplant. 15:31–35.
2017.PubMed/NCBI
|
|
5
|
Giannelli G, Koudelkova P, Dituri F and
Mikulits W: Role of epithelial to mesenchymal transition in
hepatocellular carcinoma. J Hepatol. 65:798–808. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
van Zijl F, Zulehner G, Petz M, Schneller
D, Kornauth C, Hau M, Machat G, Grubinger M, Huber H and Mikulits
W: Epithelial-mesenchymal transition in hepatocellular carcinoma.
Future Oncol. 5:1169–1179. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bottoni P, Isgro MA and Scatena R: The
epithelialmesenchymal transition in cancer: A potential critical
topic for translational proteomic research. Expert Rev Proteomics
13–115–133. 2016. View Article : Google Scholar
|
|
8
|
Yang X, Lu P, Ishida Y, Kuziel WA, Fujii C
and Mukaida N: Attenuated liver tumor formation in the absence of
CCR2 with a concomitant reduction in the accumulation of hepatic
stellate cells, macrophages and neovascularization. Int J Cancer.
118:335–345. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang XW, Qin X, Qin CY, Yin YL, Chen Y
and Zhu HL: Expression of monocyte chemoattractant protein-1 and CC
chemokine receptor 2 in non-small cell lung cancer and its
significance. Cancer Immunol Immunother. 62:563–570. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hiratsuka S, Ishibashi S, Tomita T,
Watanabe A, Akashi-Takamura S, Murakami M, Kijima H, Miyake K,
Aburatani H and Maru Y: Primary tumours modulate innate immune
signalling to create pre-metastatic vascular hyperpermeability
foci. Nat Commu. 4:18532013. View Article : Google Scholar
|
|
11
|
Zhuang H, Cao G, Kou C and Liu T:
CCL2/CCR2 axis induces hepatocellular carcinoma invasion and
epithelial-mesenchymal transition in vitro through
activation of the hedgehog pathway. Oncol Rep. 39:21–30.
2018.PubMed/NCBI
|
|
12
|
Lim SY, Yuzhalin AE, Gordon-Weeks AN and
Muschel RJ: Targeting the CCL2-CCR2 signaling axis in cancer
metastasis. Oncotarget. 7:28697–28710. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2-ΔΔCT method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dutta R and Mahato RI: Recent advances in
hepatocellular carcinoma therapy. Pharmacol Ther. 173:106–117.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wong SHM, Fang CM, Chuah LH, Leong CO and
Ngai SC: E-cadherin: Its dysregulation in carcinogenesis and
clinical implications. Crit Rev Oncol Hematol. 121:11–22. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vu T and Datta PK: Regulation of EMT in
colorectal cancer: A Culprit in metastasis. Cancers. 9:E1712017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Garg M: Epithelial plasticity and cancer
stem cells: Major mechanisms of cancer pathogenesis and therapy
resistance. World J Stem Cells. 9:118–126. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gaianigo N, Melisi D and Carbone C: EMT
and treatment resistance in pancreatic cancer. Cancers. 9:E1222017.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Qiu L, Tang Q, Li G and Chen K: Long
non-coding RNAs as biomarkers and therapeutic targets recent
insights into hepatocellular carcinoma. Life Sci. 191:273–282.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Giannelli G, Koudelkova P, Dituri F and
Mikulits W: Role of epithelial to mesenchymal transition in
hepatocellular carcinoma. J Hepatol. 65:798–808. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hu J, Li X, Guo X, Guo Q, Xiang C, Zhang
Z, Xing Y, Xi T and Zheng L: The CCR2 3′UTR functions as a
competing endogenous RNA to inhibit breast cancer metastasis. J
Cell Sci. 130:3399–3413. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cai J, Wang M, Zhu M, Zhang Q, Zhang X,
Yan Y, Qian H and Xu W: N-methyl-N-nitro-N'-nitrosoguanidine
induces the expression of CCR2 in human gastric epithelial cells
promoting CCL2-mediated migration. Mol Med Rep. 13:1083–1090. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rao Q, Chen Y, Yeh CR, Ding J, Li L, Chang
C and Yeh S: Recruited mast cells in the tumor microenvironment
enhance bladder cancer metastasis via modulation of ERβ/CCL2/CCR2
EMT/MMP9 signals. Oncotarget. 7:7842–7855. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Moisan F, Francisco EB, Brozovic A, Duran
GE, Wang YC, Chaturvedi S, Seetharam S, Snyder LA, Doshi P and
Sikic BI: Enhancement of paclitaxel and carboplatin therapies by
CCL2 blockade in ovarian cancers. Mol Oncol. 8:1231–1239. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Izumi K, Fang LY, Mizokami A, Namiki M, Li
L, Lin WJ and Chang C: Targeting the androgen receptor with siRNA
promotes prostate cancer metastasis through enhanced macrophage
recruitment via CCL2/CCR2-induced STAT3 activation. EMBO Mol Med.
5:1383–1401. 2013. View Article : Google Scholar : PubMed/NCBI
|