Introduction
Cancer is a deadly disease in humans that is
characterised by the dysregulation of the cell cycle leading to
uncontrolled cell division and continuing growth. Unfortunately,
current chemo/radiation therapies are not sufficiently effective,
but instead are mostly palliative. Thus, the patient survival rate
is very poor for most diagnosed cancers. Therefore, there is an
urgent need for a new line of therapeutics which offer more
selective, effective and curative prospects such as
molecular-targeted therapies that have shown promise in both in
vitro and in vivo models (1).
The epidermal growth factor receptor (EGFR) is part
of the receptor tyrosine kinase family ErbB. The EGFR is important
in the signalling pathway for the control of fundamental cellular
functions including cell growth and survival (2). The receptor, when activated by its
ligand (EGF), leads to autophosphorylation of a number of tyrosine
residues, leading to the activation of downstream Ras/MAPK and
PI3K/AKT proteins which promote cell survival and proliferation.
The deregulation and overexpression of the EGFR has been shown to
be a hallmark of several neoplastic malignancies (3). Thus, generating new anticancer agents
that selectively interfere with specific signalling pathways
critical to a malignant phenotype, metastasis and tumour
progression such as EGFR would block or slow the growth of
EGFR-positive cancers, while minimising harm to other normal cells.
Current leading agents are monoclonal antibodies (mAbs) and small
chemical inhibitors (SCIs) that target EGFR (3).
There are two EGFR-directed monoclonal antibodies
(cetuximab and panitumumab) currently in clinical use for cancer
patients. These monoclonal antibodies function extracellularly to
block the EGFR and potential receptor activation (4). However, mutations in the EGFR and the
downstream effector KRAS have given rise to the resistance to these
forms of therapies (5,6).
ETA is an extremely potent exotoxin released from
Pseudomonas aeruginosa, a common Gram-negative, rod-shaped
bacterium, which is comprised of three domains: a receptor binding
domain (domain I), translocation domain (domain II) and catalysis
domain (domain III). ETA inhibits ADP-ribosylation of eEF2
(eukaryotic elongation factor 2), arresting protein synthesis and
leading to cell death (7,8). In a previous research, a truncated
fragment of Pseudomonas aeruginosa exotoxin A (ETA) lacking
the receptor binding domain was fused with the variable domain of a
single chain antibody fragment (425scFv) specific to EGFR and was
found to be effective in human Hodgkin's lymphoma in a SCID-mouse
model (9). Furthermore, we have
previously shown that CLDN-4 targeted ETA (CPE-ETA) specifically
inhibited the growth of cancer cells overexpressing the CLDN-4
receptor (10).
In this study, a chimeric molecule was constructed
by fusing EGF and ETA. Its N-terminal domain has ADP-ribosylation
activity and induces apoptosis predominantly (11). We then characterised the ability of
EGF-ETA to specifically bind EGFR-expressing cancer cells and
subsequently induce cell-specific death in vitro. This
receptor-facilitated molecular-directed therapy warrants additional
research in order to ascertain its therapeutic potential for a
variety of EGFR-expressing cancers.
Materials and methods
Cell culture conditions
The following human cell lines were used in the
present study: head and neck squamous carcinoma HN5 cells, breast
ductal carcinoma MCF-7 cells, non-small cell lung cancer A549
cells, colorectal cancer cell lines HCT116, HT29, SW480 and
epidermoid carcinoma A431 cells. All cells were cultured in a
complete medium containing: 500 ml Dulbecco's modified Eagle's
medium (DMEM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA), 12.5 ml HEPES buffer solution (1 M), 10% fetal bovine serum
(FBS) and penicillin (5,000 U)/streptomycin (5,000 µg) (all are
from Gibco; Thermo Fisher Scientific, Inc.). The cells were
cultured in a humidified atmosphere at 37°C and 5% CO2.
Routine methods were used for the culturing of cell lines (12). The cell lines were obtained from the
American Type Culture Collection (ATCC; Manassas, VA, USA), except
for HN5, which was provided by Dr Hong-Jian Zhu, University of
Melbourne, Australia.
Construction of EGF-ETA
To isolate the EGF gene, mRNA was isolated from
human FHC cells and subsequently converted to cDNA using the
Invitrogen™ SuperScript III cDNA synthesis kit (Thermo Fisher
Scientific, Inc.). Primers incorporating NdeI (EGF forward:
CCCATATGAATAGTGACTCCTGAATGTCCCCTGTCC) and NotI (EGF reverse:
GGGCGGCCGCGCGCAGTTCCCACCACTTCAG) were designed to isolate the
mature EGF coding sequence. After amplification, the EGF PCR
product was cloned into the NdeI and NotI sites of
the p425-ScFv-ETA vector (13) to
give p452-EGF-ETA. This cloning step removes the ScFv and replaces
it with EGF, resulting in the 10His-EGF-ETA in-frame
fusion protein which lacks the native ETA receptor binding domain
(9). The plasmid was sequenced to
verify the correct insertion and sequence of EGF-ETA.
Expression and purification of
EGF-ETA
After p10His-EGF-ETA was transferred to
E. coli BL21 Al by heat shock, cells were cultured in LB
media containing 50 µg/ml kanamycin. For purification, 15 ml of
starter culture was used to inoculate a 300 ml LB liquid medium
with 50 µg/ml kanamycin. After the OD600 reached 0.4, 3
ml of 20% L-arabinose and 1 ml isopropyl thiogalactoside (IPTG)
(100 mM) (Sigma-Aldrich, St. Louis, MO, USA) were used to induce
protein expression in the cells. The cells were further cultured
for another 2 h at 30°C before harvesting. Centrifugation was used
to pellet the cells and they were stored at −80°C until further
use.
Before purification, the cell pellet was resuspended
in 15 ml lysis buffer (Qiagen GmbH, Hilden, Germany) with 75 µl
PMSF (200 mM) and 300 µl protease inhibitor cocktail. The cells
were lysed by sonication 7 times at 30-sec bursts each time on ice.
The mixture was then centrifuged at 20,000 × g for 25 min to remove
insoluble proteins. The supernatant containing the recombinant
protein was processed using the Ni-NTA Fast Start kit (Qiagen)
according to the protocols provided. Purified EGF-ETA was dialysed
in phosphate-buffered saline (PBS) using ultrafiltration (Amicon
Ultra-15 30 kDa cut-off) (Merck Millipore, Billerica, MA, USA). The
purified protein was stored at −80°C in PBS buffer containing 20%
glycerol, and protein concentrations were determined by DC protein
assay (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
MTT cell proliferation assays
MTT cell proliferation assays measure the metabolic
activity of a cell via the conversion of the tetrazolium dye, MTT,
to the insoluble purple dye formazan. This insoluble dye can be
detected colorimetrically and used to determine the effects of
drugs on cells. To perform MTT assays, cells were seeded in 96-well
plates at a density of 104 cells/well and incubated for
24 h. EGF-ETA (1–10,000 ng/ml) or cetuximab (100 or 1,000 ng/ml)
was introduced into the wells and the cells were incubated for a
further 48 h. Cell proliferation was measured by MTT assays as
described previously (14). The
absorbance of the samples was quantified by using a
spectrophotometer (POLARstar Omega; BMG Labtech, Ortenberg,
Germany). Absorbance readings were transformed into percentage of
proliferation relative to the control PBS group.
Binding specificity assays
To determine the affinity of EGF-ETA towards the EGF
receptor, an anti-EGF monoclonal antibody (cat. no. ab10409, 100
ng/ml) (Abcam, Cambridge, MA, USA) was used to block the binding of
EGF-ETA to EGFR. Briefly, 104 A431 cells were seeded in
a 96-well plate and incubated overnight. The cells were treated
according to the following regimes: PBS, 100 ng/ml EGF-ETA, 100
ng/ml anti-EGF mAB, and 100 ng/ml EGF-ETA pre-incubated with 100
ng/ml of anti-EGF mAB for 1 h. Treated cells were incubated for 48
h and MTT assays were used to determine cell proliferation.
Western blot analysis
The total soluble protein was extracted from cells
using a RIPA cell extraction buffer (Invitrogen; Thermo Fisher
Scientific, Inc.) following the instructions provided. Equal
amounts of proteins (25 µg) were run on 8–16% Mini-PROTEAN TGX
Precast Protein Gels (Bio-Rad Laboratories, Gladesville, NSW,
Australia) and subsequently transferred to polyvinylidene fluoride
(PDVF) membranes for immunoblot detection. The primary antibodies
used were human anti-EGFR (1:500 dilution; cat. no. ab131498;
Abcam) and loading control human anti-GAPDH (1:2,500 dilution; cat.
no. ab9485; Abcam). The secondary antibody used was mouse
anti-rabbit IgG HRP-linked (1:5,000 dilution; cat. no. ab99697;
Abcam). The bands that reacted were detected using the Pierce ECL
chemiluminescent detection kit (Thermo Fisher Scientific, Inc.).
Imaging and analysis were performed with the VersaDoc MP 4000
imaging system (Bio-Rad Laboratories).
Real-time quantitative PCR
Total RNA was extracted from cancer cells using
TRIzol reagent according to the instructions provided (Invitrogen;
Thermo Fisher Scientific, Inc.) and quantified using a NanoDrop
2000 spectrophotometer (Thermo Fisher Scientific, Inc.). Total RNA
(1 µg) was converted to cDNA using the Superscript III reverse
transcription kit (Invitrogen; Thermo Fisher Scientific, Inc.)
following the manufacturer's instructions. The primers used were
human EGFR (NM_005228, HP208404) and human control GAPDH
(NM_002046, HP205798) (OriGene Technologies, Inc., Beijing, China).
Quantitative PCR reactions were carried out with iQ SYBR Green
Supermix according to the instructions provided (Bio-Rad
Laboratories). The Bio-Rad iQ5 cycler was used for the
quantification and analysis of the PCR reactions.
Statistical analysis
ANOVA with Tukey's multiple comparison test was used
to determine the significance between the treatment groups. All
statistical tests were conducted using the statistical programme
GraphPad Prism version 6 (GraphPad Software, Inc., La Jolla, CA,
USA). P<0.05 was considered to indicate a statistically
significant difference between groups. All experiments were in
triplicate, and the results are shown as means with standard
errors.
Results
Confirmation of EGFR expression in
cancer cells
Western blot analysis showed that head and neck
cancer HN5 cells and melanoma A431 cells showed a significantly
higher expression of EGFR. Furthermore, the non-small cell lung
cancer cell line A549 was found to express a significantly low
level of EGFR compared to the high expressing lines, while EGFR
expression in the breast cancer cell line MCF-7 was completely
absent (Fig. 1A). Real-time PCR
analysis of the mRNA expression of EGFR was consistent with the
western blot results, except for MCF-7 transcripts which were
slightly higher compared to A549 (Fig.
1B).
Construction and expression of
EGF-ETA
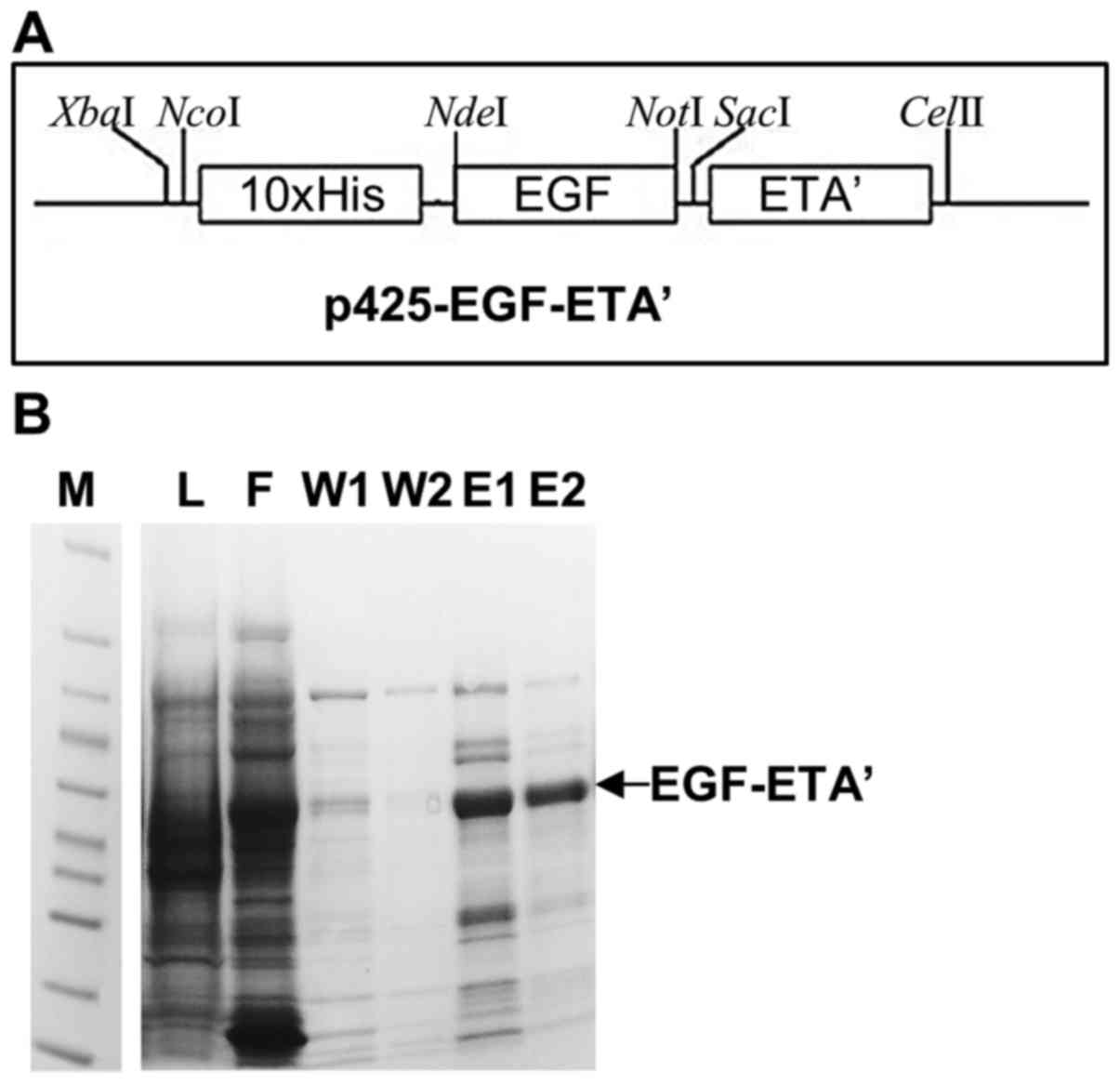
Fig. 2A shows the
map of the EGF-ETA constructed in the p425-ScFv-ETA backbone. This
plasmid allows for the expression of EGF-ETA with an N-terminal
10×His-tag for easy protein purification. Therefore, EGF-ETA was
purified by the immobilized metal ion affinity chromatography
(IMAC) using the 10×His-tag (Fig.
2A). SDS-PAGE analysis of the purified protein resolved a
protein of the predicted size of 47 kDa (Fig. 2B). A total of 5 mg of protein was
isolated and subsequently used in the in vitro cell toxicity
assays.
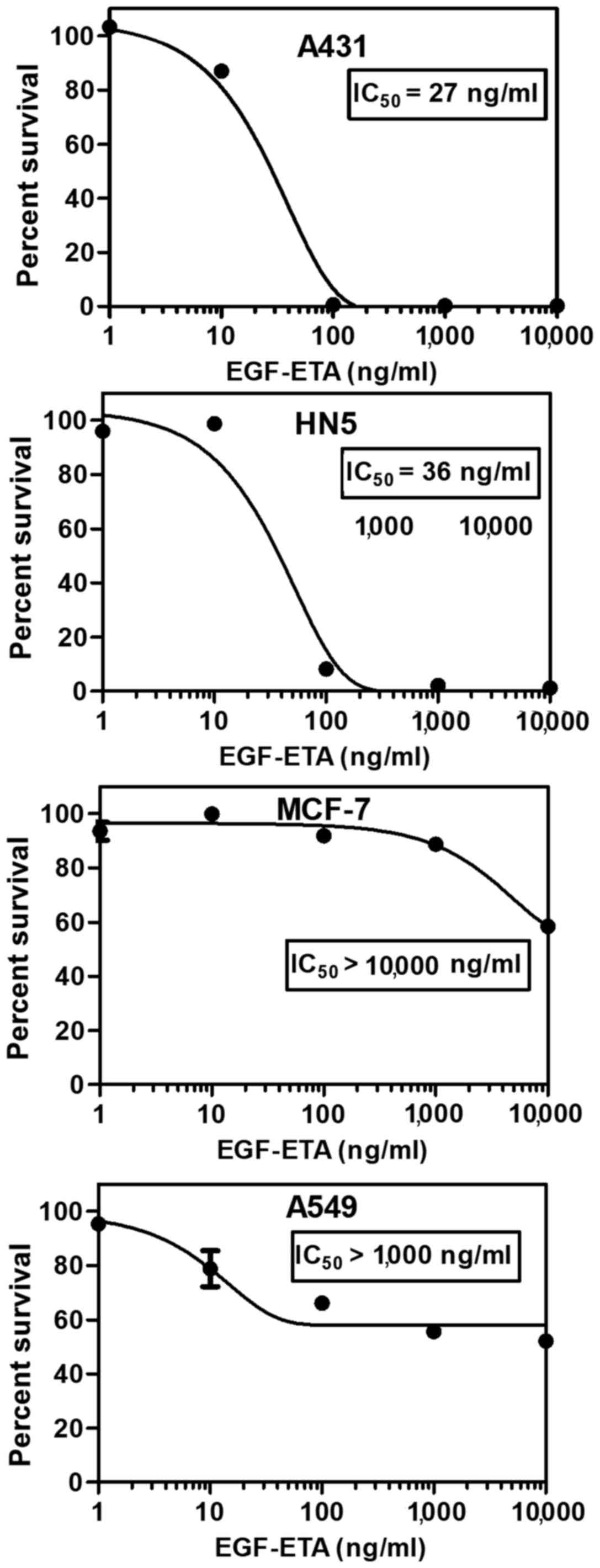
EGF-ETA inhibits the proliferation of
EGFR-expressing cells
The toxicity of purified EGF-ETA was tested using
MTT proliferation assays (14).
Living cells convert MTT to formazan, which can be measured
spectrophotometrically. EGF-ETA was added to the cultured cells in
the range of 0 to 10,000 ng/ml to provide a dose response for
calculating the 50% inhibitory concentration (IC50)
(Fig. 3). PBS containing 20%
glycerol was used as a control and DMEM media served as a blank for
the MTT assay. EGF-ETA was found to trigger rapid cell death in HN5
and A431 cancer cells with an IC50 of 26–37 ng/ml
(Fig. 3). However, A549 cancer
cells exhibited low sensitivity towards EGF-ETA (IC50
>1,000 ng/ml), while the EGFR-negative cell line MCF-7 was the
least responsive (IC50 >10,000 ng/ml).
Anti-EGF antibody abrogates the
effects of EGF-ETA
To test the specific binding of EGF-ETA to the EGFR,
the immunotoxin was pre-incubated with an anti-EGF antibody prior
to its addition to HN5 cells. MTT analysis was used to determine
the effect of the immunotoxin on HN5 cells. It was found that the
pre-treated EGF-ETA with the anti-EGF antibody had little effect on
cell survival when compared to the cells that were exposed to
EGF-ETA alone which had a significant inhibitory effect (Fig. 4A). Furthermore, the EGF antibody
alone did not elicit cell death when compared with the PBS control.
Microscopic analysis confirmed the MTT results (Fig. 4B).
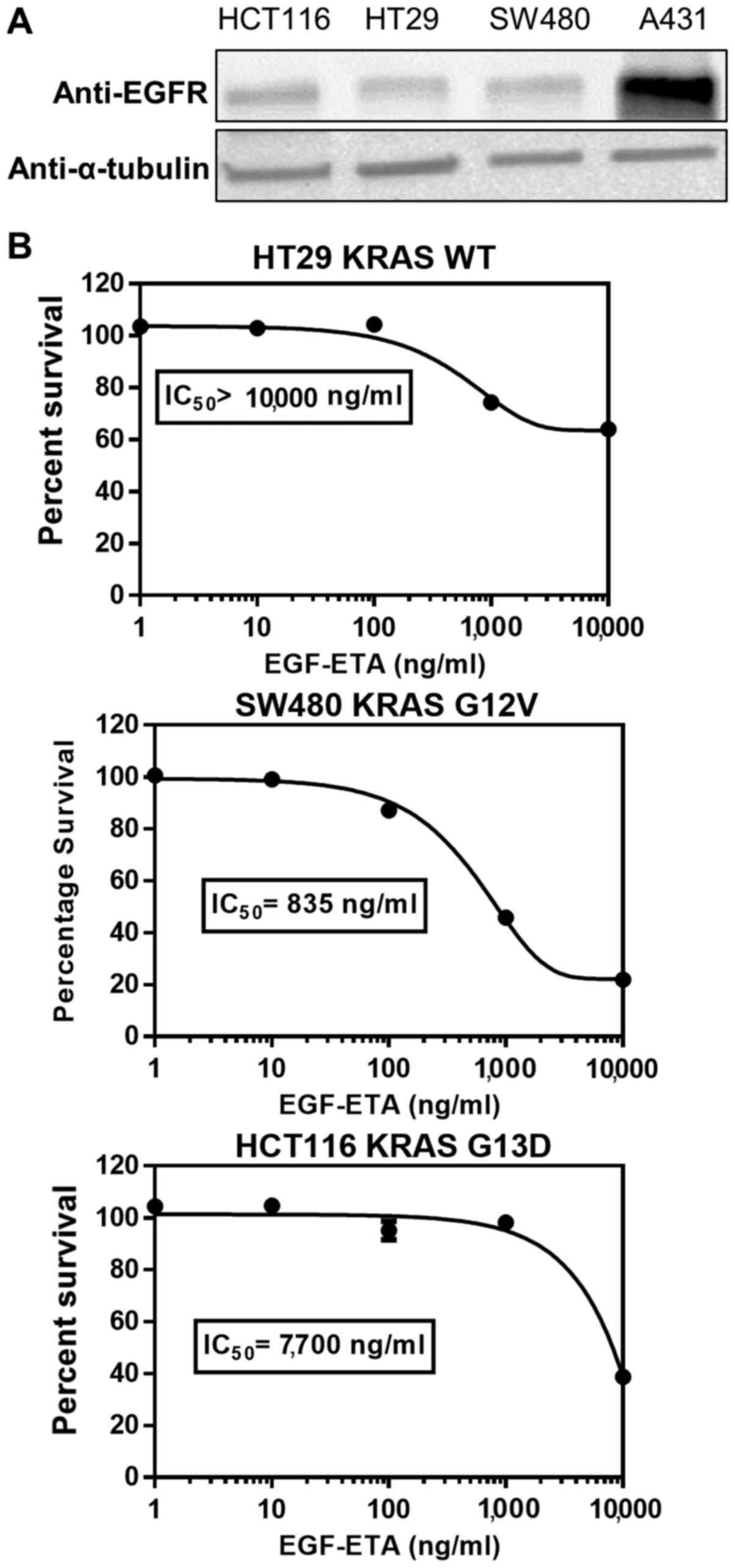
EGF-ETA inhibits the growth of cancer
cells harbouring KRAS mutations
To ascertain the effect of KRAS-hyperactivating
mutations on the efficacy of EGF-ETA, HCT116 (KRAS G13D) and SW480
(KRAS G12V) colorectal cancer cell lines were used. HT29 colorectal
cancer cells served as KRAS wild-type controls. Western blot
analysis showed that HCT116, SW480 and HT29 cells expressed similar
levels of EGFR, but at lower levels than A431 cells (Fig. 5A). EGF-ETA was most effective
against SW480 cells (IC50 835 ng/ml), while the
proliferation inhibitory effect on HT29 (IC50
>10,000) and HCT116 (IC50 7,700) was of a lesser
extent (Fig. 5B).
Efficacy of EGF-ETA vs. cetuximab
The efficacy of EGF-ETA was compared to the
FDA-approved monoclonal anti-EGFR antibody, cetuximab. Treatment of
EGFR-positive cells with EGF-ETA at concentrations 100 and 1,000
ng/ml reduced the viability of the cells to 30–40% in relation to
that of the PBS-treated cells (Fig.
6). However, when the cells were exposed to 100 ng/ml of
cetuximab, there was no significant change in the viability of the
cells. Increasing the dose of cetuximab to 1,000 ng/ml
significantly reduced the viability of the cells to 90% of that of
the PBS controls. Morphologically, cells treated with 1,000 ng/ml
of EGF-ETA were rounded and detached, while those exposed to 1,000
ng/ml of cetuximab were mostly attached to the culture plates.
Discussion
Cancerous cells arise from deregulated cellular
signalling pathways due to accumulated mutations in the genome.
Upregulation of EGFR leads to the activation of multiple branching
pathways including, but not limited to, the
Ras/Raf/mitogen-activated protein kinase, phosphatidylinositol
3-kinase/Akt and Src kinase pathways, leading to cell
proliferation, migration, adhesion, angiogenesis and survival
(15). Therefore, targeting the
EGFR provides an excellent opportunity to disrupt the survival and
progression of cancer cells. In the present study, we synthesised a
recombinant targeted chimera of EGF (ligand of EGFR) with a
truncated version of the well-known bacterial toxin (ETA) lacking
the cell binding domain (16).
The expression of EGFR was analysed in four
different cancer cell lines at the protein and gene level. Our data
confirmed the overexpression of EGFR in head and neck squamous
carcinoma HN5 cells and epidermoid carcinoma A431 cells (17), while non-small cell lung cancer A549
cells had minimal EGFR expression. The breast ductal carcinoma
MCF-7 cell line was found to be negative at the protein level,
while the transcript level was similar to A549.
Suppression of EGFR activity in cancers can occur
through two regimens. Tyrosine kinase inhibitors (TKIs) such as
gefitinib or erlotinib inhibit the catalytic domain of EGFR, while
monoclonal antibodies (mAb) such as cetuximab target the EGFR
extracellular domain, resulting in downregulation (18). These treatment regimens have been
found to be functionally restricted from being effective as a
result of the mutations developed in the EGFR signalling pathway
(19). The uniqueness of ETA is
that it ADP-ribosylates the eukaryotic elongation factor 2 (eEF-2)
of the host cells and as a consequence abrogates protein synthesis
(11). Thus, the targeted delivery
of ETA to the tumour microenvironment through EGF would selectively
target cancer cells harbouring increased levels of the EGFR.
Furthermore, the novelty of using EGF as the targeting moity is due
to its small size (6.2 kDa) and human origin (20). By contrast, single chain variable
fragments of monoclonal antibodies are of murine origin and are
significantly larger in size (26 kDa) (21).
Cell proliferation assays were performed to
ascertain the inhibitory effects of EGF-ETA on various cancer cells
overexpressing the EGFR. The EGFR-positive cells HN5 and A431 were
selectively inhibited by the chimeric EGF-ETA protein, indicating
its binding capacity and potency, while having little effect on
EGFR-negative MCF-7 cells. Furthermore, our results showed that
EGF-ETA was significantly more effective than cetuximab. Previous
studies have shown that when heparin-binding epidermal growth
factor (HBEGF) was fused with the plant toxin saporin (SAP), it was
effective in killing EGFR-positive cancer cells (22). Yang et al showed that a
diphtheria toxin-epidermal growth factor chimera inhibited urinary
bladder cancer cells in vitro and in vivo (23). Furthermore, Liu et al showed
that diphtheria toxin-epidermal growth factor inhibited
glioblastoma multiforme subcutaneous tumours in nude mice (24).
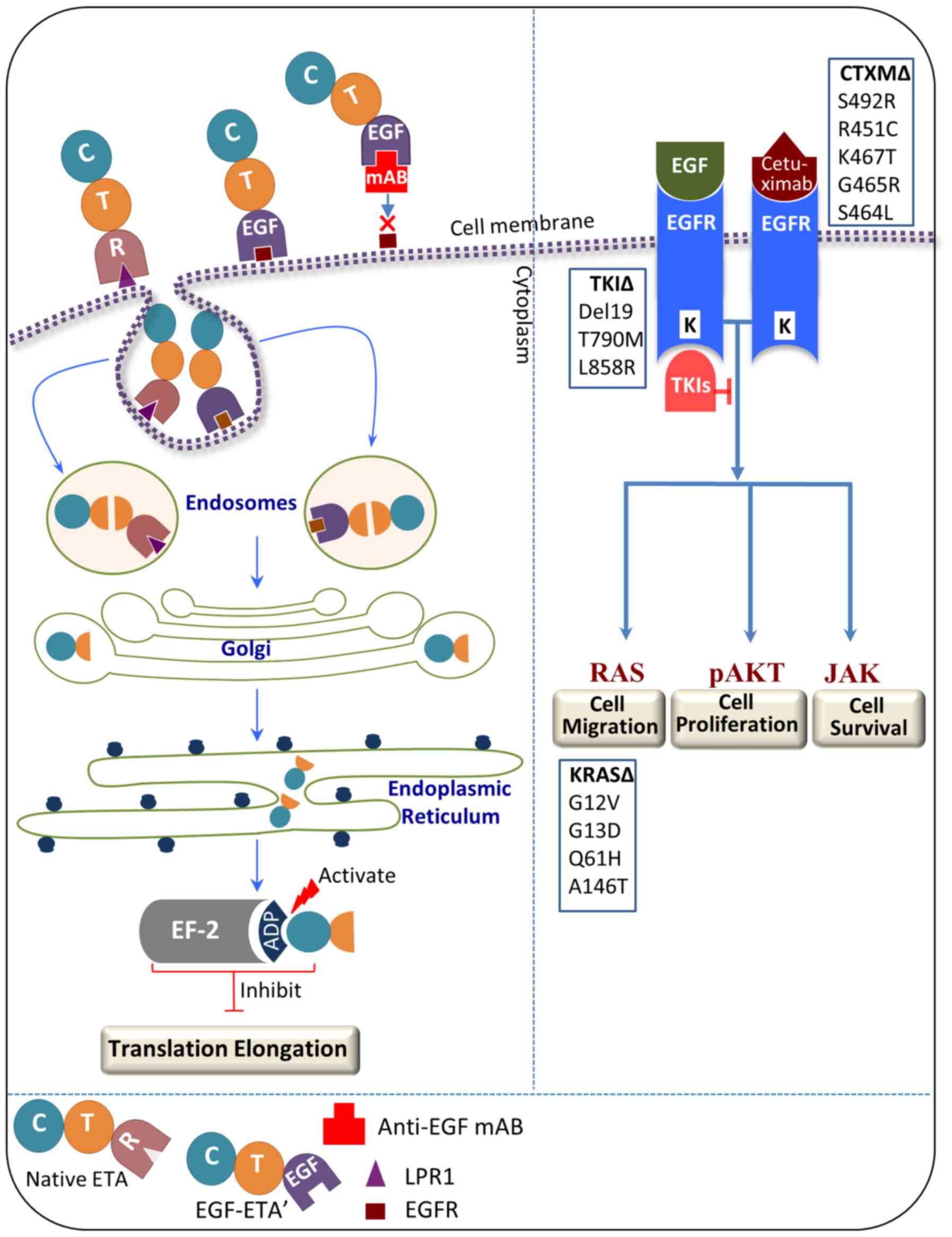
Antibody-mediated inference confirmed the binding
capacity of EGF-ETA. EGF-ETA pre-incubated with a monoclonal
anti-EGF antibody showed minimal inhibition of EGFR-positive cells,
indicating that EGF binding to the EGFR was critical. Since EGF-ETA
contains the translocation domain and ADP-ribosylation domain of
Pseudomonas aeruginosa exotoxin A (ETA), it is expected
that, upon binding to the EGFR, the chimeric protein will be
internalised and translocated to the cytoplasm, cleaved inside the
endosomes and transferred by the Golgi apparatus to the endoplasmic
reticulum (Fig. 7), subsequently
binding to elongation factor-2 (EF-2) and inhibiting protein
synthesis (16). By contrast, the
monoclonal antibody cetuximab targets the EGFR by competing for EGF
ligand binding, subsequent internalisation and downregulation
(18). However, recent studies have
found that colorectal cancer cells acquire resistance to cetuximab
due to mutations of the extracellular domain of EGFR at S492R,
R451C, K467T, G465R and S464L (5,6). The
presence of mutations in KRAS, a known downstream signalling
effector of EGFR, is a predictor for resistance to this antibody.
Thus, tumour cells can evade downregulation of EGFR by the
anti-EGFR antibody through constitutive activation of the KRAS
pathway (5). Notably, Arena et
al showed that EGF was not perturbed in binding the
cetuximab-resistant EGFR mutants as shown by EGFR phosphorylation
upon exposure to EGF (5). This is a
significant finding since the mechanism of action for EGF-ETA is
different to cetuximab, and binding of the EGF moiety is sufficient
for the action of EGF-ETA (Fig.
7).
Tumour cells have been shown to harbour resistance
against TKIs as a result of mutations in EGFR and/or its downstream
pathway effector, KRAS (25). A
study conducted on lung carcinoma cells has reported that KRAS is
the main reason for resistance to gefitinib and erlotinib (26). It has been claimed that KRAS
mutations which mediate TKI resistance in non-small cell lung
carcinoma are not targetable with the current treatment regimens
(27). By contrast, the cancer
killing capacity of EGF-ETA is not expected to be perturbed by
mutations in the KRAS pathway or in TKI-resistant EGFR mutants, as
only binding to EGFR is required (Fig.
7). Furthermore, our results show that EGF-ETA is effective in
hyperactivating KRAS mutations in HCTT116 (KRAS G13D) and SW480
(KRAS G12V) colorectal cancer cells when compared to HT29 wild-type
colorectal cancer cells.
Collectively, the findings of the present study
confirm that EGF specifically delivers the ETA toxin to
EGFR-positive cancer cells and has the potential to circumvent the
mechanisms which cause cetuximab and TKI resistance. Furthermore,
in vivo studies are required to ascertain the effective
killing capacity of EGF-ETA in the tumour microenvironment and
determine its ability to overcome cancer cells resistant to
anti-EGFR drugs.
Acknowledgements
We would like to thank the other members of the Wei
Laboratory for their support and helpful comments.
Funding
This research project was supported by the Deanship
of Scientific Research, Imam Abdulrahman Bin Faisal University
(project no. PYSS-224-2016).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
SMH conceived, designed and performed the
experiments. BG, NA and FA performed the experiments and
contributed to the writing of the manuscript. SMH and MQW reviewed
and edited the manuscript. MQW was also involved in the conception
of the study. All authors read and approved the manuscript and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lucas R and Keisari Y: Innovative cancer
treatments that augment radiotherapy or chemo-therapy by the use of
immunotherapy or gene therapy. Recent Pat Anticancer Drug Discov.
1:201–208. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Herbst RS: Review of epidermal growth
factor receptor biology. Int J Radiat Oncol Biol Phys. 59 Suppl
2:S21–S26. 2004. View Article : Google Scholar
|
|
3
|
Zhang H, Berezov A, Wang Q, Zhang G,
Drebin J, Murali R and Greene MI: ErbB receptors: From oncogenes to
targeted cancer therapies. J Clin Invest. 117:2051–2058. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Agelaki S and Georgoulias V: Epidermal
growth factor receptor inhibitors in the treatment of non-small
cell lung cancer. Expert Opin Emerg Drugs. 10:855–874. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Arena S, Bellosillo B, Siravegna G,
Martínez A, Cañadas I, Lazzari L, Ferruz N, Russo M, Misale S,
González I, et al: Emergence of multiple EGFR extracellular
mutations during cetuximab treatment in colorectal cancer. Clin
Cancer Res. 21:2157–2166. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Montagut C, Dalmases A, Bellosillo B,
Crespo M, Pairet S, Iglesias M, Salido M, Gallen M, Marsters S,
Tsai SP, et al: Identification of a mutation in the extracellular
domain of the epidermal growth factor receptor conferring cetuximab
resistance in colorectal cancer. Nat Med. 18:221–223. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pier GB, Boyer D, Preston M, Coleman FT,
Llosa N, Mueschenborn-Koglin S, Theilacker C, Goldenberg H, Uchin
J, Priebe GP, et al: Human monoclonal antibodies to Pseudomonas
aeruginosa alginate that protect against infection by both mucoid
and nonmucoid strains. J Immunol. 173:5671–5678. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yates SP, Taylor PL, Jørgensen R, Ferraris
D, Zhang J, Andersen GR and Merrill AR: Structure-function analysis
of water-soluble inhibitors of the catalytic domain of exotoxin A
from Pseudomonas aeruginosa. Biochem J. 385:667–675. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Barth S, Huhn M, Matthey B, Schnell R,
Tawadros S, Schinköthe T, Lorenzen J, Diehl V and Engert A:
Recombinant anti-CD25 immunotoxin RFT5(SCFV)-ETA' demonstrates
successful elimination of disseminated human Hodgkin lymphoma in
SCID mice. Int J Cancer. 86:718–724. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hashimi SM, Yu S, Alqurashi N, Ipe DS and
Wei MQ: Immunotoxin-mediated targeting of claudin-4 inhibits the
proliferation of cancer cells. Int J Oncol. 42:1911–1918. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wolf P and Elsasser-Beile U: Pseudomonas
exotoxin A: From virulence factor to anti-cancer agent. Int J Med
Microbiol. 299:161–176. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Phelan MC: Basic techniques in mammalian
cell tissue culture. Curr Protoc Cell Biol Chapter. 1:Unit 1.1.
2007.
|
|
13
|
Bruell D, Stöcker M, Huhn M, Redding N,
Küpper M, Schumacher P, Paetz A, Bruns CJ, Haisma HJ, Fischer R, et
al: The recombinant anti-EGF receptor immunotoxin 425(scFv)-ETA'
suppresses growth of a highly metastatic pancreatic carcinoma cell
line. Int J Oncol. 23:1179–1186. 2003.PubMed/NCBI
|
|
14
|
Mosmann T: Rapid colorimetric assay for
cellular growth and survival: Application to proliferation and
cytotoxicity assays. J Immunol Methods. 65:55–63. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Scaltriti M and Baselga J: The epidermal
growth factor receptor pathway: A model for targeted therapy. Clin
Cancer Res. 12:5268–5272. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Michalska M and Wolf P: Pseudomonas
Exotoxin A: Optimized by evolution for effective killing. Front
Microbiol. 6:9632015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kwok TT and Sutherland RM: Differences in
EGF related radiosensitisation of human squamous carcinoma cells
with high and low numbers of EGF receptors. Br J Cancer.
64:251–254. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Harari PM: Epidermal growth factor
receptor inhibition strategies in oncology. Endocr Relat Cancer.
11:689–708. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kuan CT, Wikstrand CJ and Bigner DD: EGF
mutant receptor vIII as a molecular target in cancer therapy.
Endocrine-Related Cancer. 8:83–96. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Carpenter G and Cohen S: Epidermal growth
factor. J Biol Chem. 265:7709–7712. 1990.PubMed/NCBI
|
|
21
|
Schmidt M, Vakalopoulou E, Schneider DW
and Wels W: Construction and functional characterization of
scFv(14E1)-ETA-a novel, highly potent antibody-toxin specific for
the EGF receptor. Br J Cancer. 75:1575–1584. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chandler LA, Sosnowski BA, McDonald JR,
Price JE, Aukerman SL, Baird A, Pierce GF and Houston LL: Targeting
tumor cells via EGF receptors: Selective toxicity of an HBEGF-toxin
fusion protein. Int J Cancer. 78:106–111. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang X, Kessler E, Su LJ, Thorburn A,
Frankel AE, Li Y, La Rosa FG, Shen J, Li CY, Varella-Garcia M, et
al: Diphtheria toxin-epidermal growth factor fusion protein
DAB389EGF for the treatment of bladder cancer. Clin Cancer Res.
19:148–157. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu TF, Hall PD, Cohen KA, Willingham MC,
Cai J, Thorburn A and Frankel AE: Interstitial diphtheria
toxin-epidermal growth factor fusion protein therapy produces
regressions of subcutaneous human glioblastoma multiforme tumors in
athymic nude mice. Clin Cancer Res. 11:329–334. 2005.PubMed/NCBI
|
|
25
|
Cree IA and Charlton P: Molecular chess?
Hallmarks of anti-cancer drug resistance. BMC Cancer. 17:102017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pao W, Wang TY, Riely GJ, Miller VA, Pan
Q, Ladanyi M, Zakowski MF, Heelan RT, Kris MG and Varmus HE:
KRAS mutations and primary resistance of lung
adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2:e172005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shea M, Costa DB and Rangachari D:
Management of advanced non-small cell lung cancers with known
mutations or rearrangements: Latest evidence and treatment
approaches. Ther Adv Respir Dis. 10:113–129. 2016. View Article : Google Scholar : PubMed/NCBI
|