Introduction
Macrophage colony-stimulating factor (M-CSF), also
known as colony-stimulating factor-1 (CSF-1), promotes monocyte and
macrophage cell growth, proliferation, and differentiation as well
as maintenance of the biological functions of monocytes and
macrophages. Notably, M-CSF is also expressed in many tumour
tissues and cancer cells. The expression of M-CSF is markedly
enhanced in various cancers (1–3).
Increased nuclear expression of M-CSF was revealed to be correlated
with poor prognosis and the metastatic potential of breast cancer
cells (4). Aharinejad et al
found that the high expression of cytoplasmic M-CSF in MDA-MB-231
breast cancer cells contributes to the invasion and metastasis of
tumours in a mouse model (2).
Similarly, M-CSF was revealed to play an important role in the
resistance of 5-FU in U87MG glioblastoma (5). In addition, an M-CSF antibody was
revealed to reverse the chemoresistance of MCF-7 cells (6). In our previous study, it was revealed
that M-CSF induced drug and apoptosis resistance in MCF-7 cells.
Therefore, M-CSF is a tumour marker since it is related to
anti-apoptosis and drug resistance in tumour cells.
Apoptosis is a common form of programmed cell death,
and its deregulation has been associated with tumour initiation,
progression, and metastasis in various cancers including breast
cancer (7). HIF-1 has been
demonstrated to be involved in glycolysis, angiogenesis and
migration, and to regulate invasion factors that are important for
tumour progression and metastasis (8). HIF-1 activity depends on the
expression level of HIF-1α. HIF-1α expression is maintained at low
levels under normoxic conditions, however it is significantly
induced by hypoxia (9). HIF-1α
induces various transcriptional programs, some of which include
pluripotency factors in hypoxic conditions (10). A recent study revealed that HIF-1α
regulated anti-apoptotic genes, which ultimately led to increased
tumour growth and drug resistance (11). Murine double minute 2 (MDM2), is an
oncogene that is upstream of HIF-1α and regulates the expression of
HIF-1α (12,13). M-CSF was revealed to directly
decrease the expression of MDM2, further contributing to drug
resistance in tumour treatments (5). M-CSF may modulate the expression of
HIF-1α, however, the mechanism is still unclear.
BNIP3 is a proapoptotic member of the Bcl-2 family
and is a downstream target protein of HIF-1α (14). BNIP3 has a key role in the
pathogenesis of many diseases, and it binds anti-apoptotic
proteins, including Bcl-2 and BCL-XL, which inhibits their
anti-apoptotic activity (15). When
BNIP3 binds anti-apoptotic proteins to form heterodimers, it
activates pro-apoptotic proteins, such as Bax and Bak, resulting in
pro-apoptotic effects (16). A
recent study revealed that apoptosis was upregulated after
transfection of BNIP3 into MCF-7 cells (17) and rat fibroblasts (18). BNIP3 was activated by the ATPase
inhibitor, bafilomycin, in MCF-7 and MDA-MB-231 breast cancer
cells, resulting in apoptosis (19). Thus, BNIP3 induced apoptosis in
breast cancer cells, indicating that it may be an effective tumour
therapeutic target.
Our previous results revealed that cytoplasmic
macrophage colony-stimulating factor induced adriamycin-resistance
(20). Moreover, antineoplastic
agents play an important role in inducing cancer cell apoptosis,
and the anti-apoptosis mechanism in cancer cells is vital for
tumour multidrug resistance (21).
However, anti-apoptotic mechanisms have not been clearly
elucidated. Therefore, our hypothesis indicated that M-CSF
inhibited the expression of HIF-1α, which decreased BNIP3, further
reducing the binding of anti-apoptotic proteins, such as Bax, to
suppress the apoptotic effect. Experiments based on the
aforementioned hypothesis were performed, to elucidate the
mechanism of cytoplasmic M-CSF-induced cancer cell anti-apoptosis
and multidrug resistance mechanisms.
Materials and methods
Cell lines and reagents
MCF-7, a human breast cancer cell line, was obtained
from the American Type Culture Collection (ATCC; Manassas, VA,
USA). MCF-7-M cells were transfected with M-CSF, and MCF-7-C cells
were transfected with a control plasmid (empty vector). MCF-7,
MCF-7-C and MCF-7-M cells were cultured in RPMI-1640 medium
(Gibco-BRL; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
containing 10% newborn calf serum (NBCS) and antibiotics (ExCell,
Shanghai, China) at 37°C in a humidified atmosphere containing 95%
air and 5% CO2. Adriamycin was purchased from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany).
Stable transfection
In this experiment, the cytoplasmic positioning and
recombination vector pCMV/myc/cyto-M-CSF, was constructed in this
laboratory (Department of Pharmacology, Hunan University of
Medicine, Huaihua, China) and was used for the present study. This
vector contained a cytoplasmic positioning sequence, which forced
M-CSF localization in the cytoplasm. The M-CSF molecule in the
recombinant vector had a deleted exocytosine signal peptide
consisting of 32 amino acids at the N-terminus, which prevented
M-CSF secretion outside of the cell, thereby blocking its function
as a signal molecule. The pCMV/myc/cyto-M-CSF recombinant vector
was used in our previous research (20). In the present study, in order to
confirm the efficiency of stable transfection, M-CSF expression was
determined by western blot analysis in MCF-7 cells.
MCF-7 cells were seeded in 6-well plates at a
density of 1×105 cells/well in RPMI-1640 medium
containing 10% FBS for 24 h. Cells were then stably transfected
with either pCMV/cyto/myc-M-CSF (cytoplasmic M-CSF gene
overexpressed) or pCMV/cyto/myc vector (empty vector) using
Lipofectamine 2000 reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol. The transfection
mixtures were replaced with RPMI-1640 medium containing 10% FBS.
Cells were harvested at 48 h post-transfection.
Bioinformatics analysis of protein
interaction
Using the online STRING database (https://string-db.org/), which is a biological
database and web resource for known and predicted PPIs, we
developed a network of DEG-encoded proteins and PPIs.
Western blot analysis
Cells were washed with cold PBS and mechanically
homogenized in RIPA lysis buffer (Beyotime Institute of
Biotechnology, Haimen, China). Total protein samples (60 mg/well)
were separated on 10 or 15% SDS-PAGE gels. Proteins were then
transferred to PVDF membranes (EMD Millipore, Billerica, MA, USA).
After blocking with 5% non-fat dried milk for 2 h, the membranes
were washed for 10 min in TBST (0.1% Tween-20, TBS) three times.
The membranes were then incubated with primary antibodies against
HIF-1α (dilution 1:1,000; cat. no. 3716; Cell Signaling Technoogy,
Inc., Danvers, MA, USA), BNIP3 (Ana40) (dilution 1:1,000; cat. no.
ab10433; Abcam, Cambridge, UK) Bax (dilution 1:1,000; cat. no.
D2E11; Cell Signaling Technoogy, Inc.), Bcl-2 (E17) (dilution
1:800; cat. no. ab32124; Abcam) and β-actin (dilution 1:2,000; cat.
no. 66009-1-lg; Proteintech Group, Inc., Wuhan, China) overnight at
4°C. Subsequently, the membranes were then incubated with secondary
antibodies [goat anti-rabbit IgG-HRP (dilution 1:4,000; cat. no.
SA00001-2; Proteintech Group, Inc.), goat anti-mouse IgG-HRP
(dilution 1:4,000; cat. no. SA00001-1; Proteintech Group, Inc.) and
rabbit anti-goat IgG-HRP (dilution 1:4,000; cat. no. SA00001-4;
Proteintech Group, Inc.) for 1 h at room temperature. Signals were
detected by Western Chemiluminescence HRP Substrate (ECL) solution
(Beyotime Institute of Biotechnology). Protein bands relative to
β-actin were quantified using Glyko BandScan 5.0 software (Glyko
Inc., Novato, CA, USA).
Annexin V-fluorescein isothiocyanate
apoptosis assay
An Annexin V-FLUO Staining Kit (Boehringer-Mannheim;
Roche Diagnostics, Mannheim, Germany) was used to evaluate
doxorubicin-induced apoptosis. Cells were cultured in a 6-well
plate and exposed to 0.5 µM ADM for 24 h. Cells were collected in a
10-ml centrifuge tube and stained with Annexin V-FLUOS and PI for
15 min. Apoptosis was immediately analysed with a flow cytometer
(Beckman Coulter, Inc., Fullerton, CA, USA) at a wavelength of 488
nm.
Hoechst 33342 staining for the
apoptosis assay
Hoechst 33342 dye is cell permeable and binds to DNA
in live or dead cells. However, PI is cell membrane impermeable and
excluded from viable cells, and is typically used to identify dead
cells. MCF-7 cells (5×104 cells/well in 1 ml) were
seeded in 24-well plates and cultured for 24 h at 37°C under a
humidified atmosphere of 5% CO2. Thereafter, serum-free
medium was replaced with the same medium containing 0, 0.5, 1, 2, 4
and 8 µM ADM. After 24 h of drug incubation, the medium was removed
and Immunol Staining Fix Solution (Beyotime Institute of
Biotechnology) was added (0.5 ml/well) for 20 min at 4°C. Plates
were then washed two times for 3 min in PBS. After washing, Hoechst
33342 staining solution (Beyotime Institute of Biotechnology) was
added (0.5 ml/well) and incubated for 20 min at 37°C. Plates were
then washed two times for 3 min in PBS. Hoechst-positive cells
exhibited blue fluorescence, while PI-positive cells exhibited red
fluorescence. Apoptotic cells were Hoechst-positive and
demonstrated characteristic features of apoptosis, such as,
condensed or fragmented nuclei. Staining was analysed by morphology
and fluorescence.
Co-immunoprecipitation analysis
Cells were divided into six groups according to
different processing factors as follows: MCF-7, MCF-7-C, MCF-7-M,
MCF-7+ADM, MCF-7-C+ADM and MCF-7-M+ADM. Cells were cultured for 24
h before adding 200 µl of IP lysis buffer (containing 2 µl of PMSF,
2 µl of protease inhibitor and 2 µl protein phosphatase inhibitor),
which was 5-fold the total volume of cells. Cells were suspended
and lysed on ice for 30 min. Cells were lysed and then incubated
overnight with 1 µg of Bcl-2 antibody at 4°C, and 1 µg of rabbit
normal lgG was used as the negative control group. Lysates were
then incubated for 4 h with 150 µl of a 10% suspension of protein
A-sepharose beads (Sigma-Aldrich, Poole, UK) at 4°C.
Immunocomplexes were then collected for western blotting to detect
the expression of Bcl-2, Bax and BNIP3.
Statistical analysis
All results were expressed as the mean ± standard
deviation (SD). Data analysis was performed using SPSS 18.0 (SPSS,
Inc., Chicago, IL, USA). Groups were compared using Student's
t-test or two-way ANOVA. Multiple comparison between the groups was
performed using the S-N-K method at a significance level of α=0.05.
P<0.05 was considered to indicate a statistically significant
difference.
Results
M-CSF expression is upregulated in
overexpressed trans-fectants of MCF-7 cells
To determine the efficiency of M-CSF stable
transfection, the expression of M-CSF was assessed in MCF-7,
MCF-7-C and MCF-7-M cells using western blotting (Fig. 1). The results revealed that the
expression of M-CSF was not significantly different in MCF-7-C
cells and MCF-7 cells. There was a much higher expression of M-CSF
protein in MCF-7-M cells compared to the MCF-7 and MCF-7-C
cells.
 | Figure 1.The efficiency of M-CSF stable
transfection. M-CSF protein expression was determined by western
blotting in MCF-7, MCF-7-C, and MCF-7-M cells. Band densitometry
analysis of M-CSF expression in MCF-7, MCF-7-C, and MCF-7-M cells
normalized to β-actin, respectively. **P<0.01, MCF-7-M vs. MCF-7
or MCF-7-C. M-CSF, macrophage colony-stimulating factor; MCF-7-M,
MCF-7 cells transfected with M-CSF; MCF-7-C, MCF-7 cells
transfected with control plasmid. M-CSF, macrophage
colony-stimulating factor. |
Cytoplasmic M-CSF regulates the
expression of HIF-1α in MCF-7 cells
The bioinformatics online analysis software, STRING,
was used to analyse the interaction of M-CSF and HIF-1α. M-CSF and
its receptor interacted with MDM2, which resulted in MDM2 and
HIF-1α regulating each other (Fig.
2). Previous research demonstrated that M-CSF directly
decreased the expression of MDM2, further leading to tumour drug
resistance. Additionally, MDM2 upregulated HIF-1α in a
p53-independent manner. Thus, these results indicated that M-CSF
decreased the expression of HIF-1α by regulating MDM2.
Cytoplasmic M-CSF suppresses the
expression of HIF-1α, BNIP3 and Bax in MCF-7 cells treated with
ADM
As aforementioned, M-CSF is associated with tumour
cell anti-apoptosis and drug resistance. HIF-1α, BNIP3 and Bax play
an important role in cell apoptosis. To determine if M-CSF has a
regulatory effect on HIF-1α, BNIP3 and Bax in MCF-7 cells, western
blotting was performed to analyse the expression of these proteins
in MCF-7, MCF-7-C and MCF-7-M cells before and after treatment with
ADM. The expression of HIF-1α and BNIP3 was lower in MCF-7-M cells
compared to MCF-7 cells or MCF-7-C cells without ADM (Fig. 3A and B). Bax protein expression had
no significant difference in MCF-7, MCF-7-C and MCF-7-M cells
treated without ADM (Fig. 3C).
Compared to MCF-7 and MCF-7-C cells, the expression of HIF-1α,
BNIP3 and Bax was strongly decreased after ADM treatment (Fig. 3A-C). Moreover, HIF-1α, BNIP3 and Bax
protein expression decreased in MCF-7-M cells treated with ADM
compared to untreated MCF-7-M (Fig.
3A-C). Collectively, these data revealed that cytoplasmic M-CSF
inhibited the expression of HIF-1α, BNIP3 and Bax in MCF-7cells
treated with ADM and that ADM enhanced the inhibitory effect of
M-CSF in MCF-7 cells.
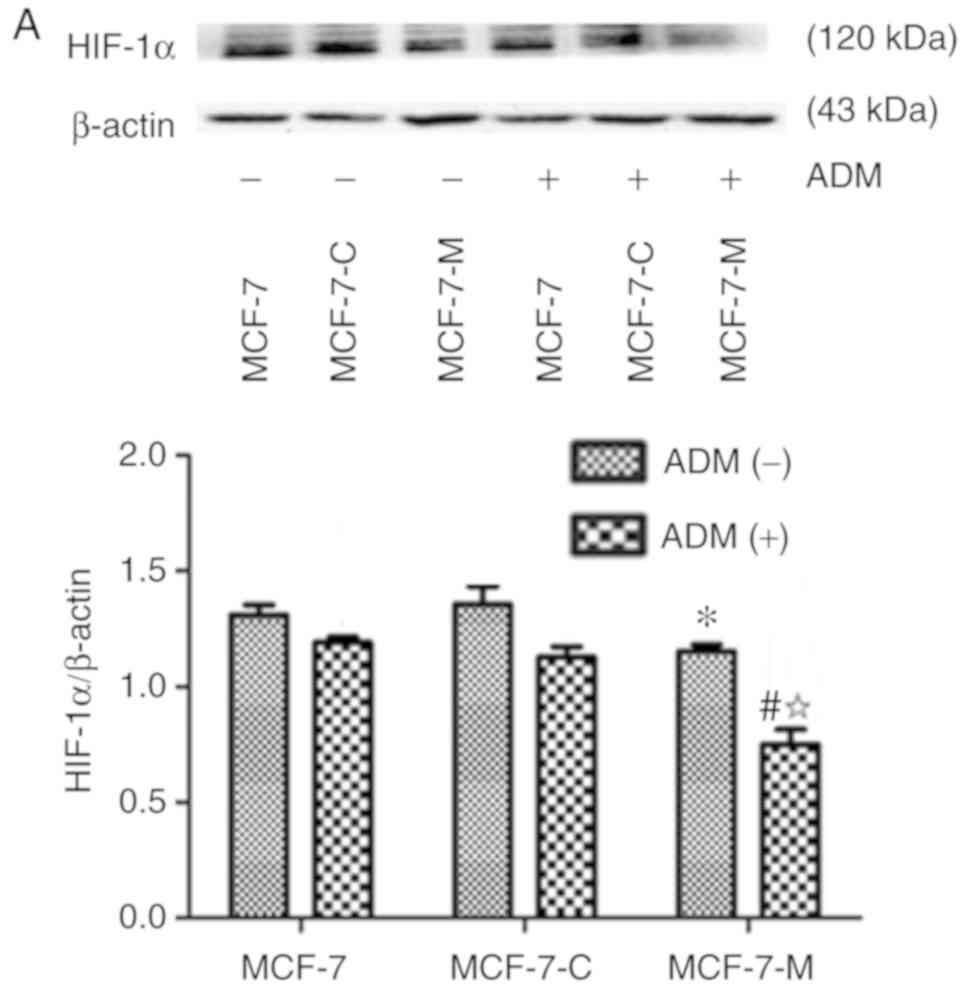 | Figure 3.Effect of cytoplasmic M-CSF on the
expression of HIF-1α, BNIP3 and Bax in MCF-7 cells treated with or
without ADM. (A-C) HIF-1α, BNIP3 and Bax expression in MCF-7,
MCF-7-C, and MCF-7-M cells with or without ADM (2 µM) treatment as
analysed by western blotting. Band densitometry analysis of HIF-1α,
BNIP3 and Bax expression normalized to β-actin in MCF-7, MCF-7-C
and MCF-7-M cells with or without ADM treatment. (A and B)
*P<0.05, MCF-7-M vs. MCF-7 or MCF-7-C; #P<0.01,
MCF-7-M+ADM vs. MCF-7+ADM or MCF-7-C+ADM; and
☆P<0.01, MCF-7-M+ADM vs. MCF-7-M. (C) *P>0.05,
MCF-7-M vs. MCF-7 or MCF-7-C; #P<0.01, MCF-7-M+ADM
vs. MCF-7+ADM or MCF-7-C+ADM; and ☆P<0.01,
MCF-7-M+ADM vs. MCF-7-M. M-CSF, macrophage colony-stimulating
factor; ADM, adriamycin; HIF-1α, hypoxia-inducible factor-1α;
BNIP3, Bcl-2/adenovirus E1B 19 kDa-interacting protein 3; MCF-7-M,
MCF-7 cells transfected with M-CSF; MCF-7-C, MCF-7 cells
transfected with control plasmid. |
Cytoplasmic M-CSF reduces the binding
of Bcl-2 to BNIP3 but increases Bcl-2 binding to Bax in MCF-7 cells
after treatment with ADM
Previous research has demonstrated that Bcl-2 is an
anti-apoptotic protein and that BNIP3 and Bax competitively bind to
Bcl-2. The present study revealed that M-CSF suppressed the
expression of BNIP3 and Bax. Thus, M-CSF significantly decreased
Bax expression in MCF-7 cells by inhibiting the binding of BNIP3 to
Bcl-2 but increasing the binding of Bax to Bcl-2, blocking
apoptosis in MCF-7 cells. Co-immunoprecipitation analysis was
performed to analyse the state of Bcl-2 binding to BNIP3 and Bax
protein using Bcl-2 as the antibody in MCF-7, MCF-7-C and MCF-7-M
cells incubated with ADM (2 µM) for 24 h. There was no significant
difference in the amount of BNIP3 that Bcl-2 bound in MCF-7,
MCF-7-C and MCF-7-M cells (Fig. 4A and
B). The amount of Bcl-2 binding to Bax was greater in MCF-7-M
cells than in MCF-7 or MCF-7-C cells without ADM treatment
(Fig. 4A and B). Treatment with ADM
caused a significantly lower amount of BNIP3 binding to Bcl-2 in
MCF-7-M cells compared to MCF-7 or MCF-7-C cells (Fig. 4A and B). The amount of Bax binding
to Bcl-2 in MCF-7-M cells was higher than that in MCF-7 cells and
MCF-7-C cells (Fig. 4A and B).
Collectively, these results indicated that cytoplasmic M-CSF
induced anti-apoptosis by inhibiting the binding of Bcl-2 to BNIP3
protein and by increasing the binding of Bcl-2 to Bax protein in
MCF-7 cells.
 | Figure 4.Effect of cytoplasmic M-CSF on the
interaction between Bcl-2 and BNIP3 or between Bcl-2 and Bax in
MCF-7 cells treated with or without ADM. (A) Cells were divided
into a control group (Input group) and positive group (IP)
according to the presence of the Bcl-2 antibody. BNIP3, Bax and
Bcl-2 protein expression was analysed by western blotting in MCF-7,
MCF-7-C and MCF-7-M cells with or without ADM treatment (2 µM). (B)
Band densitometry analysis of BNIP3, Bax and Bcl-2 expression in
MCF-7, MCF-7-C and MCF-7-M cells with or without ADM treatment.
*P>0.05, MCF-7-M vs. MCF-7 or MCF-7-C; #P<0.01,
MCF-7-M+ADM vs. MCF-7+ADM or MCF-7-C+ADM; ☆P<0.01,
MCF-7-M+ADM vs. MCF-7-M. M-CSF, macrophage colony-stimulating
factor; ADM, adriamycin; HIF-1α, hypoxia-inducible factor-1α;
BNIP3, Bcl-2/adenovirus E1B 19 kDa-interacting protein 3; MCF-7-M,
MCF-7 cells transfected with M-CSF; MCF-7-C, MCF-7 cells
transfected with control plasmid. |
Cytoplasmic M-CSF increases the
capability of anti-apoptosis
Hoechst 33342 staining detection of
cell apoptosis
MCF-7, MCF-7-C, and MCF-7-M cells were plated and
cultured in 24-well plates for 12 h followed by incubation with ADM
at 0, 0.5, 1.0, 2.0, 4.0 and 8.0 µM for 24 h. Cells apoptosis was
then analysed using Hoechst 33342 staining. MCF-7, MCF-7-C, and
MCF-7-M cells apoptosis significantly increased with increasing
drug concentration, but the number of nuclear MCF-7-M cells was
decreased in comparison to that of MCF-7 and MCF-7-C cells treated
with the same concentration of ADM (Fig. 5A and B). Collectively, these results
indicated that M-CSF enhanced the anti-apoptotic ability of MCF-7
cells.
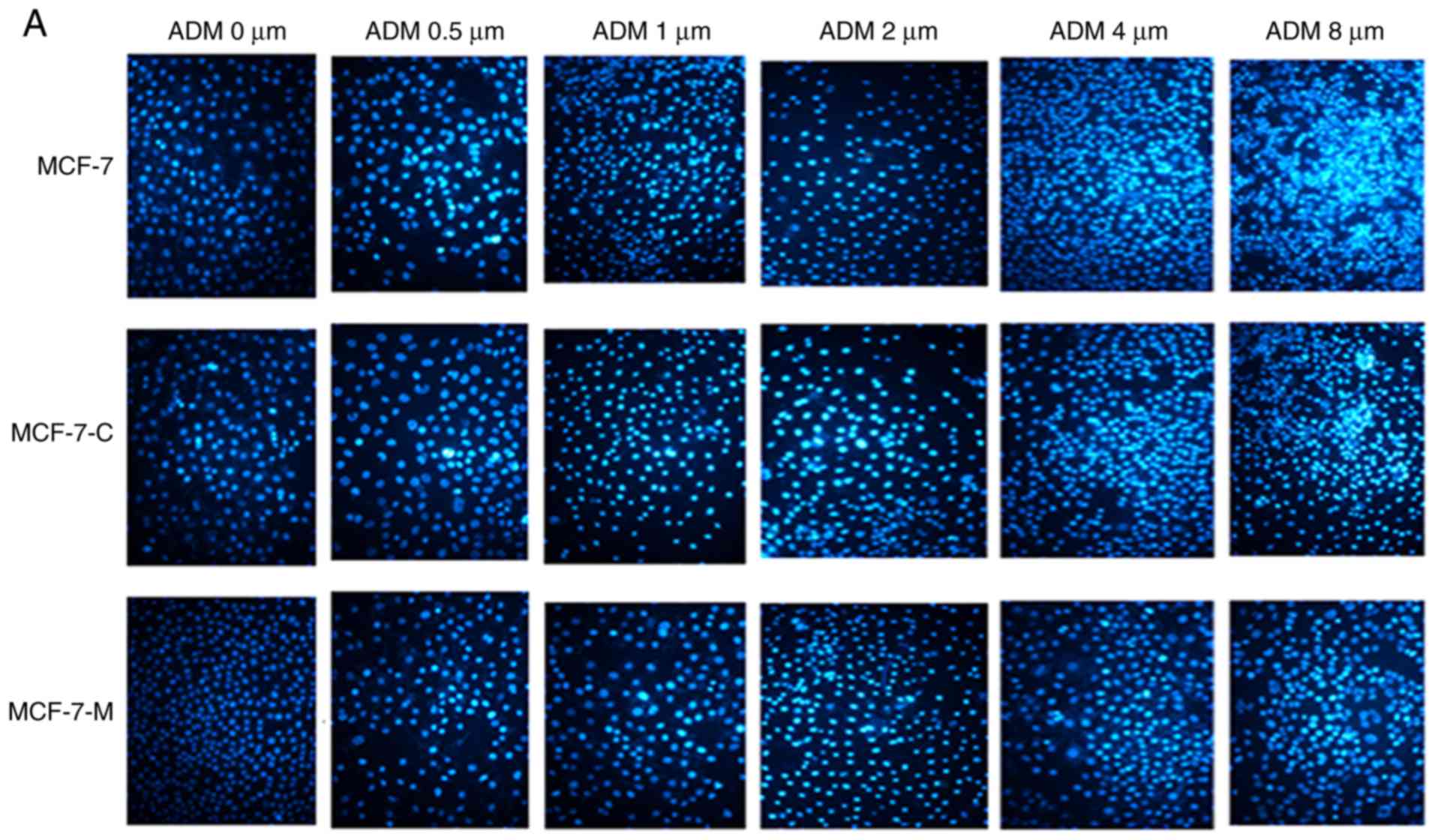 | Figure 5.Cytoplasmic M-CSF inhibits apoptosis
in MCF-7 cells treated with ADM (Hoechst 33342 staining). (A)
Apoptosis analysis by Hoechst 33342 staining in 0, 0.5, 1.0, 2.0,
4.0 and 8.0 µM ADM-treated MCF-7, MCF-7-C and MCF-7-M cells. (B)
Data analysis of the rate of Hoechst 33342-positive nuclei
(*P<0.05, MCF-7-M vs. MCF-7 or MCF-7-C; n=6). M-CSF, macrophage
colony-stimulating factor; ADM, adriamycin; MCF-7-M, MCF-7 cells
transfected with M-CSF; MCF-7-C, MCF-7 cells transfected with
control plasmid. |
Apoptosis analysis by flow
cytometry
To further determine if M-CSF influences the
anti-apoptotic capability in MCF-7 cells, MCF-7 cell apoptosis was
assessed using flow cytometry. A significant reduction of
ADM-induced apoptosis was observed in MCF-7-M cells compared to
MCF-7 and MCF-7-C cells (Fig. 6A and
B). Collectively, these data revealed that M-CSF inhibited
ADM-induced apoptosis in MCF-7 cells.
 | Figure 6.Cytoplasmic M-CSF inhibits apoptosis
in MCF-7 cells treated with ADM. (A-a, -b, -c) Flow cytometric
apoptosis analysis of MCF-7, MCF-7-C and MCF-7-M cells treated with
or without ADM (0.5 µM). (B) Band densitometry analysis of the
apoptosis rate represented in A. Flow cytometry, error bars
represent ± standard deviation, n=3; *P<0.05, MCF-7-M vs. MCF-7
or MCF-7-C; #P<0.01, MCF-7-M+ADM vs. MCF-7+ADM or
MCF-7-C+ADM; ☆P<0.01, MCF-7-M+ADM vs. MCF-7-M; n=3).
M-CSF, macrophage colony-stimulating factor; ADM, adriamycin;
MCF-7-M, MCF-7 cells transfected with M-CSF; MCF-7-C, MCF-7 cells
transfected with control plasmid. |
Cytoplasmic M-CSF decreases the
expression of caspase-3 and caspase-9
The expression of caspase-3 and caspase-9 was
assessed in MCF-7, MCF-7-C and MCF-7-M cells incubated in the
presence or absence of ADM (2 µM) for 24 h using western blotting.
The expression of caspase-3 and caspase-9 was not significantly
different in untreated MCF-7, MCF-7-C and MCF-7-M cells (Fig. 7A-C), however ADM treatment,
significantly reduced caspase-3 and caspase-9 in MCF-7-M cells in
comparison to MCF-7 or MCF-7-C cells (Fig. 7A-C). The expression levels of
caspase-3 and caspase-9 in MCF-7-M cells treated with ADM were
significantly lower than those in untreated MCF-7-M cells (Fig. 7A-C). These results indicated that
cytoplasmic M-CSF suppressed ADM-induced caspase-3 and caspase-9
protein expression in MCF-7 cells.
 | Figure 7.Effect of cytoplasmic M-CSF on the
expression of caspase-3 and caspase-9 in MCF-7 cells treated with
ADM. (A) Caspase-3 and caspase-9 protein expression was analysed by
western blotting in MCF-7, MCF-7-C and MCF-7-M cells treated with
or without ADM (2 µM). (B and C) Band densitometry analysis of
caspase-3 and caspase-9 expression normalized to β-actin in MCF-7,
MCF-7-C and MCF-7-M cells treated with or without ADM. *P>0.05,
MCF-7-M vs. MCF-7 or MCF-7-C; #P<0.01, MCF-7-M+ADM
vs. MCF-7+ADM or MCF-7-C+ADM; ☆P<0.01, MCF-7-M+ADM
vs. MCF-7-M. M-CSF, macrophage colony-stimulating factor; ADM,
adriamycin; caspase-3/9, cysteinyl aspartate-specific proteinase
3/9; MCF-7-M, MCF-7 cells transfected with M-CSF; MCF-7-C, MCF-7
cells transfected with control plasmid. |
Discussion
Breast cancer is a serious threat to the health of
women and is the major cause of death in 40- to 55-year-old women.
Globally, breast cancer accounted for the highest number of new
cancer cases in 2015 (22). Nearly
30% of newly diagnosed patients with early stage breast cancer
develop a distant metastasis despite receiving therapy (23). Current therapy options for breast
cancer include surgery, hormonal therapy, immunotherapy,
chemotherapy, radiation therapy, or a combination of these
treatments (24). The main
treatment method for breast cancer is radical surgery combined with
postoperative chemotherapy and radiotherapy. However, the use of
chemotherapeutic drugs is usually accompanied by deleterious side
effects, and the development of drug resistance occurs when applied
for a longer period. Drug resistance is related to tumour cell
apoptosis, but the mechanism is unclear.
The growth of tumour cells is regulated by various
factors. Many growth factors and cytokines are involved in the
regulation of the tumour microenvironment in the immune system, and
their function in immune surveillance and immune clearance. For
example, M-CSF, which is known as CSF-1, has a vital role in the
biological function of mononuclear macrophages as well as in tumour
invasion, metastasis, drug resistance and prognosis (25). M-CSF is expressed in
tumour-associated macrophages (TAMs) (26,27).
In recent years, several studies have reported high expression of
cytoplasmic M-CSF in type II papillary renal cell carcinoma
(28), breast (29,30),
ovarian (30,31), endometrial (32), colorectal (33), pancreatic (34), prostate and head and neck cancer
(35). Additionally, a study
revealed that the overexpression of M-CSF and its receptor was
associated with a poor prognosis (36). M-CSF also promoted tumour cell
proliferation (37,38) and non-small cell lung cancer bone
metastases (39). Lin et al
discovered that overexpression of cytoplasmic M-CSF was responsible
for the invasion and metastases of cancer cells in a mouse breast
cancer model (40). An M-CSF gene
null mutation in rats resulted in decreased malignancy and
metastasis of tumours (41).
Collectively, these findings indicated that M-CSF plays a vital
role in the development of diverse tumours. Although, the
mechanisms may be different in these tumours, M-CSF ultimately
results in tumour development and chemoresistance. Hence, M-CSF may
act as a factor to induce tumour cell anti-apoptosis in MCF-7
breast cancer. The specific effects of M-CSF in cancer cells were
increased by stable transfection of cytoplasmic M-CSF into MCF-7
cells. In the presence of ADM, cytoplasmic M-CSF led to an increase
in the anti- apoptosis capability of MCF-7 cells.
Considerable attention has been paid to the
contribution of the tumour microenvironment. For example, hypoxia
is an important component of the microenvironment of various types
of solid tumours (42), including
breast cancer. Hypoxia increases ‘stemness’, EMT, migratory
capabilities and invasive capabilities (20). HIF-1 is a transcription factor that
plays an important role in the response to hypoxia. Under hypoxic
conditions, HIF-1 has a corresponding physiological function via
binding target proteins, including vascular endothelial growth
factor (VEGF), nitric oxide synthase (NOS), p53, growth factors and
inflammatory factors. A previous study revealed that
hypoxia-inducible factor-dependent signalling promoted
M-CSF-induced macrophage recruitment in triple-negative breast
cancer cells and mesenchymal stem cells (43). MDM2 is located upstream of HIF-1α
and has been revealed to regulate the expression of HIF-1.
Moreover, M-CSF has been demonstrated to reduce the protein
expression of MDM2. These findings indicated that M-CSF may induce
tumour cell proliferation and drug resistance by regulating the
expression of HIF-1α. The present study determined that M-CSF was
related to the expression of HIF-1α through bioinformatics analysis
and that cytoplasmic M-CSF suppressed the expression of HIF-1α in
MCF-7 cells treated with ADM.
Apoptosis is a cell death process that uses
specialized machinery for self-destruction. If the apoptotic
process is dysregulated, tumour tissue develops rapidly, leading to
malignancy. The anti-apoptotic Bcl-2 protein has been revealed to
be increased in breast cancer cells, indicating the imbalance
between apoptosis and anti-apoptosis (44). BNIP3 is a target protein of HIF-1α
and is a proapoptotic member of the Bcl-2 family of proteins, and
HIF-1α has been demonstrated to bind to the HRE-2 site of the BNIP3
promoter. BNIP3 binds anti-apoptotic proteins, such as Bcl-2 and
Bcl-xl, to form heterodimers, which activate pro-apoptotic proteins
(45). Elevated BNIP3 expression
was revealed to be associated with poor prognosis (46). These results indicated that
cytoplasmic M-CSF induced anti-apoptosis in breast cancer by
regulating HIF-1α/BNIP3/Bax. The present study also revealed that
cytoplasmic M-CSF induced cell anti-apoptosis by inhibiting the
binding of Bcl-2 to BNIP3 protein and by increasing the binding of
Bcl-2 to Bax protein in MCF-7 cells treated with ADM.
Collectively, these results indicated that
cytoplasmic M-CSF suppresses cells apoptosis by inhibiting
HIF-1α/BNIP3/Bax signalling in MCF-7 cells. Bioinformatics analysis
revealed that M-CSF not only directly regulated the expression of
HIF-1 only MDM2, but also indirectly regulated HIF-1α protein
through p53. A previous study revealed that M-CSF-induced 5-FU
resistance was mediated by decreasing the expression of MDM2 and
ABCB1. In addition, MDM2 induced upregulation of HIF-1α protein in
a p53-independent manner (13).
Therefore, M-CSF suppressed the expression of HIF-1α through a
p53-independent pathway. A model was generated by stably
transfecting MCF-7 cells with M-CSF to explore the relationship
between cytoplasmic M-CSF and MCF-7 cell death. Hoechst 33342
staining and flow cytometry was performed to demonstrate that the
antineoplastic agent-induced rate of apoptosis was significantly
decreased in MCF-7-M cells in comparison with control groups.
Western blotting revealed a significant reduction of HIF-1α, BNIP3,
Bax, caspase-3 and caspase-9 protein expression in MCF-7-M cells
treated with ADM compared to control groups. To further explore the
specific mechanism of M-CSF-mediated low expression of
pro-apoptotic Bax protein, the relationship of Bcl-2 binding to
BINP3 and Bax was analysed using co-immunoprecipitation. The
binding rate of BNIP3 to Bcl-2 was decreased, but the binding rate
of Bcl-2 to Bax was increased, thereby, leading to the reduction of
free Bax. Thus, cytoplasmic M-CSF suppressed cell apoptosis by
inhibiting HIF-1α/BNIP3/Bax signalling in human MCF-7 breast cancer
cells due to decreased binding of BNIP3/Bcl-2 and increased binding
of Bcl-2 to Bax, which resulted in low free Bax and ultimately
apoptosis resistance.
The present study reported for the first time, to
the best of our knowledge, that apoptosis was regulated through the
M-CSF/HIF-1α/BNIP3/Bax signalling pathway in MCF-7 breast cancer
cells, which provided a new target for breast cancer therapy. This
regulation is a new p53-independent pathway, which plays an
important role in the therapy and prognosis of breast cancer.
However, it remains unknown which pathway is involved in the
M-CSF-induced reduction of HIF-1α expression in MCF-7 breast cancer
cells. M-CSF may regulate HIF-1α protein expression via MDM2. Since
HIF-1α regulates angiogenic factors, M-CSF/HIF-1α may be associated
with tumour angiogenesis. Several studies have revealed that BNIP3
is related to autophagy. Thus, M-CSF-mediated autophagy may be
induced by the HIF-1α/BNIP3/beclin1 pathway in breast cancer cells,
and excessive autophagy induces apoptosis.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Hunan
Provincial Science and Technology Plan Key Projects of China
(2017SK2183), the Science and Technology Plan Key Projects of Hunan
Province (10A087) and the Hunan Provincial Natural Science Key
Foundation of Hunan (09JJ3060).
Availability of data and materials
All data generated or analysed during this study are
included in this published article.
Authors' contributions
ST designed the experiments. MZ, QL, LL, JN, JT, XL
performed all the experimental procedures; ST and ZM performed the
statistical analysis; MZ, QL and LL prepared the first draft of the
manuscript. All authors read and approved the manuscript and agree
to be accountable for all aspects of the research in ensuring that
the accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mroczko B and Szmitkowski M:
Macrophage-colony stimulating factor (M-csf) in diagnostic and
monitoring of non-small-cell lung cancer (NSCLC). Pol Arch Med
Wewn. 105:203–209. 2001.(In Polish). PubMed/NCBI
|
|
2
|
Aharinejad S, Paulus P, Sioud M, Hofmann
M, Zins K, Schäfer R, Stanley ER and Abraham D: Colony-stimulating
factor-1 blockade by antisense oligonucleotides and small
interfering RNAs suppresses growth of human mammary tumor
xenografts in mice. Cancer Res. 64:5378–5384. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ding J, Guo C, Hu P, Chen J, Liu Q, Wu X,
Cao Y and Wu J: CSF1 is involved in breast cancer progression
through inducing monocyte differentiation and homing. Int J Oncol.
49:2064–2074. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Scholl SM, Pallud C, Beuvon F, Hacene K,
Stanley ER, Rohrschneider L, Tang R, Pouillart P and Lidereau R:
Anti-colony-stimulating factor-1 antibody staining in primary
breast adenocarcinomas correlates with marked inflammatory cell
infiltrates and prognosis. J Natl Cancer Inst. 86:120–126. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chockalingam S and Ghosh SS: Amelioration
of cancer stem cells in macrophage colony stimulating
factor-expressing U87MG-human glioblastoma upon 5-fluorouracil
therapy. PLoS One. 8:e838772013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Paulus P, Stanley ER, Schäfer R, Abraham D
and Aharinejad S: Colony-stimulating factor-1 antibody reverses
chemoresistance in human MCF-7 breast cancer xenografts. Cancer
Res. 66:4349–4356. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sharma S, Patnaik PK, Aronov S and
Kulshreshtha R: Apoptomirs of breast cancer: Basics to clinics.
Front Genet. 7:1752016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schmid T, Zhou J and Brune B: HIF-1 and
p53: Communication of transcription factors under hypoxia. J Cell
Mol Med. 8:423–431. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang GL, Jiang BH, Rue EA and Semenza GL:
Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS
heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci
USA. 92:5510–5514. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang C, Samanta D, Lu H, Bullen JW, Zhang
H, Chen I, He X and Semenza GL: Hypoxia induces the breast cancer
stem cell phenotype by HIF-dependent and ALKBH5-mediated
m6A-demethylation of NANOG mRNA. Proc Natl Acad Sci USA.
113:E2047–E2056. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lin SC, Liao WL, Lee JC and Tsai SJ:
Hypoxia-regulated gene network in drug resistance and cancer
progression. Exp Biol Med. 239:779–792. 2014. View Article : Google Scholar
|
|
12
|
Nieminen AL, Qanungo S, Schneider EA,
Jiang BH and Agani FH: Mdm2 and HIF-1alpha interaction in tumor
cells during hypoxia. J Cell Physiol. 204:364–369. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lehman JA, Hauck PM, Gendron JM, Batuello
CN, Eitel JA, Albig A, Kadakia MP and Mayo LD: Serdemetan
antagonizes the Mdm2-HIF1α axis leading to decreased levels of
glycolytic enzymes. PLoS One. 8:e747412013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tan EY, Campo L, Han C, Turley H, Pezzella
F, Gatter KC, Harris AL and Fox SB: BNIP3 as a progression marker
in primary human breast cancer; Opposing functions in in situ
versus invasive cancer. Clin Cancer Res. 13:467–474. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Siddiqui WA, Ahad A and Ahsan H: The
mystery of BCL2 family: Bcl-2 proteins and apoptosis: An update.
Arch Toxicol. 89:289–317. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yuan C, Pu L, He Z and Wang J:
BNIP3/Bcl-2-mediated apoptosis induced by cyclic tensile stretch in
human cartilage endplate-derived stem cells. Exp Ther Med.
15:235–241. 2018.PubMed/NCBI
|
|
17
|
Zhao L, Man Y and Liu S: Long non-coding
RNA HULC promotes UVB-induced injury by up-regulation of BNIP3 in
keratinocytes. Biomed Pharmacother. 104:672–678. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen Y, Decker KF, Zheng D, Matkovich SJ,
Jia L and Dorn GW II: A nucleus-targeted alternately spliced
Nix/Bnip3L protein isoform modifies nuclear factor κB
(NFκB)-mediated cardiac transcription. J Biol Chem.
288:15455–15465. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Graham RM, Thompson JW and Webster KA:
Inhibition of the vacuolar ATPase induces Bnip3-dependent death of
cancer cells and a reduction in tumor burden and metastasis.
Oncotarget. 5:1162–1173. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang M, Zhang H, Tang F, Wang Y, Mo Z,
Lei X and Tang S: Doxorubicin resistance mediated by cytoplasmic
macrophage colony-stimulating factor is associated with switch from
apoptosis to autophagic cell death in MCF-7 breast cancer cells.
Exp Biol Med. 241:2086–2093. 2016. View Article : Google Scholar
|
|
21
|
Douzono M, Suzu S, Yamada M, Yanai N,
Kawashima T, Hatake K and Motoyoshi K: Augmentation of cancer
chemotherapy by preinjection of human macrophage colony-stimulating
factor in L1210 leukemic cell-inoculated mice. Jpn J Cancer Res.
86:315–321. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Global Burden of Disease Cancer
Collaboration, ; Fitzmaurice C, Allen C, Barber RM, Barregard L,
Bhutta ZA, Brenner H, Dicker DJ, Chimed-Orchir O, Dandona R,
Dandona L, et al: Global, Regional, and national cancer incidence,
mortality, years of life lost, years lived with disability, and
disability-adjusted life-years for 32 cancer groups, 1990 to 2015:
A systematic analysis for the global burden of disease study. JAMA
Oncol. 3:524–548. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Morry J, Ngamcherdtrakul W, Gu S, Reda M,
Castro DJ, Sangvanich T, Gray JW and Yantasee W: Targeted treatment
of metastatic breast cancer by PLK1 siRNA delivered by an
antioxidant nanoparticle platform. Mol Cancer Ther. 16:763–772.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Parvani JG and Jackson MW: Silencing the
roadblocks to effective triple-negative breast cancer treatments by
siRNA nanoparticles. Endocr Relat Cancer. 24:R81–R97. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li Y, Cai L, Wang H, Wu P, Gu W, Chen Y,
Hao H, Tang K, Yi P, Liu M, et al: Pleiotropic regulation of
macrophage polarization and tumorigenesis by formyl peptide
receptor-2. Oncogene. 30:3887–3899. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Qian BZ and Pollard JW: Macrophage
diversity enhances tumor progression and metastasis. Cell.
141:39–51. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pollard JW: Tumour-educated macrophages
promote tumour progression and metastasis. Nat Rev Cancer. 4:71–78.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Behnes CL, Bremmer F, Hemmerlein B,
Strauss A, Ströbel P and Radzun HJ: Tumor-associated macrophages
are involved in tumor progression in papillary renal cell
carcinoma. Virchows Arch. 464:191–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kacinski BM, Scata KA, Carter D, Yee LD,
Sapi E, King BL, Chambers SK, Jones MA, Pirro MH and Stanley ER:
FMS (CSF-1 receptor) and CSF-1 transcripts and protein are
expressed by human breast carcinomas in vivo and in vitro.
Oncogene. 6:941–952. 1991.PubMed/NCBI
|
|
30
|
Ramakrishnan S, Xu FJ, Brandt SJ, Niedel
JE, Bast RC Jr and Brown EL: Constitutive production of macrophage
colony-stimulating factor by human ovarian and breast cancer cell
lines. J Clin Invest. 83:921–926. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kacinski BM, Carter D, Mittal K, Yee LD,
Scata KA, Donofrio L, Chambers SK, Wang KI, Yang-Feng T and
Rohrschneider LR: Ovarian adenocarcinomas express fms-complementary
transcripts and fms antigen, often with coexpression of CSF-1. Am J
Pathol. 137:135–147. 1990.PubMed/NCBI
|
|
32
|
Kacinski BM: CSF-1 and its receptor in
ovarian, endometrial and breast cancer. Ann Med. 27:79–85. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mroczko B, Groblewska M,
Wereszczyńska-Siemiatkowska U, Okulczyk B, Kedra B, Łaszewicz W,
Dabrowski A and Szmitkowski M: Serum macrophage-colony stimulating
factor levels in colorectal cancer patients correlate with lymph
node metastasis and poor prognosis. Clin Chim Acta. 380:208–212.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Groblewska M, Mroczko B,
Wereszczyńska-Siemiatkowska U, Myśliwiec P, Kedra B and Szmitkowski
M: Serum levels of granulocyte colony-stimulating factor (G-CSF)
and macrophage colony-stimulating factor (M-CSF) in pancreatic
cancer patients. Clin Chem Lab Med. 45:30–34. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
McDermott RS, Deneux L, Mosseri V,
Védrenne J, Clough K, Fourquet A, Rodriguez J, Cosset JM, Sastre X,
Beuzeboc P, et al: Circulating macrophage colony stimulating factor
as a marker of tumour progression. Eur Cytokine Netw. 13:121–127.
2002.PubMed/NCBI
|
|
36
|
Chambers SK, Kacinski BM, Ivins CM and
Carcangiu ML: Overexpression of epithelial macrophage
colony-stimulating factor (CSF-1) and CSF-1 receptor: A poor
prognostic factor in epithelial ovarian cancer, contrasted with a
protective effect of stromal CSF-1. Clin Cancer Res. 3:999–1007.
1997.PubMed/NCBI
|
|
37
|
Wang ZE, Myles GM, Brandt CS, Lioubin MN
and Rohrschneider L: Identification of the ligand-binding regions
in the macrophage colony-stimulating factor receptor extracellular
domain. Mol Cell Biol. 13:5348–5359. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Stein J, Borzillo GV and Rettenmier CW:
Direct stimulation of cells expressing receptors for macrophage
colony-stimulating factor (CSF-1) by a plasma membrane-bound
precursor of human CSF-1. Blood. 76:1308–1314. 1990.PubMed/NCBI
|
|
39
|
Hung JY, Chang WA, Tsai YM, Hsu YL, Chiang
HH, Chou SH, Huang MS and Kuo PL: Tricetin, a dietary flavonoid,
suppresses benzo(a)pyreneinduced human nonsmall cell lung cancer
bone metastasis. Int J Oncol. 46:1985–1993. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lin EY, Nguyen AV, Russell RG and Pollard
JW: Colony-stimulating factor 1 promotes progression of mammary
tumors to malignancy. J Exp Med. 193:727–740. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lewis CE and Pollard JW: Distinct role of
macrophages in different tumor microenvironments. Cancer Res.
66:605–612. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu L, Liu W, Wang L, Zhu T, Zhong J and
Xie N: Hypoxia-inducible factor 1 mediates intermittent
hypoxia-induced migration of human breast cancer MDA-MB-231 cells.
Oncol Lett. 14:7715–7722. 2017.PubMed/NCBI
|
|
43
|
Chaturvedi P, Gilkes DM, Takano N and
Semenza GL: Hypoxia-inducible factor-dependent signaling between
triple-negative breast cancer cells and mesenchymal stem cells
promotes macrophage recruitment. Proc Natl Acad Sci USA.
111:E2120–E2129. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhao YT, Yan JY, Han XC, Niu FL, Zhang JH
and Hu WN: Anti-proliferative effect of digoxin on breast cancer
cells via inducing apoptosis. Eur Rev Med Pharmacol Sci.
21:5837–5842. 2017.PubMed/NCBI
|
|
45
|
Kothari S, Cizeau J, McMillan-Ward E,
Israels SJ, Bailes M, Ens K, Kirshenbaum LA and Gibson SB: BNIP3
plays a role in hypoxic cell death in human epithelial cells that
is inhibited by growth factors EGF and IGF. Oncogene. 22:4734–4744.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lin A, Yao J, Zhuang L, Wang D, Han J, Lam
EW and Gan B: The FoxO-BNIP3 axis exerts a unique regulation of
mTORC1 and cell survival under energy stress. Oncogene.
33:3183–3194. 2014. View Article : Google Scholar : PubMed/NCBI
|















