Introduction
RFamide peptides comprise a family of neuropeptides
which are characterized by a common carboxy-terminal motif
consisting of an arginine (R) and an amidated phenylalanine (F)
(1). Vertebrate RFamides are
categorized into five groups, namely i) the neuropeptide FF (NPFF);
ii) the prolactin-releasing peptide (PrRP); iii) the
gonadotropin-inhibitory hormone (GnIH); iv) the kisspeptin (also
known as metastin); and v) the 26RFa/QRFP group (2,3). The
latter was initially identified in frog brain (4), with the N-extended longest form of the
glutamine RF-amide peptide (QRFP) consisting of 43 amino acids,
while due to several processing sites of this peptide a 26 (26RFa),
6 (26RFa20-26) and 9 (9RFa) amino acid form can also be produced
(4–7). QRFP has been identified as the cognate
ligand of the previously identified human orphan G protein-coupled
receptor (GPCR) GPR103 (8,9). Notably, GPR103 shares 48 and 47%
protein sequence homology with the two orexin receptors, OX1R and
OX2R, respectively (8).
In the human brain, the QRFP gene has been found to
be almost exclusively expressed in certain hypothalamic
areas/nuclei, such as the lateral hypothalamic area (LHA), the
ventromedial hypothalamic nucleus (VMH), the arcuate nucleus (Arc)
and the paraventricular nucleus (PVN), which are involved in the
regulation of the feeding behaviour (5,7).
Outside the central nervous system (CNS), in humans, QRFP is also
expressed in various endocrine glands (e.g., in the testis and the
adrenal, thyroid and parathyroid glands), as well as in the
prostate gland, where its expression is higher compared to that in
the hypothalamus (7,8,10).
According to its widespread expression, QRFP appears to be
implicated in a number of biological functions/systems, including
the regulation of feeding behaviour (11) and the control of the gonadotropic
axis (12,13). Notably, the central administration
of QRFP in mice has been demonstrated to result in increased
arterial blood pressure (BP) and heart rate (HR), as well as in
increased stress activity levels based on grooming behaviour
(6).
In addition, 26RFa has been shown to be expressed in
human prostate cancer and to stimulate the neuroendocrine
differentiation and migration of androgen-independent DU145
prostate cancer cells (14).
Overall, prostate cancer is the fourth most common cancer globally,
whilst it constitutes a leading cause of cancer-related mortality
and the second most frequently diagnosed cancer among men,
following only lung cancer (15,16).
More than 1.1 million new prostate cancer cases were diagnosed
worldwide in 2012 (17), whilst the
global burden of prostate cancer is expected to keep increasing in
the next decades in parallel to the increasingly ageing population
(18). Notably, androgen
deprivation/ablation therapy is the mainstay treatment option for
advanced prostate cancer, which is initially effective in slowing
the disease progression, since androgens stimulate prostate cancer
growth (19). However, prostate
cancer often progresses eventually to an androgen-independent state
which is characterized by a poor prognosis (19).
Given the existing evidence indicating that 26RFa
and GPR103 are present in prostate carcinomatous foci which exhibit
neuroendocrine differentiation (14), in the present study, we aimed to
further explore the role of both QRFP and GPR103 in prostate
carcinogenesis by studying their expression in human prostate
cancer samples and using two androgen-independent prostate cancer
cells lines (PC3 and DU145) as in vitro experimental
models.
Materials and methods
Prostate cancer cell lines
cultures
The human androgen-independent prostate cancer cell
lines, DU145 and PC3, were purchased from the American Type Culture
Collection (ATCC, Manassas, VA, USA) and were cultured in 75
cm2 cell culture flasks, in Ham's F12 (Sigma-Aldrich,
Gillingham, UK) and RPMI-1640 media (Invitrogen, Paisley, UK),
respectively, supplemented with 10% fetal calf serum (FCS) (Thermo
Fisher Scientific, Loughborough UK) and 5 ml of 100X
antibiotic-antimycotic (Thermo Fisher Scientific). All flasks were
incubated in a humidified incubator at 37°C in 5% CO2,
and were routinely passaged at approximately 70–80% confluency. Of
note, although the PC3 cell line has been assigned twice under NCBI
(catalogue number C427), this does not affect the outcomes of our
study.
In vitro treatments
For the phosphorylation analyses, both the PC3 and
DU145 cell lines were treated with QRFP (Phoenix Peptides,
Burlingame, CA, USA; 100 nM) for up to 60 min at the following
time-points: 0 (no supplement), 5, 15, 30 and 60 min. For the cell
invasion assays, cells were treated with QRFP for 8 h at 1, 10 and
100 nM. Epidermal growth factor (EGF; Sigma-Aldrich), at 50 ng/ml
was also used as a positive control. For the same experiment, we
used the PI3K inhibitor, LY294002 (Sigma-Aldrich), and the MAPK
inhibitor, U0126 (Sigma-Aldrich), both at 10 mM, in the presence or
absence of QRFP. For the effects of adipokines, the PC3 cells were
treated with leptin (100 nM), adiponectin (10 nM), and chemerin (1
nM) (R&D Systems, Abingdon, UK) for 24 h prior to assessing the
levels of QRFP and GP103 by RT-qPCR. The same concentrations of
LY294002 and U0126 were used.
Prostate tissue samples
Human prostate tissue samples (benign prostatic
hyperplasia, n=5 and malignant, n=5) were obtained from men
undergoing various prostate procedures, such as radical retro-pubic
prostatectomy (RRP), transurethral prostate resection (TURP) or
transrectal ultrasound (TRUS) and prostate biopsy, at the
University Hospitals Coventry and Warwickshire (UHCW) NHS Trust
(date range of recruitment/sample collection: November, 2010 to
October, 2015). The present study was approved by the National
Research Ethics Committee and the Research and Development
department of the UHCW NHS Trust (16/11/2010-1/10/2015) and was
conducted according to the principles of good clinical practice and
the recommendations of the Declaration of Helsinki. Men undergoing
prostate surgery for either benign or malignant conditions or
undergoing a prostate biopsy for suspected cancer either with an
elevated age-specific PSA or with an abnormal prostate
investigation were approached for potential inclusion into the
study. Patients were presented with a full explanation of the
nature of the study, including the potential benefits and risks of
taking part. All patients recruited into the study provided
informed signed consent. All collected prostate tissue samples for
the present study were immediately snap-frozen in liquid nitrogen
and stored at −80°C until use. Radical prostatectomy specimens were
removed en bloc and were formalin-fixed, paraffin-embedded
as per standard hospital practice. The tissue samples were
available for use in the present study after the hospital
pathologist had issued a final pathology report on each specimen
for grading/staging purposes.
Western blot analysis
Cell and tissue lysates were extracted using RIPA
cell lysis buffer (Sigma-Aldrich) and a cell scraper or a
homogeniser respectively, according to the manufacturers'
guidelines. Protein concentrations were determined calorimetrically
using the bicinchoninic acid (BCA) protein assay kit according to
manufacturer's instructions (Sigma-Aldrich). Samples were
subsequently prepared for gel electrophoresis prepared by the
addition of 2X Laemmli buffer (Sigma-Aldrich), and boiled for 5
min. The proteins were separated by SDS-PAGE 8–10%, and transferred
to polyvinylidene difluoride (PVDF) membranes at 100 V for 1 h in a
transfer buffer containing 20 mM Tris, 150 mM glycine and 20%
methanol. PVDF membranes were blocked in Tris-buffered saline (TBS)
containing 0.1% Tween-20 and 5% BSA for 1 or 2 h and were incubated
with the relevant anti-rabbit primary antibodies overnight at 4°C.
On the following day, these membranes were washed thoroughly four
times in 60 min with TBS-0.1% Tween, before incubation with the
appropriate secondary anti-rabbit antibody buffer (1:2,000
dilution; Sigma-Aldrich), for 1 h at room temperature. The
antibodies (all anti-rabbit) used were: GPR103 (1:3,000 dilution;
Acris Antibodies, Herford, Germany), QRFP-43 (1:4,000 dilution;
Phoenix Pharmaceuticals, Belmont, CA), ERK1/2, p38, JNK, AKT (all
1:1,000 dilution; both phospho and total; Cell Signaling
Technology, Leiden, Netherlands), MMP2 (1:1,000 dilution) and GAPDH
(1:5,000 dilution; Sigma-Aldrich, Gillingham, UK) at concentrations
recommended by the manufacturers. Antibody complexes were
visualized using ECL-Plus (Thermo Fisher Scientific), according to
the manufacturer's instructions. The appropriate positive and
negative controls were used. All densities were determined using a
scanning densitometer coupled to scanning ImageQuant™ software 7.0
(GE Healthcare Life Sciences, Little Chalfont, UK).
RNA isolation, cDNA synthesis and
reverse transcription- quantitative PCR (RT-qPCR)
Total RNA was extracted from the human prostate
cancer tissue samples and cell lines using the Qiagen RNeasy plus
Mini kit (Qiagen, Manchester, UK). RNA samples were then treated by
RNase-free DNase to eliminate genomic DNA contamination. The
extracted RNA purity and quantity was assessed by a NanoDrop
spectrophotometer (Thermo Fisher Scientific). In addition, 1
µg of total RNA was reverse transcribed into cDNA, by using
Moloney Murine Leukemia Virus (M-MuLV) Reverse Transcriptase and
random hexamers primers (Thermo Fisher Scientific).
The relative expression of the genes of interest was
assessed by quantitative polymerase chain reaction (qPCR) on an
ABI7500 instrument (Applied Biosystems; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) using SYBR®−Green-PCR reaction
mixture (Sigma Aldrich; Merck KGaA, Darmstadt, Germany). The
primers used in the present study were as follows: for QRFP sense,
5′-AGGCAGGACGAAGGCAGTGA-3′ and antisense,
5′-GACCGAAGCGGAAGCTGAAG-3′; for GPR103 sense,
5′-CCAGTCTACCGCTGTTGTGA-3′ and antisense 5′-GCCAGACCACACCTAGCATT-3′
and; for GAPDH (as a reference gene) sense 5′-TGCACCACCAACTGCTTA-3′
and antisense, 5′-GATGCAGGGATGATGTTC-3′. The thermocycling
conditions were as follows: Hotstart 95°C for 10 min (1 cycle);
amplification at 95°C for 15 sec followed by at 60°C for 60 sec (40
cycles) and dissociation curve at 60–95°C for 1 min (1 cycle).
Negative controls for all the reactions included preparations
lacking cDNA or RNA-lacking reverse transcriptase in place of the
cDNA. RNAs were assayed from two to three independent biological
replicates. RNA levels were expressed as a ‘relative
quantification’ using the housekeeping gene GAPDH value. The
2−ΔCt method was employed for comparing relative
expression results between treatments in qPCR (20).
xCELLigence migration and invasion
assays
Real-time cell proliferation, migration and invasion
experiments were performed using the xCELLigence system (ACEA
Biosciences, San Diego, CA, USA), consisting of the Real-Time Cell
Analyzer Dual Purpose (RTCA-DP) instrument placed in a humidified
incubator maintained at 5% CO2 and 37°C, and cell
invasion and migration (CIM) plates for cell migration and invasion
according to the manufacturer's protocol. The RTCA software 1.2
(Roche, Basel, Switzerland) monitored cell proliferation, reporting
changes in the Cell Index (CI) following the treatment of the
cells.
Statistical analysis
All results presented are expressed as the mean ±
SD. A Student's t-test was used to difference between 2 groups.
Comparisons between more than 2 groups were analysed by ANOVA
(non-parametric) analysis followed by post hoc Tukey's multiple
comparison test. Values were considered to be statistical
significance set at either P<0.05, P<0.01 or P<0.001. All
statistical analyses were performed using Graph Pad software 5.0
(La Jolla, CA, USA).
Results
Expression of QRFP and GPR103 in human
prostate cancer clinical samples and cell lines
QRFP and GPR103 mRNA levels in human prostate tissue
samples were determined by RT-qPCR, which revealed that the gene
expression of both QRFP and GPR103 was significantly higher in the
prostate cancer samples (n=5) compared to the samples from benign
prostate hyperplasia patients (BPH; n=5) (Fig. 1A). Furthermore, this statistically
significant difference was also detected at the protein level, with
both QRFP and GPR103 being significantly upregulated in prostate
cancer tissue samples compared to BPH (Fig. 1B). Using human prostate cancer
tissue lysate from one of the patients described above as a
positive control, we also revealed that both the
androgen-insensitive prostate cancer cell lines (PC3 and DU145),
which we used for in vitro experiments in the present study,
expressed QRFP and GPR103 at the protein level (Fig. 1C).
Effects of QRFP on the phosphorylation
status of prostate cancer cell lines
To examine the signalling pathways which may be
involved in the effects of QRFP on prostate cancer cells, the
phosphorylation status of ERK1/2, p38, JNK and Akt was assessed in
the two androgen-insensitive prostate cancer cell lines of the
present study following QRFP treatment. The PC3 and DU145 cells
were treated with QRFP (100 nM) for up to 60 min (0, 5, 15, 30 and
60 min time-points). This QRFP treatment resulted in the
statistically significant activation of ERK1/2 in the treated PC3
and DU145 cells at 5 min (P<0.01 and P<0.001, respectively)
(Fig. 2A and B). In addition, the
significantly increased p38 phosphorylation was observed at
different time-points in these two cell lines. In PC3 cells, the
significant activation of p38 was noted at 30 min (P<0.01), as
well as at 15 and 60 min (P<0.05), compared to the basal levels.
In DU145 cells, maximal activation of p38 was reached at 60 min
(P<0.01) compared to basal levels (Fig. 2C and D). Furthermore, maximal JNK
phosphorylation occurred at 30–60 min in PC3 cells (P<0.05;
Fig. 2E), whilst in DU145 cells, a
biphasic response was noted at 5 and 60 min (P<0.01 and
P<0.05, respectively; Fig. 2F).
Finally, Akt phosphorylation increased significantly in PC3 cells
from 15 to 60 min of QRFP treatment (P<0.01; Fig. 2G), whilst a similar trend was also
noted in the QRFP-treated DU145 cells, with maximal Akt
phosphorylation at 30 and 60 min (P<0.05 and P<0.01,
respectively; Fig. 2H).
 | Figure 2.Representative western blots and
quantitative analysis of signalling pathway-related molecules in
androgen-independent (A, C, E and G) PC3 and (B, D, F and H) DU145
prostate cancer cells, demonstrating significantly increased
phosphorylation status of (A and B) ERK1/2, (C and D) p38, (E and
F) JNK, and (G and H) Akt. Cells were treated for 5, 15, 30 and 60
min with 100 nM QRFP (***P<0.001, **P<0.01, *P<0.05). |
To demonstrate the specificity of these responses,
we also employed a siRNA approach for the GPR103 receptor, using
the PC3 cells and Akt phosphorylation as our experimental paradigm.
Following siRNA transfection, the GPR103 mRNA levels significantly
decreased in the transfected PC3 cells with increasing
concentrations of siRNA, with the maximum reduction observed at the
10 nM siRNA concentration. Furthermore, QRFP-induced Akt
phosphorylation was significantly decreased in the PC3 cells
transfected with GPR103 siRNA compared to the control PC3 cells
(data not shown).
Effects of QRFP on cell migration and
invasion in prostate cancer cell lines
Following the effects of QRFP treatment on the
phosphorylation of key kinases in the two prostate cancer cell
lines in our study, we hypothesised that this would further affect
cell migration and invasion. To explore such effects, we used the
xCELLigence system, and EGF 50 ng/ml as a positive control.
Our experiments revealed that there was a
significant and concentration-dependent increase in cell migration
(10 nM, P<0.05; and 100 nM, P<0.01, compared to basal) in
QRFP-treated PC3 cells at 8 h of treatment (Fig. 3A). In DU145 cells statistical
significance compared to basal levels was reached only at the
highest QRFP concentration (100 nM; P<0.01; Fig. 3C). In both cell lines, only a PI3K
inhibitor (LY294002; 10 mM) significantly inhibited the effect of
the applied QRFP treatment. The use of a MAPK inhibitor (U0126; 10
mM) revealed a similar trend; however its effects did not reach to
statistical significance in either PC3 (Fig. 3B) or DU145 (Fig. 3D) QRFP-treated cells.
Similarly, 8 h of QRFP treatment significantly
increased the invasive ability of PC3 cells (10 nM; P<0.05 and
100 nM; P<0.01, compared to basal; Fig. 4A). In DU145 cells, a significant
effect on invasion was noted only at the highest used QRFP
concentration (100 nM; P<0.05, compared to basal; Fig. 4B). Despite the effects of QRFP on
cell migration and invasion, there was no apparent effect on
overall cell proliferation in either cell line (data not
shown).
Since there is strong evidence of the role of matrix
metalloproteinases (MMPs) in the remodelling, including
angiogenesis, of the extracellular matrix (ECM), in the present
study, we also tested the hypothesis that MMP2 is involved in the
effects of cellular invasion. The PC3 and DUP145 cells were
incubated with 100 nM QRFP for up to 12 h and the expression of
MMP2 was assessed at regular time-points (1, 2, 4, 6, 10 and 12 h)
by western blotting. In PC3 cells, QRFP treatment significantly
induced MMP2 protein expression at 1 to 6 h (all P-values <0.05,
compared to basal; Fig. 4C). In
QRFP-treated DUP145 cells, the highest significant increase was
noted at 1 h (P<0.01), whilst significantly increased levels
were also documented at 4 to 10 h (all P-values <0.05, compared
to basal; Fig. 4D).
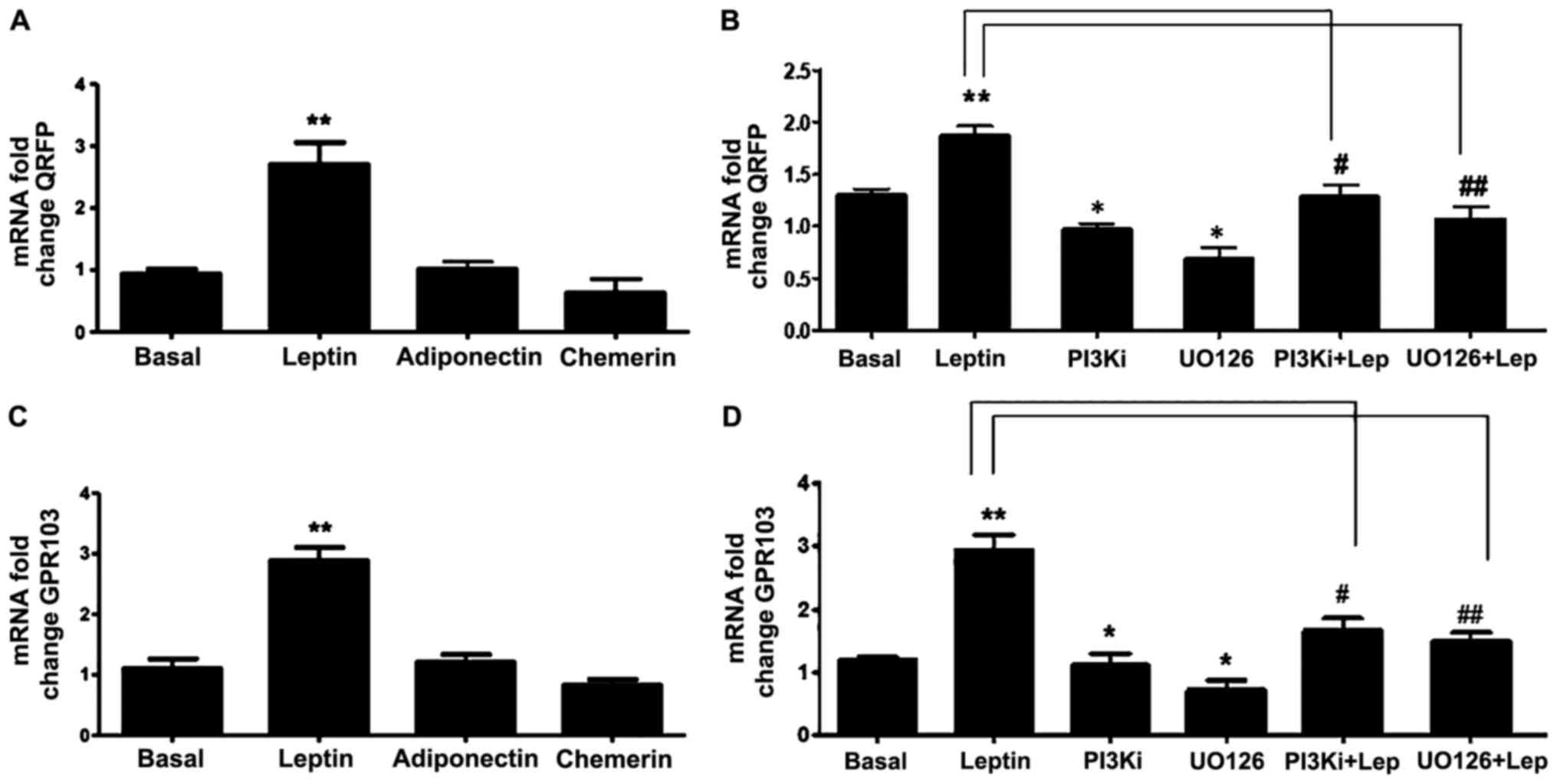
Effects of leptin on QRFP and GPR103
gene expression in PC3 prostate cancer cells
Several studies have revealed an association between
adipokines with prostate cancer progression. Thus, in this study,
we also investigated whether QRFP and GPR103 gene expression in PC3
cells is regulated by certain key adipokines, namely leptin,
adiponectin, and chemerin. Based on these experiments, only leptin
significantly induced the expression of QRFP and GPR103 (Fig. 5A and C). This leptin-induced effect
was time-dependent with a significant increase at 12 and 24 h (data
not shown). Using specific inhibitors of PI3K (LY294002) and MAPK
(U0126), we were also able to demonstrate that this leptin-induced
effect was mediated via the PI3K and MAPK pathways (Fig. 5B and D).
Discussion
In the present study, we revealed that QRFP and its
cognate receptor GPR103 were expressed in two androgen-independent
human prostate cancer cell lines (PC3 and DU145), and that their
expression was upregulated in human prostate cancer tissue samples
compared to that in samples from benign prostate hyperplasia
patients. These findings were in line with the data from Alonzeau
et al (14), indicating that
26RFa was expressed in human prostate cancer, stimulating the
neuroendocrine differentiation and migration of the DU145 cells.
Based on these findings, we could not exclude the autocrine effects
of this peptide, along with QRFP acting in an endocrine manner.
Our data also revealed that QRFP induced the
phosphorylation of ERK1/2, p38, JNK and Akt in both prostate cancer
cell lines used in the present study. It is already known that the
MAPK regulates different cellular processes in prostate cancer, and
that this signalling pathway is overexpressed in human prostate
cancer compared to normal prostate tissue (21,22).
Autocrine and paracrine-acting growth factors can induce the
increased expression of the Ras/Raf/MEK/ERK pathway, which has been
associated with progressive prostate cancer (21). Furthermore, Ras signalling has been
implicated in cancer cell invasion and metastasis, whilst the
activation of the EGFR-ERK1/2 pathway promotes the migration and
invasion of prostate cancer cells (23). Conversely, there are data indicate
the activated ERK-dependent apoptosis of prostate cancer cell lines
(24). It should also be noted
that, Ras/Raf/MEK/ERK levels are differentially expressed amongst
cancers (25). Hence, while it is
plausible that the activation of the Ras/Raf/MEK/ERK cascade could
contribute to prostate cancer development, further research is
required to elucidate these mechanisms in androgen-independent
prostate cancer lines which exhibit low expression levels of this
cascade.
Unlike ERK1/2, p38 exhibits weak activation by
mitogens, although it is strongly activated in response to various
stressors, including inflammatory cytokines, UV radiation, and
osmotic and heat shock (26).
Notably, TNFα (a pro-inflammatory cytokine which is known to
activate the MAPK stress response) has been shown to induce the
apoptosis of the androgen-dependent LNCaP prostatic cancer cell
line, but not that of androgen-independent PC3 cells, while p38
appears to exert protective effects against this TNFα-induced
apoptosis of LNCaP cells (27). Of
note, the activation of p38 in prostate cancer may be a result of
upregulated upstream kinases (MKK3/6) combined with downregulated
MAPK phosphatases (28–30). Furthermore, p38 phosphorylation has
been demonstrated in prostate cancer cell lines exposed to toxic
agents, and this activation has been implicated in apoptosis
(31,32). Furthermore, it has been demonstrated
that prostate cancer cell invasion was mediated via the p38 MAPK
pathway, leading to phosphorylation of the heat shock protein 27
(HSP27) that in turn regulated MMP2 activation and cell invasion
(33). In addition, Chen et
al (34) revealed that the
stimulation of the G protein-coupled P2Y purinoceptor (metabotropic
GPCR family) can induce prostate cancer cell invasion, which was
regulated via the activation of the p38 pathway (34). It becomes evident that the
activation of p38 plays a significant role in prostate cancer, and
additional research is clearly needed to further explore the exact
implications of the QRFP-induced p38 phosphorylation that was
observed in the two androgen-independent prostatic cancer cell
lines in the present study.
Furthermore, in the present study, we revealed the
QRFP-mediated the activation of the JNK pathway in
androgen-independent prostate cancer cells (PC3 and DU145). It is
known that JNK is activated by certain growth factors and stressors
(e.g., UV radiation) (35). In
turn, this JNK activation frequently results in cell death via the
activation of the mitochondrial apoptotic pathway in various cell
types, including prostate cancer PC3 and DU145 cells, where
JNK-initiated Fas-mediated apoptotic signals are considered to play
a significant role in chemosensitivity (35,36).
It has also been revealed that, depending on the cell type and
stimulus, JNK can activate several transcription factors (e.g.,
c-Jun, c-Fos, Elk-1, c-My, ATF-2 and p53), as well as various
members of the Bcl-2 family (37,38).
JNK appears to regulate apoptosis via two distinct mechanisms: i)
By promoting the phosphorylation of c-Jun and ATF-2 which results
in the activation of AP-1 and the expression of Fas/FasL signalling
pathway-related proteins, which further mediates the activation of
certain caspases (caspase-8 and −3) that trigger apoptosis; and ii)
by mediating the phosphorylation of the anti-apoptotic proteins,
Bcl-2/Bcl-xL, thus altering mitochondrial membrane potential and
resulting in the release of cytochrome c and the activation
of caspase-9 and −3 to induce apoptosis (39).
In the present study, we also observed a marked
promoting effect of QRFP on the activation of Akt signalling in
both human prostate cell lines used, with a higher degree of
phosphorylation in PC3 compared to DU145 cells. The latter finding
may be attributed to the higher expression of Akt in PC3 cells
(40). Using siRNA for the cognate
GPR103 receptor, we further demonstrated that the QRFP-induced
response was receptor-specific. Notably, the Akt signalling pathway
can be activated by several cytokines, growth factors and oncogenes
(41), whilst the phosphorylation
of PI3K/Akt may contribute to the induction of tumour invasiveness
and cancer development. The PI3K/Akt pathway activation has been
more frequently associated with prostate cancer progression toward
resistant/metastatic disease (42).
Therefore, it has been considered that the PI3K/Akt pathway plays a
role in the progression of prostate cancer, with the inhibition of
the Akt pathway significantly affecting the EGFR-induced prostate
cancer cell migration (43).
Collectively, the PI3K/Akt/mTOR signalling pathway
has been revealed to regulate multiple cellular processes, such as
cell survival, metabolism, proliferation, migration and
angiogenesis. Accordingly, the ERK and PI3K/Akt pathways are
critical for the regulation of cancer cell survival and
proliferation. Indeed, in prostate cancer, activated ERK and Akt
translocate to the nucleus, inducing various downstream effects
(e.g., cell proliferation, migration, invasion and angiogenesis)
(44). Furthermore, the activation
of ERK1/2 promotes cell migration and invasion in prostate cancer
cells (37,45). Of note, in this study, QRFP
significantly induced the migration and invasion of both PC3 and
DU145 cells. The inhibition of the PI3K/Akt pathway significantly
reduced the effects of QRFP (100 nM) on the migration of both PC3
and DU145 cells. On the other hand, the inhibition of MAPK/ERK with
U0126 inhibited only partially the effect of QRFP, without reaching
statistical significance. Although there was no effect of QRFP on
cell proliferation, in a recent study by Ljujic et al
(46), it was evident that the
addition of both inhibitors to PC3 cells on their own significantly
reduced cell viability. Similarly, in another study by Rybak et
al (47), using DU145 cells,
treatment with U0126 resulted in the reduction of cell propagation.
Therefore, we could not exclude the direct effects of these two
inhibitors. Future studies using a wider repertoire of inhibitors
should provide a better insight into the involvement of MAPK and
PI3K in these responses.
Notably the expression of MMPs is associated with
the migration, invasiveness and metastatic potential of prostate
cancer cell lines (48). Recent
data have also indicated a connection between the ERK1/2 signalling
activation and MMPs (49). Indeed,
the upregulated expression of MMPs has been linked to MAPK (ERK,
p38, JNK) and Akt pathways (50).
Furthermore, it has been also shown that the p38 MAPK pathway is
required for the TGF-β-mediated MMP2 induction and increased cell
invasion in prostate cancer (51).
In the present study, we focused primarily on MMP2, since an
increase in its expression has been reported in prostate cancer and
appears to correlate with a larger tumour size (52). A recent study indicated that the
expression of MMP2 was observed in metastatic cancer, but not in
micro-metastasis, strongly proposing that increased MMP2 expression
was related with prostate cancer development and metastasis
(53). However, we acknowledge that
activated MMP2 can activate other MMPs, such as MMP-9 through
enzymatic cleavage. Therefore, future studies are warranted to also
concentrate on other MMPs, as well as the use of gelatin zymography
to determine their effect.
It is plausible therefore, that the activation of
MMP2 and the subsequent induction of cell invasion involved the
MAPK/PI3K/Akt signalling pathways in accordance with the findings
of the cell migration and invasion experiments in the present
study. In the study by Ljujic et al, alpha-1-antitrypsin
antagonized cisplatin-induced cytotoxicity in PC3 cells, an effect
that was differentially mediated by ERK or Akt inhibitors (46). Furthermore, as displayed in Fig. 5, the effect of the inhibitors on
QRFP and GPR103 expression was similar in terms of leptin
inhibition, but in terms of cell migration, treatment with U0126
failed to reach statistical significance. It is possible therefore,
that overlapping and distinct pathways are operating in these cells
to perform specific functions. For example, there is an abundance
of evidence that mTOR signalling is implicated in prostate cancer.
Upstream of mTORC1, TSC1/TSC2 complexes can be phosphorylated by
either Akt or ERK1/2. Thus, there is a possibility of convergence
of those two signalling pathways at this point.
Finally, there are data indicating that obesity may
be associated with a higher risk of advanced/aggressive prostate
cancer, potentially through the effects of adipose-tissue derived
factors/hormones, collectively termed adipokines (e.g,. leptin
which constitutes the prototype adipokine) (54–57).
Although available data on leptin and the expression of leptin
receptor in human prostate and relevant prostate cell lines are
contradictory, the existing evidence indicates that this
pleiotropic, pro-inflammatory adipokine may exert varying effects
on prostate cancer at different stages of its progression (56,58).
Of note, it has been revealed that in the androgen-resistant PC3
and DU145 prostate cancer cell lines, leptin can increase cell
growth in a dose-dependent manner and induce ERK1/2 phosphorylation
and JNK activation, whereas these leptin-induced effects are less
prominent or absent in the androgen-sensitive LNCaP prostate cancer
cells (59,60). In the experiments of the present
study, only leptin, but neither adiponectin nor chemerin, was
demonstrated to significantly induce the gene expression of both
QRFP and its cognate GP103 receptor in the PC3 androgen-independent
prostate cancer cell line. Notably, recent data indicated that the
leptin-induced stimulatory effects on the proliferative activity of
prostate cancer cell lines depend on the expression of the variant
1 isoform of the leptin receptor (LEPR var 1; OB-R) (58). Previous data have also indicated
that JNK mediated the leptin-stimulated cell proliferation of
androgen-independent prostate cancer cells through STAT3 and Akt
(61). In our experiments, we
demonstrated that the inhibition of both the MAPK/ERK and PI3K/Akt
pathways significantly abated the leptin-induced effect on the
expression of QRFP and GPR103 in the androgen-independent PC3
prostate cancer cell line.
Collectively, the present study provided novel
insight into the effects of QRFP in human prostate cancer. Our
present findings also indicated that the adipokine, leptin,
modulated the expression of QRFP and GPR103 in an
androgen-independent human prostate cancer cell line via a PI3K-
and a MAPK-dependent mechanism, thus providing a potential link
between adiposity and prostate cancer.
Acknowledgements
Not applicable.
Funding
This study was funded by the UHCW NHS Trust and the
Libyan Embassy in the UK.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HSR and EK were involved in the conception and
design of the study. MABK was involved in the development of the
methodology. MABK, MR, KW and JJ were involved in the acquisition
of the data. MABK, HSR, EK and IK were involved in the analysis and
interpretation of the data. HSR, EK and IK were involved in the
writing, reviewing and/or revision of the manuscript. HSR
supervised the study. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the National
Research Ethics Committee and the hospital Research and Development
department and was conducted according to the principles of good
clinical practice and the recommendations of the Declaration of
Helsinki. All patients who were recruited in the study provided
informed written consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
GRFP
|
glutamine RF-amide peptide
|
|
GPR103
|
G protein-coupled receptor 103
|
|
GPCR
|
G protein-coupled receptor
|
References
|
1
|
Findeisen M, Rathmann D and Beck-Sickinger
AG: RFamide peptides: Structure, function, mechanisms and
pharmaceutical potential. Pharmaceuticals. 4:1248–1280. 2011.
View Article : Google Scholar :
|
|
2
|
Ukena K, Vaudry H, Leprince J and Tsutsui
K: Molecular evolution and functional characterization of the
orexigenic peptide 26RFa and its receptor in vertebrates. Cell
Tissue Res. 343:475–81. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sandvik GK, Hodne K, Haug TM, Okubo K and
Weltzien FA: RFamide peptides in early vertebrate development.
Front Endocrinol. 5:2032014. View Article : Google Scholar
|
|
4
|
Chartrel N, Dujardin C, Anouar Y, Leprince
J, Decker A, Clerens S, Do-Régo JC, Vandesande F, Llorens-Cortes C,
Costentin J, et al: Identification of 26RFa, a hypothalamic
neuropeptide of the RFamide peptide family with orexigenic
activity. Proc Natl Acad Sci USA. 100:15247–52. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bruzzone F, Lectez B, Tollemer H, Leprince
J, Dujardin C, Rachidi W, Chatenet D, Baroncini M, Beauvillain JC,
Vallarino M, et al: Anatomical distribution and biochemical
characterization of the novel RFamide peptide 26RFa in the human
hypothalamus and spinal cord. J Neurochem. 99:616–627. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Takayasu S, Sakurai T, Iwasaki S,
Teranishi H, Yamanaka A, Williams SC, Iguchi H, Kawasawa YI, Ikeda
Y, Sakakibara I, et al: A neuropeptide ligand of the G
protein-coupled receptor GPR103 regulates feeding, behavioral
arousal, and blood pressure in mice. Proc Natl Acad Sci USA.
103:7438–7443. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Leprince J, Bagnol D, Bureau R, Fukusumi
S, Granata R, Hinuma S, Larhammar D, Primeaux S, Sopkova-de
Oliveiras, Santos J, Tsutsui K, et al: The Arg-Phe-amide peptide
26RFa/glutamine RF-amide peptide and its receptor: IUPHAR Review
24. Br J Pharmacol. 174:3573–3607. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jiang Y, Luo L, Gustafson EL, Yadav D,
Laverty M, Murgolo N, Vassileva G, Zeng M, Laz TM, Behan J, et al:
Identification and characterization of a novel RF-amide peptide
ligand for orphan G-protein-coupled receptor SP9155. J Biol Chem.
278:27652–27657. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fukusumi S, Yoshida H, Fujii R, Maruyama
M, Komatsu H, Habata Y, Shintani Y, Hinuma S and Fujino M: A new
peptidic ligand and its receptor regulating adrenal function in
rats. J Biol Chem. 278:46387–46395. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Baribault H, Danao J, Gupte J, Yang L, Sun
B, Richards W and Tian H: The G-protein-coupled receptor GPR103
regulates bone formation. Mol Cell Biol. 26:709–717. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Moriya R, Sano H, Umeda T, Ito M,
Takahashi Y, Matsuda M, Ishihara A, Kanatani A and Iwaasa H:
RFamide peptide QRFP43 causes obesity with hyperphagia and reduced
thermogenesis in mice. Endocrinology. 147:2916–2922. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Navarro VM, Fernández-Fernández R,
Nogueiras R, Vigo E, Tovar S, Chartrel N, Le Marec O, Leprince J,
Aguilar E, Pinilla L, et al: Novel role of 26RFa, a hypothalamic
RFamide orexigenic peptide, as putative regulator of the
gonadotropic axis. J Physiol. 573:237–249. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Patel SR, Murphy KG, Thompson EL,
Patterson M, Curtis AE, Ghatei MA and Bloom SR: Pyroglutamylated
RFamide peptide 43 stimulates the hypothalamic-pituitary-gonadal
axis via gonadotropin-releasing hormone in rats. Endocrinology.
149:4747–4754. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Alonzeau J, Alexandre D, Jeandel L, Courel
M, Hautot C, El Yamani FZ, Gobet F, Leprince J, Magoul R, Amarti A,
et al: The neuropeptide 26RFa is expressed in human prostate cancer
and stimulates the neuroendocrine differentiation and the migration
of androgeno-independent prostate cancer cells. Eur J Cancer.
49:511–519. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ørsted DD, Bojesen SE, Nielsen SF and
Nordestgaard BG: Association of clinical benign prostate
hyperplasia with prostate cancer incidence and mortality revisited:
A nationwide cohort study of 3,009,258 men. Eur Urol. 60:691–698.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ervik M, Lam F, Ferlay J, Mery L,
Soerjomataram I and Bray F: Cancer Today. Lyon, France:
International Agency for Research on Cancer. Cancer Today 2016.
http://gco.iarc.fr/todayJanuary
22–2018
|
|
17
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Center MM, Jemal A, Lortet-Tieulent J,
Ward E, Ferlay J, Brawlt O and Bray F: International variation in
prostate cancer incidence and mortality rates. Eur Urol.
61:1079–1092. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nelson EC, Cambio AJ, Yang JC, Ok JH, Lara
PN Jr and Evans CP: Clinical implications of neuroendocrine
differentiation in prostate cancer. Prostate Cancer Prostatic Dis.
10:6–14. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Foster HA, Davies J, Pink RC, Turkcigdem
S, Goumenou A, Carter DR, Saunders NJ, Thomas P and Karteris E: The
human myometrium differentially expresses mTOR signalling
components before and during pregnancy: Evidence for regulation by
progesterone. J Steroid Biochem Mol Biol. 139:166–172. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Johnson TR, Khandrika L, Kumar B, Venezia
S, Koul S, Chandhoke R, Maroni P, Donohue R, Meacham RB and Koul
HK: Focal adhesion kinase controls aggressive phenotype of
androgen-independent prostate cancer. Mol Cancer Res. 6:1639–1648.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen Y, Xin X, Li J, Xu J, Yu X, Li T, Mo
Z and Hu Y: RTK/ERK pathway under natural selection associated with
prostate cancer. PLoS One. 8:e782542013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li WH, Qiu Y, Zhang HQ, Tian XX and Fang
WG: P2Y2 receptor and EGFR cooperate to promote prostate cancer
cell invasion via ERK1/2 pathway. PLoS One. 10:e01331652015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ghosh PM, Malik SN, Bedolla RG, Wang Y,
Mikhailova M, Prihoda TJ, Troyer DA and Kreisberg JI: Signal
transduction pathways in androgen-dependent and -independent
prostate cancer cell proliferation. Endocr Relat Cancer.
12:119–134. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
McCubrey JA, Steelman LS, Abrams SL,
Bertrand FE, Ludwig DE, Bäsecke J, Libra M, Stivala F, Milella M,
Tafuri A, et al: Targeting survival cascades induced by activation
of Ras/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for
effective leukemia therapy. Leukemia. 22:708–722. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tibbles LA and Woodgett JR: The
stress-activated protein kinase pathways. Cell Mol Life Sci.
55:1230–1254. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ricote M, García-Tuñón I, Fraile B,
Fernández C, Aller P, Paniagua R and Royuela M: P38 MAPK protects
against TNF-alpha-provoked apoptosis in LNCaP prostatic cancer
cells. Apoptosis. 11:1969–1975. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Koul HK, Pal M and Koul S: Role of p38 MAP
Kinase signal transduction in solid tumors. Genes Cancer.
4:342–359. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lotan TL, Lyon M, Huo D, Taxy JB, Brendler
C, Foster BA, Stadler W and Rinker-Schaeffer CW: Up-regulation of
MKK4, MKK6 and MKK7 during prostate cancer progression: An
important role for SAPK signalling in prostatic neoplasia. J
Pathol. 212:386–394. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Magi-Galluzzi C, Mishra R, Fiorentino M,
Montironi R, Yao H, Capodieci P, Wishnow K, Kaplan I, Stork PJ and
Loda M: Mitogen-activated protein kinase phosphatase 1 is
overexpressed in prostate cancers and is inversely related to
apoptosis. Lab Invest. 76:37–51. 1997.PubMed/NCBI
|
|
31
|
Boldt S, Weidle UH and Kolch W: The role
of MAPK pathways in the action of chemotherapeutic drugs.
Carcinogenesis. 23:1831–1838. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xia Z, Dickens M, Raingeaud J, Davis RJ
and Greenberg ME: Opposing effects of ERK and JNK-p38 MAP kinases
on apoptosis. Science. 270:1326–1331. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xu L, Chen S and Bergan RC: MAPKAPK2 and
HSP27 are downstream effectors of p38 MAP kinase-mediated matrix
metalloproteinase type 2 activation and cell invasion in human
prostate cancer. Oncogene. 25:2987–2998. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen L, He HY, Li HM, Zheng J, Heng WJ,
You JF and Fang WG: ERK1/2 and p38 pathways are required for P2Y
receptor-mediated prostate cancer invasion. Cancer Lett.
215:239–247. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gururajan M, Chui R, Karuppannan AK, Ke J,
Jennings CD and Bondada S: c-Jun N-terminal kinase (JNK) is
required for survival and proliferation of B-lymphoma cells. Blood.
106:1382–1391. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shimada K, Nakamura M, Ishida E, Kishi M,
Yonehara S and Konishi N: c-Jun NH2-terminal kinase-dependent Fas
activation contributes to etoposide-induced apoptosis in
p53-mutated prostate cancer cells. Prostate. 55:265–280. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rodríguez-Berriguete G, Fraile B,
Martínez-Onsurbe P, Olmedilla G, Paniagua R and Royuela M: MAP
kinases and prostate cancer. J Signal Transduct. 2012:1691702012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Davis RJ: Signal transduction by the JNK
group of MAP kinases. Cell. 103:239–252. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sui X, Kong N, Ye L, Han W, Zhou J, Zhang
Q, He C and Pan H: p38 and JNK MAPK pathways control the balance of
apoptosis and autophagy in response to chemotherapeutic agents.
Cancer Lett. 344:174–179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Guo J, Zhu T, Chen L, Nishioka T, Tsuji T,
Xiao ZX and Chen CY: Differential sensitization of different
prostate cancer cells to apoptosis. Genes Cancer. 1:836–846. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shukla S, Maclennan GT, Hartman DJ, Fu P,
Resnick MI and Gupta S: Activation of PI3K-Akt signaling pathway
promotes prostate cancer cell invasion. Int J Cancer.
121:1424–1432. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Toren P and Zoubeidi A: Targeting the
PI3K/Akt pathway in prostate cancer: Challenges and opportunities
(Review). Int J Oncol. 45:1793–1801. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gan Y, Shi C, Inge L, Hibner M, Balducci J
and Huang Y: Differential roles of ERK and Akt pathways in
regulation of EGFR-mediated signaling and motility in prostate
cancer cells. Oncogene. 29:4947–4958. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yao Huang and Yongchang Chang: Epidermal
Growth Factor Receptor (EGFR) Phosphorylation, Signaling and
Trafficking in Prostate Cancer. Prostate Cancer-From Bench to
Bedside. Dr. Philippe E. Spiess (ed). InTech. doi: 10.5772/27021.
2011.
|
|
45
|
Ding G, Fang J, Tong S, Qu L, Jiang H,
Ding Q and Liu J: Over-expression of lipocalin 2 promotes cell
migration and invasion through activating ERK signalling to
increase SLUG expression in prostate cancer. Prostate. 75:957–968.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ljujic M, Mijatovic S, Bulatovic MZ, Mojic
M, Maksimovic-Ivanic D, Radojkovic D and Topic A:
Alpha-1-antitrypsin antagonizes cisplatin-induced cytotoxicity in
prostate cancer (PC3) and melanoma cancer (A375) cell lines. Pathol
Oncol Res. 23:335–343. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Rybak AP, Ingram AJ and Tang D:
Propagation of human prostate cancer stem-like cells occurs through
EGFR-mediated ERK activation. PLoS One. 8:e617162013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Xiao LJ, Lin P, Lin F, Liu X, Qin W, Zou
HF, Guo L, Liu W, Wang SJ and Yu XG: ADAM17 targets MMP-2 and MMP-9
via EGFR-MEK-ERK pathway activation to promote prostate cancer cell
invasion. Int J Oncol. 40:1714–1724. 2012.PubMed/NCBI
|
|
49
|
Moulik S, Pal S, Biswas J and Chatterjee
A: Role of ERK in modulating MMP 2 and MMP 9 with respect to tumour
invasiveness in human cancer cell line MCF-7 and MDA-MB-231.
Journal of Tumor. 2:87–98. 2014.
|
|
50
|
Yang JL, Lin JH, Weng SW, Chen JC, Yang
JS, Amagaya S, Funayana S, Wood WG, Kuo CL and Chung JG: Crude
extract of Euphorbia formosana inhibits the migration and invasion
of DU145 human prostate cancer cells: The role of matrix
metalloproteinase-2/9 inhibition via the MAPK signaling pathway.
Mol Med Rep. 7:1403–1408. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Huang X, Chen S, Xu L, Liu Y, Deb DK,
Platanias LC and Bergan RC: Genistein inhibits p38 map kinase
activation, matrix metalloproteinase type 2, and cell invasion in
human prostate epithelial cells. Cancer Res. 65:3470–3478. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Oguic R, Mozetič V, Cini Tešar E, Fučkar
Čupić D, Mustać E and Dorđević G: Matrix metalloproteinases 2 and 9
immunoexpression in prostate carcinoma at the positive margin of
radical prostatectomy specimens. Patholog Res Int 2014.
2621952014.
|
|
53
|
Trudel D, Fradet Y, Meyer F, Harel F and
Têtu B: Significance of MMP-2 expression in prostate cancer: An
immunohistochemical study. Cancer Res. 63:8511–8515.
2003.PubMed/NCBI
|
|
54
|
Kyrou I, Randeva HS and Weickert MO:
Clinical Problems Caused by Obesity. De Groot LJ, Chrousos G,
Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits
M, McLachlan R, New M, Purnell J, Rebar R, Singer F and Vinik A:
South Dartmouth (MA): MDText.com, Inc.; 2014, Available on Endotext
[Internet].
|
|
55
|
Kyrou I, Mattu HS, Chatha K and Randeva
HS; Chapter 7-Fat Hormones, Adipokines, . Schisler JC, Lang CH and
Willis MS: Endocrinology of the Heart in Health and Disease.
Academic Press. 2017. pp. 167–205. 2017
|
|
56
|
Alshaker H, Sacco K, Alfraidi A, Muhammad
A, Winkler M and Pchejetski D: Leptin signalling, obesity and
prostate cancer: Molecular and clinical perspective on the old
dilemma. Oncotarget. 6:35556–35563. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Baillargeon J and Rose DP: Obesity,
adipokines, and prostate cancer (Review). Int J Oncol. 28:737–745.
2006.PubMed/NCBI
|
|
58
|
Szyszka M, Tyczewska M, Milecka P, Jopek
K, Celichowski P, Malendowicz LK and Rucinski M: Effects of leptin
on leptin receptor isoform expression and proliferative activity in
human normal prostate and prostate cancer cell lines. Oncol Rep.
39:182–192. 2018.PubMed/NCBI
|
|
59
|
Hoda MR, Theil G, Mohammed N, Fischer K
and Fornara P: The adipocyte-derived hormone leptin has
proliferative actions on androgen-resistant prostate cancer cells
linking obesity to advanced stages of prostate cancer. J Oncol.
280386:2012.
|
|
60
|
Onuma M, Bub JD, Rummel TL and Iwamoto Y:
Prostate cancer cell-adipocyte interaction: Leptin mediates
androgen-independent prostate cancer cell proliferation through
c-Jun NH2-terminal kinase. J Biol Chem. 278:42660–42667. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Miyazaki T, Bub JD and Iwamoto Y: c-Jun
NH2-terminal kinase mediates leptin-stimulated
androgen-independent prostate cancer cell proliferation via signal
transducer and activator of transcription 3 and Akt. Biochim
Biophys Acta. 1782:593–604. 2008. View Article : Google Scholar : PubMed/NCBI
|