Introduction
Sulforaphane (SFN) was first isolated from broccoli
sprout and is present at high concentrations in plants which belong
to the Cruciferae family (1). SFN
is a chemopreventive agent which displays functions, including
inhibition of carcinogen-activating enzymes, such as the cytochrome
p450 isoenzyme 2E1, induction of conjugating enzymes, such as
glutathione S-transferases, and reduction of the DNA binding
ability of nuclear factor-κB (NF-κB) (2–4).
Furthermore, SFN was found to exert antiproliferative effects on
various cancer cell lines in vitro and in vivo
(5–7). According to Choi and Singh, SFN
treatment induces Bax and Bak protein expression and conformational
change and mitochondrial translocation of Bax to trigger the
release of apoptogenic molecules from the mitochondria to the
cytosol causing activation of caspases and cell death. Their
research showed that both Bax and Bak are crucial for SFN-induced
cell death (8). SFN acts as an
indirect antioxidant and inducer of antioxidant response element
(ARE) genes, and Moreover, the exposure to SFN results in a
transient reactive oxygen species (ROS) burst, for which the
duration and magnitude both depend on the SFN concentration and
exposure period (9,10). Cyclin K plays a dual role by
regulating CDKs and transcription.
Cyclin K was first discovered in a yeast screen,
which was based on its ability to restore cell cycle progression
and rescue Saccharomyces cerevisiae cells from lethality
caused by deletion of G1 cyclin (11). Cyclin K triggers Cdk9 activity by
creating a stable protein complex with Cdk9. The Cdk9/cyclin K
complex phosphorylates the carboxyl-terminal domain (CTD) of RNA
polymerase II (RNAP II). This reaction is said to be one of the
most significant steps in the transcription of many genes (12,13).
Moreover, the role of cyclin K and Cdk9 has been implied in the
pathways that provide genomic stability in response to replication
stress. Cyclin K also binds Cdk12 and Cdk13 to form two different
complexes (cyclin K/Cdk12 or cyclin K/Cdk13) in human cells.
Phosphorylation of Ser2 in the C-terminal domain of RNAP II and
expression of a small subset of human genes are regulated by the
cyclin K/Cdk12 complex as revealed in expression microarrays
(14). Decreased expression in
mainly long genes with a high number of exons is a result of
depletion of cyclin K/Cdk12. Human cells without cyclin K/Cdk12
generate spontaneous DNA damage and are sensitive to a variety of
DNA damage agents. Moreover, cyclin K/Cdk12 protects them from
genomic instability (14). The
regulation of Cdk13 activity is currently not well understood as
the expression levels of the corresponding cyclin subunit, cyclin
K, are rather stable. This is similar to other cyclins controlling
transcriptional CDKs, e.g., cyclin T1, but different from cell
cycle-regulating cyclins such as cyclin A (15). Cyclin D1 regulates the progression
of cells from the G1 phase to the S phase of the cell cycle. It is
involved in complexes with cyclin-dependent kinases 4 and 6
(Cdk4/6) (16).
Complexes of cyclin D1 with CDKs 4 or 6 are
essential to preserve cell homeostasis when impairments in cyclin
D1 expression can trigger tumorigenesis. Moreover, changes in
cyclin D1 expression can significantly affect cellular response to
drug treatment (17–19). Cyclin B1 and Cdk1 form a complex
called ‘mitotic promoting factor’ (MPF) that is crucial for G2/M
transition (20). Therefore,
insufficient levels of cyclin B/Cdk1 complexes, e.g. as a
consequence of the attenuation of cyclin B1 promoter by p53, are
related to cell cycle arrest at G2 phase (21). Cyclin B1 and Cdk1 are expressed in
late S and G2 phases of the mammalian cell cycle and are active at
late G2 phase. After cyclin B1 is produced in the cytoplasm during
S phase it is transported to the nucleus at the late G2 phase and
then finally removed during anaphase via a ubiquitin-related
pathway (22). The redistribution
of cyclin B1/Cdk1 to the mitochondrial matrix shows a unique
mechanism which coordinates the mitotic events in other cellular
compartment and mitochondrial activity for G2/M progression. In
spite of the fact than cyclin B1/Cdk1 is detected in the
mitochondria of asynchronous cells in different cell cycle phases,
the timing of mitochondrial influx of cyclin B1/Cdk1 is consistent
with the accumulation of G2/M cells. What is important, the kinase
activity of Cdk1 is also maximized at the G2/M which indicates that
cyclin B1/Cdk1-mediated mitochondrial bioenergetics is integrated
into the overall process of G2/M transition (23). There are reports suggesting that
changes in the regulation of cyclin expression may be connected not
only with unrestrained cell growth and malignant transformation,
but also in tumor suppressor mechanisms (24).
The aim of the present study was to evaluate cyclin
B1, cyclin D1 and cyclin K expression in H1299 cells treated with
SFN as well as to investigate a potential involvement of these
cyclins in the therapeutic outcome of SFN treatment.
Materials and methods
Cell culture and SFN treatment
The human non-small cell lung carcinoma cell line
H1299 was purchased from the American Type Culture Collection
(ATCC; Manassas, VA, USA). The cells were cultured in monolayers at
37°C in a humidified CO2 incubator (5% CO2)
in Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal
bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc) and 50
µg/ml of gentamycin (Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany). Twenty-four hours after seeding, the cells were treated
with SFN (Sigma-Aldrich; Merck KGaA) (5, 10 and 15 µM) for 48 h,
and the following experimental procedures were performed.
MTT assay
Twenty-four hours prior to SFN treatment, the H1299
cells were seeded in 12-well plates. The cells were then treated
with appropriate SFN concentrations (5, 10 and 15 µM) for 24 and 48
h. Following the treatment, cells were washed with
phosphate-buffered saline (PBS) and 1 ml of DMEM without phenol red
and 100 µl of the thiazolyl blue tetrazolium bromide (MTT) working
solution (Sigma-Aldrich; Merck KGaA; 5 mg/ml in PBS) were added to
each well and incubated for 3 h. Formazan crystals were diluted in
isopropanol and the absorbance was read at 570 nm in a
spectrophotometer (Spectra Academy, K-MAC, Daejeon, Korea).
Annexin V/propidium iodide (PI)
binding assay
To assess the mode of cell death, the Alexa Fluor
488 Annexin V/Dead Cell Apoptosis kit (Invitrogen; Thermo Fisher
Scientific, Inc.) was used according to the manufacturer's
instructions. In short, after the SFN treatment, the cells were
collected from 6-well plates using trypsin-EDTA solution,
centrifuged at 300 × g for 8 min, resuspended in ABB (Annexin
binding buffer) and incubated with Annexin V Alexa
Fluor® 488 and propidium iodide (PI) at room temperature
in the dark for 20 min. The cells were examined using a Guava
6HT-2L Cytometer (Merck KGaA). The data were analyzed by InCyte
software (version 3.2; Merck KGaA) and expressed as the percentage
of cells in each population (viable, Annexin
V−/PI−; early apoptotic, Annexin
V+/PI−; late apoptotic, Annexin
V+/PI+; necrotic, Annexin
V−/PI+).
DNA content analysis
For DNA content analysis, the Guava Cell Cycle
reagent (Merck KGaA) was used according to the manufacturer's
instructions. Briefly, the cells were harvested from 6-well plates
by trypsinization, rinsed with PBS, fixed in ice-cold 70% ethanol
at 4°C and maintained at −25°C overnight. The cells were then
centrifuged at 650 × g for 5 min at room temperature (RT) and
washed with PBS. After centrifugation at 500 × g for 7 min, the
cells were resuspended in Guava Cell Cycle reagent. Following a
30-min incubation at RT in the dark, the cells were analyzed using
Guava 6HT-2L cytometer (Merck KGaA), and the percentage of cells in
each phase of the cell cycle was determined using InCyte software
(version 4.03; De Novo Software, Piscataway, NJ, USA).
Flow cytometric analysis of cyclins
B1, D1 and K
Cells grown in 6-well plates were harvested, washed
with PBS, centrifuged (for 5 min, at 300 × g) and incubated with
eBioscience Fixable Viability Dye eFluor 660 (Thermo Fisher
Scientific, Inc.) for 30 min at 4°C to exclude dead cells. Then,
the cells were fixed with Cytofix/Cytoperm solution (BD
Biosciences, San Jose, CA, USA). After incubation on ice (for 15
min in the dark) and subsequent centrifugation (for 5 min, at 300 ×
g), the cells in pellets were permeabilized by the addition of 1 ml
of ice-cold 80% (v/v) methanol (POCH) overnight at −20°C, washed
with cold Perm/Wash solution (BD Biosciences) and resuspended in 3%
bovine serum albumin (BSA; Sigma-Aldrich; Merck KGaA). For
intracellular staining, cells were incubated with the appropriate
antibody diluted in Perm/Wash solution. Following a 30-min
incubation at room temperature (RT) in the dark and washing with
Perm/Wash solution, the cells were centrifuged for 5 min, at 500 ×
g to wash off excess antibody. Cells stained with cyclin B1 and
cyclin K antibody were incubated with Alexa Fluor 488 for 30 min at
RT. Cells were resuspended in 300 µl of PBS for flow cytometric
analysis on Guava 6HT-2L Cytometer. InCyte software was used to
calculate the mean fluorescence intensity. The antibodies used in
the experiment were the following: Mouse monoclonal anti-cyclin B1
antibody (dilution 1:100; cat. no. MA5-14319; Thermo Fisher
Scientific, Inc.), FITC conjugated mouse cyclin D1 antibody (cat.
no. 554109; BD Biosciences), or mouse monoclonal anti-cyclin K
antibody (dilution 1:50; cat. no. sc-376371; Santa Cruz
Biotechnology Inc., Santa Cruz, CA, USA).
RT PCR analysis of cyclin B1, D1 and
cyclin K expression
Total RNA from H1299 cells was isolated using a
Total RNA kit (A&A Biotechnology, Gdynia, Poland) according to
the manufacturer's instruction. The concentration and purity of
RNA were determined spectrophotometrically (BioSpectrometer
Basic; Eppendorf, Hamburg, Germany). The reverse transcription and
quantitative PCR reactions were performed in a single 20-µl
LightCycler capillary (Roche Applied Science, Mannheim, Germany) as
a one-step real-time qRT-PCR with using LightCycler RNA Master
SYBR-Green I (Roche Applied Science). For each target gene, the
reactions were carried out in a 20-µl volume containing 100 ng of
RNA and 0.2 µM of each primer in addition to LightCycler RNA Master
SYBR-Green I kit components. The sequences of the primers were as
follows: CCNK forward, 5′-ACCCAAAGGAGGAAGTAATGG-3′ and
CCNK reverse, 5′-GAACTGGTATGGATGTTCTACCT-3′; CCND1
forward, 5′-TGAGGCGGTAGTAGGACAGG-3′ and CCND1 reverse,
5′-GACCTTCGTTGCCCTCTGT-3; CCNB1 forward,
5′-TTTCGCCTGAGCCTATTTTG-3′ and CCNB1 reverse,
5′-GCACATCCAGATGTTTCCATT-3′. The thermocycling conditions used for
the qRT-PCR were as follows: One cycle of reverse transcription for
20 min at 61°C, one cycle of denaturation for 1 min at 95°C, and 45
cycles of denaturation for 5 sec at 95°C, followed by annealing and
extension for 20 sec at 57–61°C (depending on the melting
temperature of the primers) and 5 sec at 72°C, respectively. The
samples were run in at least triplicate on the LightCycler 2.0
Instrument (Roche Applied Science) and evaluated with LightCycler
Software (version 4.0; Roche Applied Science). The expression of
the target gene was normalized to glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) internal control and assessed using
the ΔΔCq method (2−ΔΔCq method) (25).
Cyclin B1, cyclin D1 and cyclin K
immunofluorescence
H1299 cells growing on coverslips were briefly
washed with PBS, fixed in 4% paraformaldehyde (for 15 min at RT)
and then washed with PBS (3×5 min). After that, the cells were
incubated in permeabilization solution (0.1% Triton X-100 in PBS)
and blocked with 3% BSA. After permeabilization, the cells were
incubated with mouse monoclonal anti-cyclin B1 antibody (dilution
1:100; cat. no. MA5-14319; Thermo Fisher Scientific, Inc.) or mouse
monoclonal anti-cyclin D1 antibody (cat. no. C7464; Sigma-Aldrich;
Merck KGaA) or mouse monoclonal anti-cyclin K antibody (dilution
1:50; cat. no. sc-376371; Santa Cruz Biotechnology, Inc.),
respectively (60 min at RT), washed three times with PBS and
incubated with Alexa Fluor 488 goat anti-mouse IgG (Invitrogen;
Molecular Probes) (60 min, in the dark). Nuclear staining was
performed with DAPI (Sigma-Aldrich; Merck KGaA). Contract labeling
with Alexa Fluor 594 phalloidin (Thermo Fisher Scientific, Inc.)
was used additionally. After incubation, the cells were washed with
PBS and then mounted on microscope slides in Aqua-Poly/Mount
(Polysciences, Inc., Warrington, PA, USA). The cells were examined
using a C1 laser-scanning confocal microscopy system (Nikon, Tokyo,
Japan) with a 100× oil immersion objective. Fluorescence images
were obtained and analyzed with Nikon EZ-C1 software (ver3.80;
Nikon Instruments, Melville, NY, USA).
Western blot assay
For semi-quantitative protein expression
measurement, western blot analysis was conducted. Whole-cell
lysates were obtained from lysis in RIPA buffer (Merck KGaA).
Following normalization of the protein concentration using the BCA
protein assay kit (Thermo Fisher Scientific, Inc.), equal amounts
of protein (25 µg of total protein per lane) were separated using
4–12 or 16% NuPAGE Bis-Tris Gel (Novex/Life Technologies; Thermo
Fisher Scientific, Inc.) and transferred onto nitrocellulose
membranes using the iBlot dry western blotting system (Invitrogen;
Thermo Fisher Scientific, Inc.). The membrane was processed in
iBind Flex Western Blot system (Thermo Fisher Scientific, Inc.)
following the manufacturer's protocol. Bands were detected using
1-Step™ Ultra TMB-Blotting solution (Thermo Fisher Scientific,
Inc.).
Gene expression analysis (KM
plotter)
Kaplan-Meier database (http://kmplot.com/analysis/) was conducted to evaluate
the impact of CCNK, CDK12 and the CDK13 expression on the outcome
of adenocarcinoma patients (26).
The following probeset was used (ID Affymetrix): CCNK (219273_at),
CDK12 (225690_at), CDK13 (228991_at).
Statistical analysis
The analysis was performed using statistical
software (ver. 6.0; GraphPad Prism, San Diego, CA, USA). The data
were compared with the non-parametric Mann-Whitney U test or
non-parametric Kruskal-Wallis test with Dunn's multiple comparisons
test, and the changes were considered statistically significant at
the level of P<0.05. To analyze Kaplan-Meier survival curves,
hazard ratio and log-rank test were calculated on the KM plotter
web page.
Results
Annexin V and MTT assay
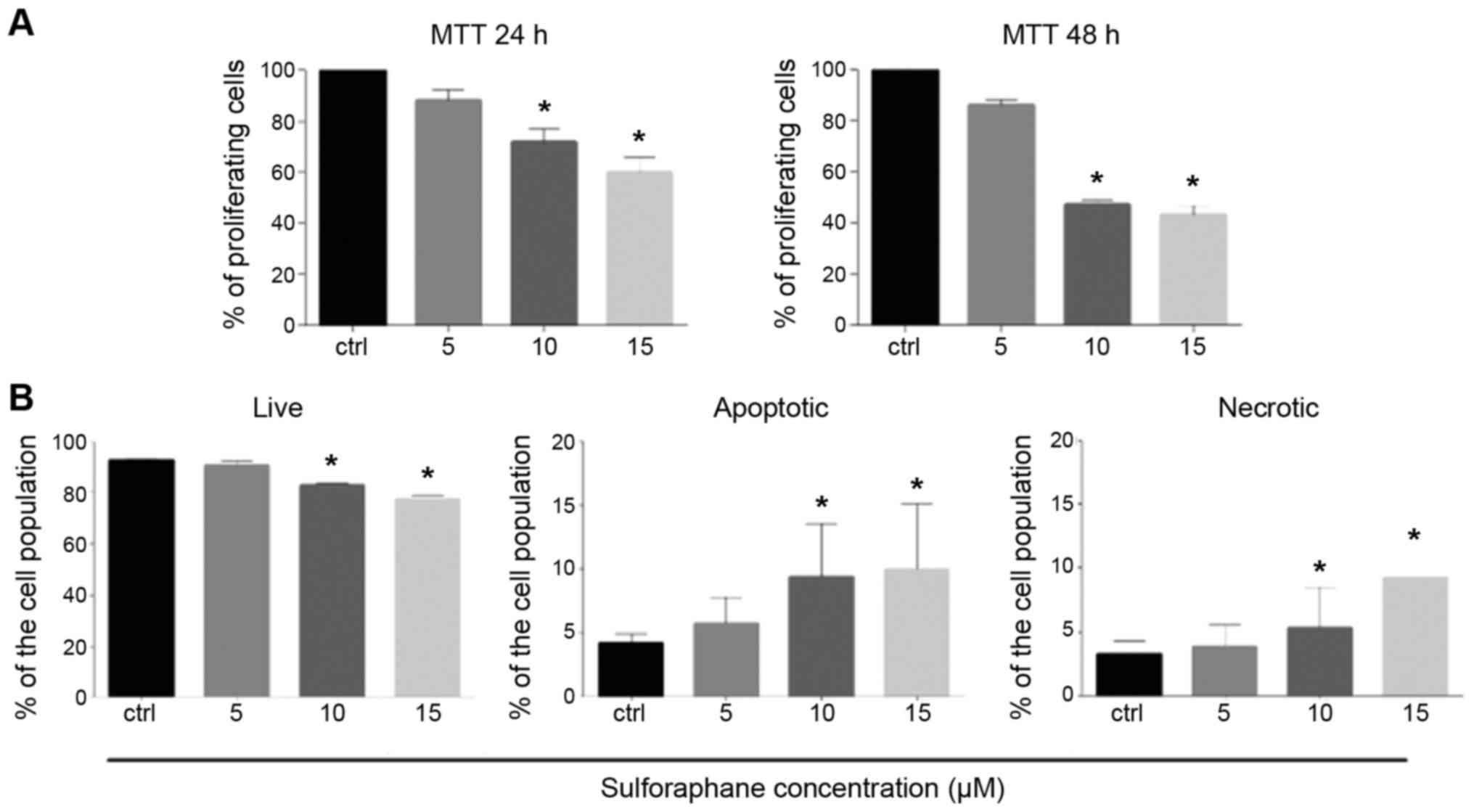
Following treatment with 5, 10 and 15 µM of SFN used
in this study, the percentage of proliferating cells was decreased
in a dose-dependent manner at both 24 and 48 h and the difference
was significant at 10 and 15 µM of SFN (P<0.05) when compared
with the control as determined by the MTT assay (Fig. 1A). In addition, as shown by the
Annexin V/propidium iodide (PI) binding assay, the percentage of
living cells was significantly decreased at 10 and 15 µM of SFN
treatment (P<0.05). A dose-dependent increase in the percentage
of apoptotic cells and a slight increase in the percentage of
necrotic cells were observed and the differences were significant
at 10 and 15 µM (Mann-Whitney U; P<0.05) (Fig. 1B). The results indicated that SFN
induces both apoptosis and necrosis in a similar extent.
DNA content analysis
Flow cytometry was conducted to assess whether SFN
treatment induced changes in cell cycle distribution in the H1299
cell line. Cell cycle analysis showed that along with the increase
in the concentration of SFN, the percentage of cells arrested at
the G2/M phase of the cell cycle was increased which was correlated
with a decrease in cells in the G0/G1 phase. These results
indicated that SFN suppressed cell cycle progression in the H1299
cell line (Fig. 2).
Expression of cyclin B1
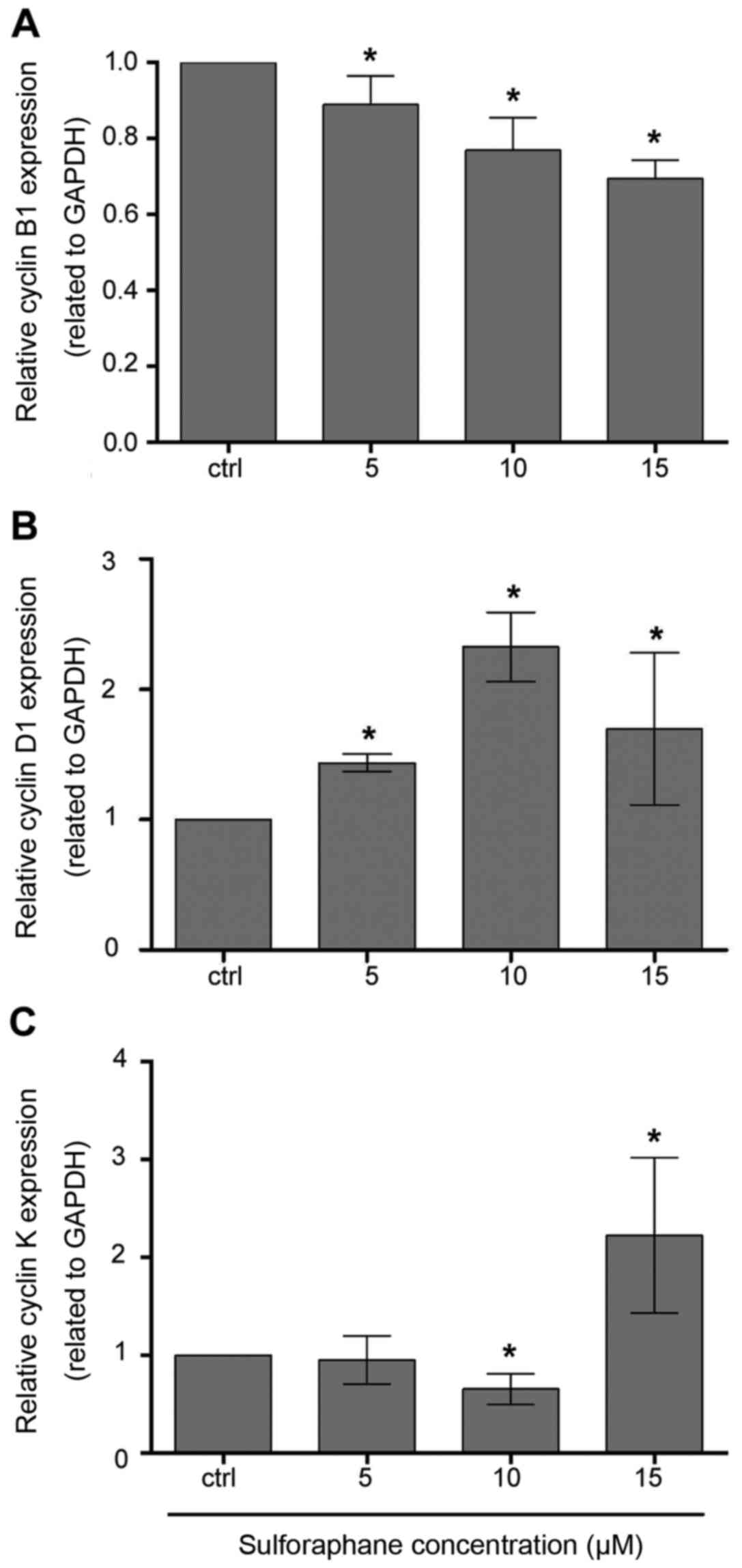
Following SFN treatment, expression of cyclin B1 was
decreased. qRT-PCR analysis showed that treatment with 5, 10 and 15
µM concentrations of SFN induced a significant decrease in cyclin
B1 mRNA (Mann-Whitney U; P<0.05) (Fig. 3A), similarly to cytometric analysis
which revealed a slight decrease at all doses of SFN (Fig. 4A). Immunofluorescent labeling of
cyclin B1 in SFN-treated cells showed a trend similar to that noted
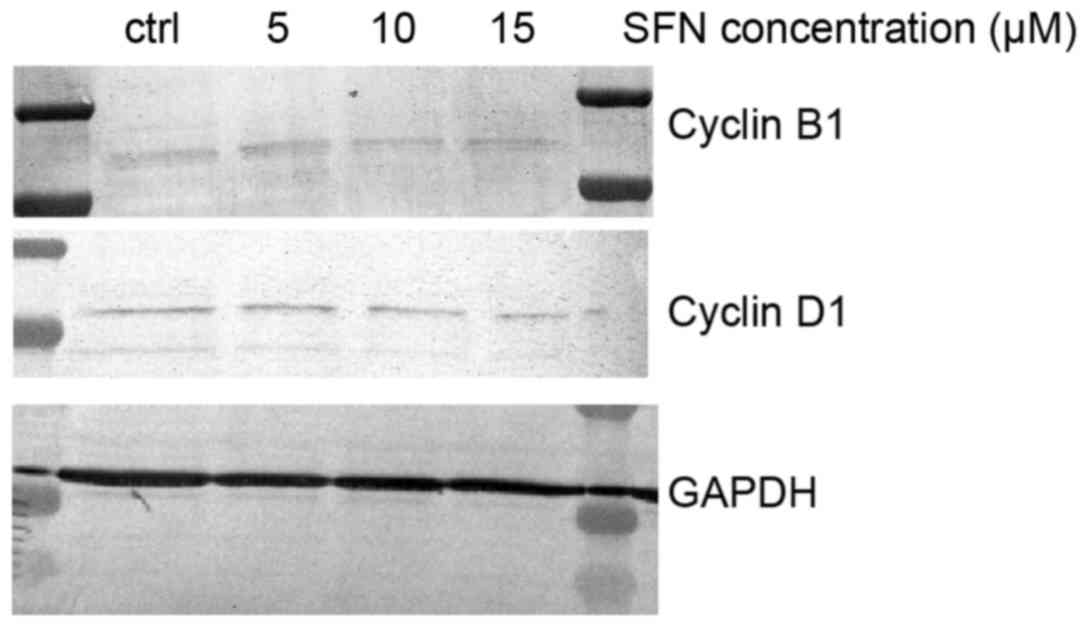
in the flow cytometric and qRT-PCR results (Fig. 5). Western blot analysis showed a
decrease in cyclin B1 expression (Fig.
6).
Expression of cyclin D1
The qRT-PCR experiment showed a significant increase
in cyclin D1 mRNA after treatment with 10 µM SFN. Slightly
increased levels resulted from the treatment with other doses, i.e.
5 and 15 µM SFN, when compared with the control (Fig. 3B). Flow cytometric measurements
indicated that the fluorescence intensity of cyclin D1 protein was
increased at all doses of SFN treatment with significant increases
at 10 and 15 µM doses (Fig. 4B).
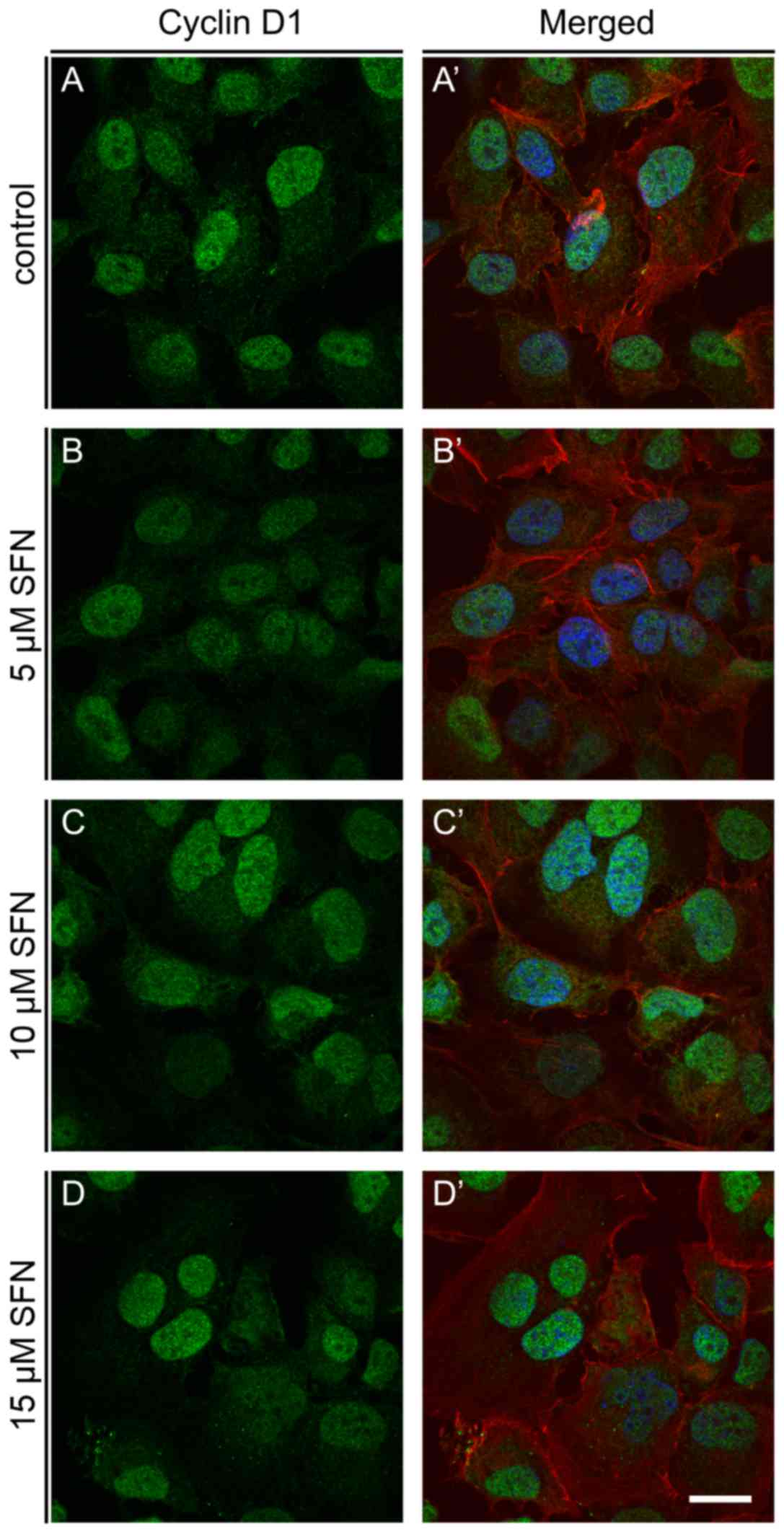
Fluorescence microscopic examination of cyclin D1 in H1299 control
and SFN-treated cells revealed the highest induction of this
protein at 10 and 15 µM SFN (Fig.
7). Western blot analysis showed a slight decrease at the
protein level following treatment with SFN, which suggests
post-transcriptional inhibition of cyclin D1 (Fig. 6).
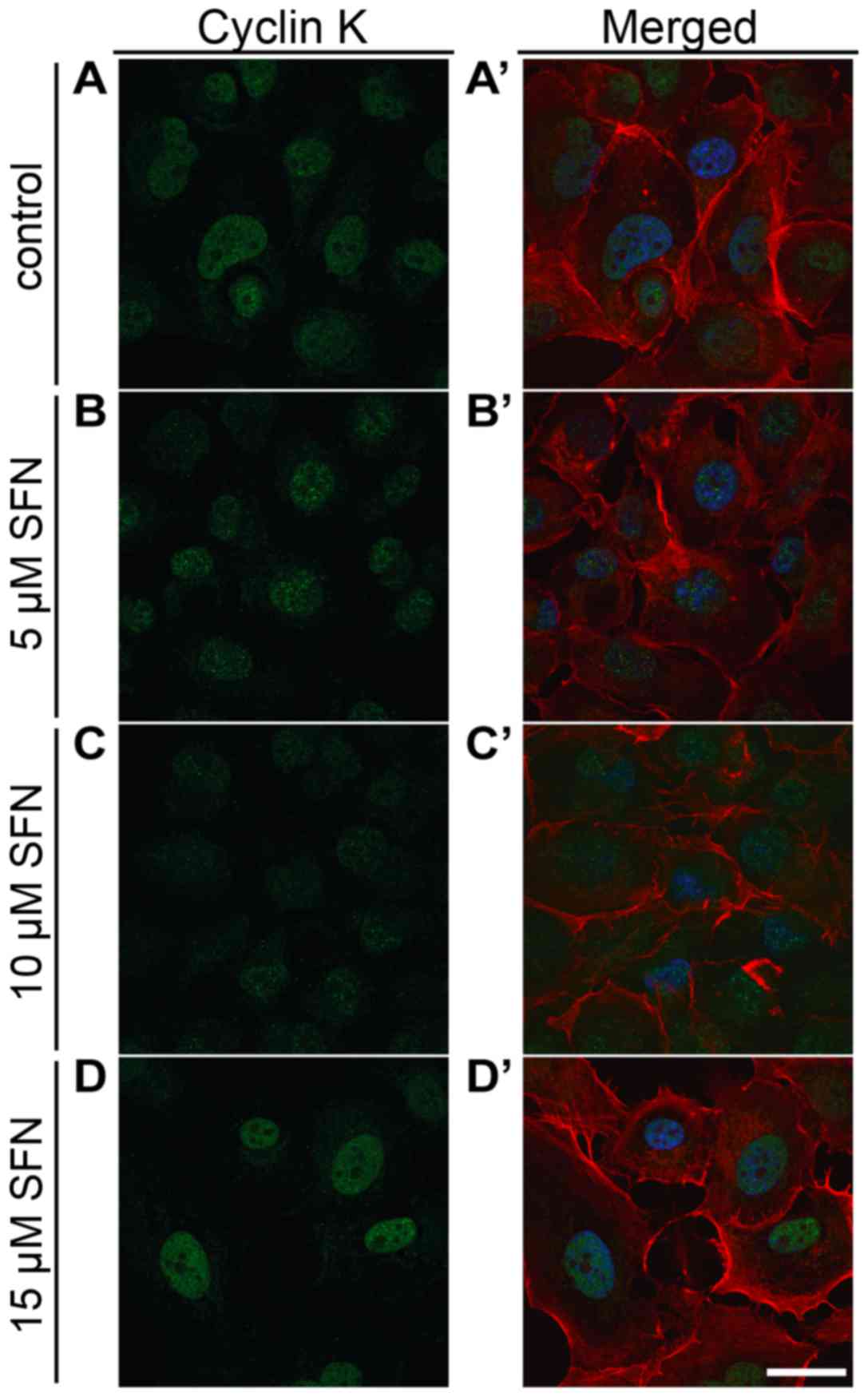
Expression of cyclin K
qRT-PCR analysis displayed that treatment of SFN at
a 15 µM concentration induced a significant increase in cyclin K
mRNA (Mann-Whitney U, P<0.05) (Fig.
3C). Flow cytometric measurements demonstrated that the
fluorescence intensity of cyclin K protein was significantly higher
comparing to the control cells (Fig.
4C). Immunofluorescent labeling of cyclin K in SFN-treated
cells showed a trend similar to that noted in the qRT-PCR results
(Fig. 8). Importantly, we observed
downregulation of cyclin K after treatment with the lowest dose of
SFN. However, to date, not enough data exist to explain this change
in expression profile. We will try to investigate this phenomenon
in the future and we hope that we can provide a more detailed
mechanism which is responsible for changes in cyclin K expression
after the treatment with a variety of drug and compounds.
Additionally, due to the very low cyclin K expression in the H1299
cell line, we failed to perform western blot analysis.
Prognostic value of cyclin K, CDK12
and CDK13
Overall survival analysis of the patients with
adenocarcinoma revealed that high expression of CDK12 and CDK13 but
no cyclin K expression (CCNK) is associated with a worse prognosis
(Fig. 9).
Discussion
Despite the significant advance in diagnosis and
treatment, cancer remains a major cause of death worldwide. One of
the deadliest cancers, which leads to a high death rate for both
women and men, is lung cancer. With a 5-year survival rate below
20%, it is still a challenge for all those involved in cancer
research (27). Phytochemicals are
a potent source of anticancer agents, which can act unassisted or
synergize with traditional drugs. Sulforaphane (SFN) is one of the
most promising natural compound which exerts anticancer activities.
This broccoli sprout-derived isothiocyanate acts through different
pathways, which are not yet fully understood. Tumor cells are often
characterized by imbalanced processes responsible for maintaining
homeostasis, regulation of cell cycle and cell death. Uncontrolled
divisions hold the danger of genetic impairments which directly
lead to malignant transformation. In the present study, we
demonstrated that SFN induces apoptosis, cell cycle arrest and
changes in the expression of proliferation markers. What is
important, the chosen doses can be attained by a normal consumption
of broccoli. Approximately 50 grams of this vegetable delivers a
sufficient amount of SFN to achieve a 20-µM concentration in urine
(28). The ability of SFN to induce
apoptosis has been demonstrated in several models in vivo
and in vitro. In the highly metastatic melanoma cell line
B16F-10 apoptosis was associated with morphological changes
involving membrane blebbing and presence of apoptotic bodies. On
the molecular level, activation of caspases-3 and −9, Bax and p53
with a simultaneous decrease in Bcl-2, caspase-8, Bid and NF-κB was
observed (29). In U251MG
glioblastoma cells, SFN induced apoptosis associated with
expression of Bad, Bax and cytochrome c and decreased levels
of survivin and Bcl-2. Moreover, cells treated with SFN were
characterized by reduced invasion capabilities connected with
increased expression of E-cadherin and lower expression of MMP-2,
MMP-9 and galectin-3 (30).
Mutations in the TP53 gene have been observed in most human
cancers and p53 protein responses to low or constitutive stress
through cell cycle arrest. When the stress drives to severe DNA
damage, p53 triggers apoptosis. The H1299 cell line is
characterized by p53 deficiency and only a few studies have shown
details for the p53-independent response following SFN treatment.
Ferreira de Oliveira et al (31) found that SFN induced DNA damage and
increased the number of nuclear and mitotic abnormalities. In
another study by Ferreira de Oliveira et al (32), the authors presented a model of SFN
involvement in ROS generation and cytotoxicity in the p53 null
osteosarcoma MG-63 cell line. SFN treatment was found to induce a
significant increase in ROS generation and subsequent changes in
mitochondrial membrane potential and eventually execution of
apoptosis in a p53-independent manner. Higher concentrations of SFN
were found to lead to inhibition of ROS-scavenging enzymes such as
SOD, Cat and GPx and contribute to lowered glutathione regeneration
and impaired antioxidative potential. Cancer cells have a lower
ratio of oxidized glutathione to reduced glutathione which confers
drug resistance (33,34). Targeting the glutathione system can
be utilized to increase the effectiveness of chemotherapy. We
showed that SFN alone has the potential to inhibit proliferation of
the H1299 cell line, thus it is possible to use SFN as a sensitizer
in the combinational therapy of tumors with dysfunctional p53
protein.
In the present study, the antiproliferative effect
of SFN was manifested by G2/M phase arrest associated with
downregulation of cyclin B1 which is a potent cell cycle regulator.
Its overexpression is observed in many different cancer types,
including lung, breast and gastric cancer (35–37).
Cyclin B1 silencing can exert antiproliferative effects on cancer
cells, thus cyclin B1 can be considered as a potential therapeutic
target. Kedinger et al downregulated cyclin B1 expression
using modified siRNAs and achieved a reduction in the growth of
melanoma xenografts and inhibition of formation and dissemination
of melanoma lung metastases (38).
In cervical cancer, G2/M arrest induced by SFN was also associated
with downregulation of cyclin B1. Moreover, the cellular response
involved upregulation of GADD45β, known as cyclin B1/Cdk1
inhibitor. The results suggest that SFN has the potential to
inhibit cancer growth via the cyclin B/GADD45β pathway (39). It is noteworthy that downregulation
of cyclin B1 is not unequivocally beneficial for cancer patients.
In colorectal cancer patients, negative or low cyclin B1 staining
was inversely correlated with lymph node metastatic potential.
Tumors with low cyclin B1 expression more frequently present with
lymphatic permeation or vessel invasion (40). Fang et al showed that cyclin
B inhibits lymph node metastasis via E-cadherin both in vivo
and in vitro. Cyclin B1 silencing resulted in decreased
E-cadherin expression, which is one of the molecular hallmarks
associated with EMT. Downregulation of cyclin B1 suppressed
p53+/+ HCT116 and p53−/− HCT116 colorectal
cancer cell lines. The same significant effect was observed in the
SW480 cell line and in a xenograft model (41). Correspondingly with a previously
described model, Wang et al showed that SFN suppressed EMT
and metastasis in human lung cancer cell lines through
miR-616-5p-mediated GSK3β/β-catenin signaling pathways. SFN
treatment was found to alter the expression of EMT-related proteins
such as β-catenin, N-cadherin, vimentin and E-cadherin (42). These data show that the impact of
cyclin B1 depends on the cellular environment and it is important
to determine the factors which promote the antimetastatic effect of
cyclin B1 and when cyclin B1 enhances EMT events.
Cyclin D1 is frequently overexpressed in many cancer
types. After treatment with SFN, we observe an increase in cyclin
D1 mRNA and protein expression. It is possible that cyclin D1
overexpression in the H1299 cell line is responsible for moderate
response to drug intervention. Our previous study showed that
cyclin D1 overexpression in the A549 cell line prevented
SFN-induced apoptosis (43).
Moreover, we also observed an increase in cyclin D1 after treatment
with the lowest dose of SFN (30 µM). It is possible that at lower
doses, SFN inhibits cyclin D1 association with cyclin-dependent
kinases and this is the cause of the increase in cyclin
fluorescence. Hagemann et al showed that SFN can upregulate
MDM-2 protein expression which is a p53 suppressor but also can act
independently of p53. MDM-2 as an oncoprotein promotes the survival
of cancer cells and contributes to drug resistance. The protective
effect of SFN can be blocked with MDM-2 inhibitor, Nutlin-3
(44). Moreover, silencing of
cyclin D1 enhances the cytostatic effect of Nutlin-3 and makes
different cancer cell lines more susceptible to cell death
(45). It is feasible that the axis
cyclin D1-MDM-2 significantly contributes to the cellular response
to SFN treatment. Treating the H1299 cell line with another
cytostatic drug, actinomycin D induced an increase in MDM2
expression. Silencing of MDM2 sensitized cells to actinomycin D and
increase the number of cells undergoing apoptosis (46). The interaction between cyclin D1 and
MDM-2 in the H1299 cell line warrants further investigations.
Cyclin-dependent kinases 12 and 13 with its
regulatory subunit cyclin K, take part in gene transcription
regulation through interaction with RNA polymerase II and
phosphorylation of C-terminal domain which is an important step in
the generation of mature mRNA (47,48).
The activity and oncogenic status of cyclin K/Cdk12 and cyclin
K/Cdk13 warrant further elucidation. To investigate how the
expression of CDK12 and CDK13 mRNA affects adenocarcinoma and the
squamous cell carcinoma outcome we employed ‘The Kaplan-Meier
plotter’ (KM plotter) database and discovered that high expression
of Cdk12 and Cdk13 mRNA is correlated with worse overall survival.
To date, limited and inconsistent data have been published, showing
both a suppressive and a promoting effect on different cancer
types. The loss of function mutations in high-grade serous ovarian
cancer probably result in deregulated expression of DDR genes and
contribute to genomic instability (49,50).
Moreover, patients bearing mutations in the CCNK gene also present
with abnormalities in BRCA1 and BRCA2 genes, well-known cancer
suppressors (51). Amplification of
the CDK12 mRNA increased protein levels, and elevated
phosphorylation was observed in HER-2 driven breast cancer samples
(52). Involvement in DNA repair
leads to believe that targeting of Cdk12/cyclin K complex can
significantly alter the drug response and can be considered as a
new strategy in therapeutic development. The cyclin K gene was
found to sensitize cancer cells to camptothecin which is a
topoisomerase inhibitor inducing DNA damage (53). The loss of function mutations in the
CDK12 gene leads to increased cisplatin sensitivity and
downregulation of CDK12 induces spontaneous cell death and
decreases resistance to different DNA-damaging agents such as
etoposide, mitomycin C and cisplatin (14,54).
In turn, the impact of CDK13 activity on cancer
development and disease outcome is still an uncharted territory.
The CKD13 gene is frequently amplified in many different cancer
cell lines. Clonogenic and invasion assays on the NIH3T3 cell line
revealed the high oncogenic potential of the CDK13 product and
showed that stable expression of CDK13 resulted in the stronger
colony forming ability and an intermediate degree of migration
activity. Additionally, cells overexpressing CDK13 exhibited
increased resistance to 5-fluorouracil and doxorubicin and a
simultaneous increase in tamoxifen susceptibility (55). These data suggest a significant role
of CDK12 and CDK13 in human cancer and response to a treatment. The
role of cyclin K in cancer development is unclear. Cyclin K plays
important roles in transcriptional regulation and cell cycle
control, but we know little about how cyclin K expression affects
drug response. Schecher et al demonstrated the potential
role of cyclin K in prostate cancer. Depletion of cyclin K in
prostate cancer cell lines led to an increase in the number of
multinucleated cells, mitotic catastrophe and eventually reduction
of proliferation or apoptotic cell death. Moreover, prostate cancer
patients with high expression of cyclin K, treated with an adjuvant
therapy had worse biochemical recurrence-free survival rates
compared to patients with low cyclin K expression. Silencing of
cyclin K resulted in a decrease in Aurora B mRNA and protein
expression. Aurora B is crucial for the formation of chromosomes
and spindle assembly and its depletion is probably caused by the
reduction of CDK12 activity rather than cyclin K itself (56). In this study, the increase in cyclin
K expression could be a part of drug response. SFN induces DNA
damage and activates various genes associated with DNA repair. The
results suggest that high expression of cyclin K and CDK12 and
CDK13 kinases should be considered as a potential obstacle in the
successful therapy of tumors. It was shown that inhibitors of Cdk12
and Cdk13 can exert an antiproliferative effect on cancer cells
thus it is reasonable to conduct further studies to investigate
CDK12, CDK13 and cyclin K as targets for cancer treatment (57,58).
In conclusion, this study revealed that SFN induced
cell cycle arrest and apoptosis in the H1299 cell line. Cell cycle
arrest was associated with downregulation of cyclin B1. Significant
changes in the expression of cyclin D1 and K suggest the role of
these proteins in response to SFN treatment. Moreover, our in
silico analysis showed that upregulated mRNA expression of the
cyclin K catalytic partners CDK12 and CDK13 is associated with
lower overall survival rates in adenocarcinoma patients. The
present study shows the urgent need for elucidating the role of
cyclin K/CDK12/CDK13 complexes in development and progression of
lung cancer.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Nicolaus
Copernicus University in Toruń, Collegium Medicum, Faculty of
Medicine (grant no. 114).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
AŻ conceived and designed the study. AK and AŻ were
responsible for the DNA content analysis and the DNA content
analysis for cytometric analysis, the immunofluorescent labeling
and the western blot analysis, they jointly designed the figures
and they wrote the study. AK was responsible for the Annexin V and
MTT assay and for the prognostic value of cyclin K, CDK12 and
CDK13. AKW was responsible for the qRT-PCR analysis. DG and AG
performed the statistical analysis; AZ, AK, AG, AKW and DG reviewed
and edited the manuscript. All authors read and approved the
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
All experiments have been approved by the Bioethics
Committee of the Nicolaus Copernicus University in Toruń
functioning at Collegium Medicum in Bydgoszcz.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Clarke JD, Dashwood RH and Ho E:
Multi-targeted prevention of cancer by sulforaphane. Cancer Lett.
269:291–304. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Barcelo S, Gardiner JM, Gescher A and
Chipman JK: CYP2E1-mediated mechanism of anti-genotoxicity of the
broccoli constituent sulforaphane. Carcinogenesis. 17:277–282.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Langouët S, Furge LL, Kerriguy N, Nakamura
K, Guillouzo A and Guengerich FP: Inhibition of human cytochrome
P450 enzymes by 1,2-dithiole-3-thione, oltipraz and its
derivatives, and sulforaphane. Chem Res Toxicol. 13:245–252. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Heiss E, Herhaus C, Klimo K, Bartsch H and
Gerhäuser C: Nuclear factor kappa B is a molecular target for
sulforaphane-mediated anti-inflammatory mechanisms. J Biol Chem.
276:32008–32015. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Parnaud G, Li P, Cassar G, Rouimi P,
Tulliez J, Combaret L and Gamet-Payrastre L: Mechanism of
sulforaphane-induced cell cycle arrest and apoptosis in human colon
cancer cells. Nutr Cancer. 48:198–206. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Singh SV, Herman-Antosiewicz A, Singh AV,
Lew KL, Srivastava SK, Kamath R, Brown KD, Zhang L and Baskaran R:
Sulforaphane-induced G2/M phase cell cycle arrest
involves checkpoint kinase 2-mediated phosphorylation of cell
division cycle 25C. J Biol Chem. 279:25813–25822. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Herman-Antosiewicz A, Johnson DE and Singh
SV: Sulforaphane causes autophagy to inhibit release of cytochrome
c and apoptosis in human prostate cancer cells. Cancer Res.
66:5828–5835. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Choi S and Singh SV: Bax and Bak Are
required for apoptosis induction by sulforaphane, a cruciferous
vegetable-derived cancer chemopreventive agent. Cancer Res.
65:2035–2043. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shankar S, Ganapathy S and Srivastava RK:
Sulforaphane enhances the therapeutic potential of TRAIL in
prostate cancer orthotopic model through regulation of apoptosis,
metastasis, and angiogenesis. Clin Cancer Res. 14:6855–6866. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Moon DO, Kim MO, Kang SH, Choi YH and Kim
GY: Sulforaphane suppresses TNF-alpha-mediated activation of
NF-kappaB and induces apoptosis through activation of reactive
oxygen species-dependent caspase-3. Cancer Lett. 274:132–142. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Edwards MC, Wong C and Elledge SJ: Human
cyclin K, a novel RNA polymerase II-associated cyclin possessing
both carboxy-terminal domain kinase and Cdk-activating kinase
activity. Mol Cell Biol. 18:4291–4300. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fu TJ, Peng J, Lee G, Price DH and Flores
O: Cyclin K functions as a CDK9 regulatory subunit and participates
in RNA polymerase II transcription. J Biol Chem. 274:34527–34530.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Napolitano G, Majello B, Licciardo P,
Giordano A and Lania L: Transcriptional activity of positive
transcription elongation factor b kinase in vivo requires the
C-terminal domain of RNA polymerase II. Gene. 254:139–145. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Blazek D, Kohoutek J, Bartholomeeusen K,
Johansen E, Hulinkova P, Luo Z, Cimermancic P, Ule J and Peterlin
BM: The Cyclin K/Cdk12 complex maintains genomic stability via
regulation of expression of DNA damage response genes. Genes Dev.
25:2158–2172. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhou Z and Fu XD: Regulation of splicing
by SR proteins and SR protein-specific kinases. Chromosoma.
122:191–207. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Baldin V, Lukas J, Marcote MJ, Pagano M
and Draetta G: Cyclin D1 is a nuclear protein required for cell
cycle progression in G1. Genes Dev. 7:812–821. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Akiyama N, Sasaki H, Katoh O, Sato T,
Hirai H, Yazaki Y, Sugimura T and Terada M: Increment of the cyclin
D1 mRNA level in TPA-treated three human myeloid leukemia cell
lines: HEL, CMK and HL-60 cells. Biochem Biophys Res Commun.
195:1041–1049. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tiemann K, Alluin JV, Honegger A, Chomchan
P, Gaur S, Yun Y, Forman SJ, Rossi JJ and Chen RW: Small
interfering RNAs targeting cyclin D1 and cyclin D2 enhance the
cytotoxicity of chemotherapeutic agents in mantle cell lymphoma
cell lines. Leuk Lymphoma. 52:2148–2154. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Danos AM, Liao Y, Li X and Du W:
Functional inactivation of Rb sensitizes cancer cells to TSC2
inactivation induced cell death. Cancer Lett. 328:36–43. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pines J and Hunter T: Human cyclin A is
adenovirus E1A-associated protein p60 and behaves differently from
cyclin B. Nature. 346:760–763. 1990. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Innocente SA, Abrahamson JL, Cogswell JP
and Lee JM: p53 regulates a G2 checkpoint through cyclin B1. Proc
Natl Acad Sci USA. 96:2147–2152. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Holloway SL, Glotzer M, King RW and Murray
AW: Anaphase is initiated by proteolysis rather than by the
inactivation of maturation-promoting factor. Cell. 73:1393–1402.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang Z, Fan M, Candas D, Zhang TQ, Qin L,
Eldridge A, Wachsmann-Hogiu S, Ahmed KM, Chromy BA, Nantajit D, et
al: Cyclin B1/Cdk1 coordinates mitochondrial respiration for
cell-cycle G2/M progression. Dev Cell. 29:217–232. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sugrue MM, Shin DY, Lee SW and Aaronson
SA: Wild-type p53 triggers a rapid senescence program in human
tumor cells lacking functional p53. Proc Natl Acad Sci USA.
94:9648–9653. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2ΔΔCT method. Methods.
25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lánczky A, Nagy Á, Bottai G, Munkácsy G,
Szabó A, Santarpia L and Győrffy B: miRpower: A web-tool to
validate survival-associated miRNAs utilizing expression data from
2178 breast cancer patients. Breast Cancer Res Treat. 160:439–446.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang Y and Callaway EC: High cellular
accumulation of sulphoraphane, a dietary anticarcinogen, is
followed by rapid transporter-mediated export as a glutathione
conjugate. Biochem J. 364:301–307. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hamsa TP, Thejass P and Kuttan G:
Induction of apoptosis by sulforaphane in highly metastatic B16F-10
melanoma cells. Drug Chem Toxicol. 34:332–340. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang Z, Li C, Shang L, Zhang Y, Zou R,
Zhan Y and Bi B: Sulforaphane induces apoptosis and inhibits
invasion in U251MG glioblastoma cells. Springerplus. 5:2352016.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ferreira de Oliveira JM, Remédios C,
Oliveira H, Pinto P, Pinho F, Pinho S, Costa M and Santos C:
Sulforaphane induces DNA damage and mitotic abnormalities in human
osteosarcoma MG-63 cells: Correlation with cell cycle arrest and
apoptosis. Nutr Cancer. 66:325–334. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ferreira de Oliveira JM, Costa M, Pedrosa
T, Pinto P, Remédios C, Oliveira H, Pimentel F, Almeida L and
Santos C: Sulforaphane induces oxidative stress and death by
p53-independent mechanism: Implication of impaired glutathione
recycling. PLoS One. 9:e929802014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen HH and Kuo MT: Role of glutathione in
the regulation of cisplatin resistance in cancer chemotherapy. Met
Based Drugs. 2010(pii): 4309392010.PubMed/NCBI
|
|
34
|
Backos DS, Franklin CC and Reigan P: The
role of glutathione in brain tumor drug resistance. Biochem
Pharmacol. 83:1005–1012. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cooper WA, Kohonen-Corish MR, McCaughan B,
Kennedy C, Sutherland RL and Lee CS: Expression and prognostic
significance of cyclin B1 and cyclin A in non-small cell lung
cancer. Histopathology. 55:28–36. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Aaltonen K, Amini RM, Heikkilä P,
Aittomäki K, Tamminen A, Nevanlinna H and Blomqvist C: High cyclin
B1 expression is associated with poor survival in breast cancer. Br
J Cancer. 100:1055–1060. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Begnami MD, Fregnani JH, Nonogaki S and
Soares FA: Evaluation of cell cycle protein expression in gastric
cancer: Cyclin B1 expression and its prognostic implication. Hum
Pathol. 41:1120–1127. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kedinger V, Meulle A, Zounib O, Bonnet ME,
Gossart JB, Benoit E, Messmer M, Shankaranarayanan P, Behr JP,
Erbacher P, et al: Sticky siRNAs targeting survivin and cyclin B1
exert an antitumoral effect on melanoma subcutaneous xenografts and
lung metastases. BMC Cancer. 13:3382013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cheng YM, Tsai CC and Hsu YC:
Sulforaphane, a dietary isothiocyanate, induces G2/M
arrest in cervical cancer cells through cyclinB1 downregulation and
GADD45β/CDC2 association. Int J Mol Sci. 17(pii): E15302016.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Korenaga D, Takesue F, Yasuda M, Honda M,
Nozoe T and Inutsuka S: The relationship between cyclin B1
overexpression and lymph node metastasis in human colorectal
cancer. Surgery. 131 Suppl 1:S114–S120. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Fang Y, Liang X, Jiang W, Li J, Xu J and
Cai X: Cyclin B1 suppresses colorectal cancer invasion and
metastasis by regulating E-cadherin. PLoS One. 10:e01268752015.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang DX, Zou YJ, Zhuang XB, Chen SX, Lin
Y, Li WL, Lin JJ and Lin ZQ: Sulforaphane suppresses EMT and
metastasis in human lung cancer through miR-616-5p-mediated
GSK3β/β-catenin signaling pathways. Acta Pharmacol Sin. 38:241–251.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Żuryń A, Litwiniec A, Safiejko-Mroczka B,
Klimaszewska-Wiśniewska A, Gagat M, Krajewski A, Gackowska L and
Grzanka D: The effect of sulforaphane on the cell cycle, apoptosis
and expression of cyclin D1 and p21 in the A549 non-small cell lung
cancer cell line. Int J Oncol. 48:2521–2533. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hagemann JH, Thomasova D, Mulay SR and
Anders HJ: Nrf2 signalling promotes ex vivo tubular epithelial cell
survival and regeneration via murine double minute (MDM)-2. Nephrol
Dial Transplant. 28:2028–2037. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yang P, Chen W, Li X, Eilers G, He Q, Liu
L, Wu Y, Wu Y, Yu W, Fletcher JA and Ou WB: Downregulation of
cyclin D1 sensitizes cancer cells to MDM2 antagonist Nutlin-3.
Oncotarget. 7:32652–32663. 2016.PubMed/NCBI
|
|
46
|
Li L, Cui D, Zheng SJ, Lou H and Tang J:
Regulation of Actinomycin D induced upregulation of Mdm2 in H1299
cells. DNA Repair. 11:112–119. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sansó M and Fisher RP: Pause, play,
repeat: CDKs push RNAP II's buttons. Transcription. 4:146–152.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Bartkowiak B, Liu P, Phatnani HP, Fuda NJ,
Cooper JJ, Price DH, Adelman K, Lis JT and Greenleaf AL: CDK12 is a
transcription elongation-associated CTD kinase, the metazoan
ortholog of yeast Ctk1. Genes Dev. 24:2303–2316. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bell D, Berchuck A, Birrer M, Chien J,
Cramer D, Dao F, Dhir R, DiSaia P, Gabra H, Glenn P, et al:
Integrated genomic analyses of ovarian carcinoma. Nature.
474:609–615. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ekumi KM, Paculova H, Lenasi T,
Pospichalova V, Bösken CA, Rybarikova J, Bryja V, Geyer M, Blazek D
and Barboric M: Ovarian carcinoma CDK12 mutations misregulate
expression of DNA repair genes via deficient formation and function
of the Cdk12/CycK complex. Nucleic Acids Res. 43:2575–2589. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Carter SL, Cibulskis K, Helman E, McKenna
A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et
al: Absolute quantification of somatic DNA alterations in human
cancer. Nat Biotechnol. 30:413–421. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Mertins P, Mani DR, Ruggles KV, Gillette
MA, Clauser KR, Wang P, Wang X, Qiao JW, Cao S, Petralia F, et al:
Proteogenomics connects somatic mutations to signalling in breast
cancer. Nature. 534:55–62. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
O'Connell BC, Adamson B, Lydeard JR, Sowa
ME, Ciccia A, Bredemeyer AL, Schlabach M, Gygi SP, Elledge SJ and
Harper JW: A genome-wide camptothecin sensitivity screen identifies
a mammalian MMS22L-NFKBIL2 complex required for genomic stability.
Mol Cell. 40:645–657. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Joshi PM, Sutor SL, Huntoon CJ and Karnitz
LM: Ovarian cancer-associated mutations disable catalytic activity
of CDK12, a kinase that promotes homologous recombination repair
and resistance to cisplatin and poly(ADP-ribose) polymerase
inhibitors. J Biol Chem. 289:9247–9253. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kim HE, Kim DG, Lee KJ, Son JG, Song MY,
Park YM, Kim JJ, Cho SW, Chi SG, Cheong HS, et al: Frequent
amplification of CENPF, GMNN and CDK13 genes in hepatocellular
carcinomas. PLoS One. 7:e432232012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Schecher S, Walter B, Falkenstein M,
Macher-Goeppinger S, Stenzel P, Krümpelmann K, Hadaschik B, Perner
S, Kristiansen G, Duensing S, et al: Cyclin K dependent regulation
of Aurora B affects apoptosis and proliferation by induction of
mitotic catastrophe in prostate cancer: Cyclin K in prostate
cancer. Int J Cancer. 141:1643–1653. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Johannes JW, Denz CR, Su N, Wu A,
Impastato AC, Mlynarski S, Varnes JG, Prince DB, Cidado J, Gao N,
et al: Structure-based design of selective noncovalent CDK12
inhibitors. ChemMedChem. 13:231–235. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhang T, Kwiatkowski N, Olson CM,
Dixon-Clarke SE, Abraham BJ, Greifenberg AK, Ficarro SB, Elkins JM,
Liang Y, Hannett NM, et al: Covalent targeting of remote cysteine
residues to develop CDK12 and CDK13 inhibitors. Nat Chem Biol.
12:876–884. 2016. View Article : Google Scholar : PubMed/NCBI
|