Introduction
Glioblastoma (GBM) is one of the most malignant
tumors in adults which is associated with severe outcomes (median
survival, <2 years) even with maximal therapy, including
surgical resection followed by radiotherapy and adjuvant
chemotherapy with temozolomide (1,2). The
majority of GBM patients have been reported to suffer
post-treatment recurrence, due to radiotherapy and chemotherapy
resistance (3). Therefore,
clarifying the mechanism for GBM treatment resistance may help to
identify a novel therapeutic target for the treatment of GBM.
Polo-like kinase 4 (PLK4) is a centrosomal kinase
which predominantly functions as a key regulator of centrosome
duplication in human cells and serves an important role in
chromosome instability (CIN) regulation, a unique genetic feature
that is observed in human cancer cells (4–6). PLK4
auto-phosphorylation has been proven to be essential for centriole
duplication and proteasomal degradation of PLK4 in the early G1
phase of cell cycles (7). In
addition, a negative feedback loop for PLK4 kinase functions
against the occurrence of centriole duplication, thus preventing
multipolar spindle formation during the cell cycle (7–10).
Furthermore, it has been reported that PLK4 kinase-dependent
centrosome amplification promotes cell proliferation, motility,
viability and treatment resistance, and therefore may be associated
with poor prognosis in breast cancer (9,11,12). A
previous study demonstrated that PLK4 expression is significantly
elevated in gastric cancer, while enriched PLK4 results in the
suppression of primary cilia formation (6). Through combined RNAi screening in
human breast cancers, it was found that PLK4 may be a promising
target for breast cancer, and a small molecule inhibitor,
CFI-400945, was proven to be effective for breast cancer in
xenograft models at well-tolerated doses (13–15).
However, the physiological role and function of PLK4 in GBM remains
unclear.
In the present study, PLK4 was identified as one of
the most upregulated kinase encoding genes in GBM and was
functionally required for both in vitro cell proliferation
and in vivo tumorigenesis. Clinically, an elevated PLK4 was
observed in high grade glioma patients and was associated with poor
prognosis. In addition, PLK4 enhanced radiotherapy resistance in
GBM, while PLK4 knockdown via lentivirus transfection significantly
increased the radiosensitivity of GBM cells. Mechanically, PLK4
expression was markedly elevated by exogenous overexpression of
ATPase family AAA domain-containing protein 2 (ATAD2) in GBM cells.
Collectively, it was shown that the ATAD2-dependent transcriptional
regulation of PLK4 promotes cell proliferation and tumorigenesis,
as well as radioresistance of GBM, thus potentially inducing tumor
recurrence. PLK4 could therefore serve as a potential therapeutic
target for GBM treatment.
Materials and methods
Ethics
The use of experimental animals was approved by the
Ethics Committee of the School of Medicine, Xi'an Jiaotong
University (Xi'an, China; approval no. 2016-085). The collection
and use of the tumor samples and patient information was approved
by the patients and the Scientific Ethics Committee of the First
Affiliated Hospital of Xi'an (approval no. 2016-18). All usage of
the human tissues was confirmed by the patients and all the
necessary consent forms were signed.
Reagents and antibodies
The following reagents and antibodies were used in
the present study: Dulbecco's modified Eagle's medium-nutrient
mixture F12 (DMEM-F12; Thermo Fisher Scientific, Inc., Waltham, MA,
USA), fetal bovine serum (FBS; Thermo Fisher Scientific, Inc.),
accutase solution (Merck KGaA, Darmstadt, Germany), alamarBlue Cell
Viability reagent (Thermo Fisher Scientific, Inc.),
radioimmunoprecipitation assay (RIPA) lysis buffer (Merck KGaA),
phosphatase inhibitor (Merck KGaA), protease inhibitor (Merck
KGaA), Bradford solution (Bio-Rad Laboratories, Inc., Hercules, CA,
USA), bovine serum albumin (BSA) standard solution (New England
BioLabs, Inc., Ipswich, MA, USA), PageRuler plus prestained protein
ladder (Thermo Fisher Scientific, Inc.), iScript Reverse
Transcription SuperMix (Bio-Rad Laboratories, Inc.), Alexa
Fluor® 488 Annexin V/Dead Cell Apoptosis kit (Thermo
Fisher Scientific, Inc.).
In vitro cell culture
GBM cell lines U138 and U251, as well as normal
human astrocytes (NHAs), were provided by the Translational
Medicine Center of the First Affiliated Hospital of Xi'an Jiaotong
University (Xi'an, China) in 2013. The U87 cell line (GBM of
unknown origin) was originally purchased from BeNa Culture
Collection (Kunshan, China). GBM cells were cultured in DMEM-F12
containing 10% FBS at 37°C with 5% CO2. The medium was
replaced every 3 days. Cells were dissociated with accutase and
seeded into new medium with a density of 106 cells/10
ml. After 24 h culture at 37°C with 5% CO2, radiotherapy
was performed in vitro using X-RAD 320 from Precision X-Ray
at a dose of 12 Gy.
Lentivirus transduction
pGFP-shPLK4 lentivirus particles were purchased from
OriGene Technologies, Inc. (cat. no. TL320644V; Beijing, China).
pLenti-GIII-CMV ATAD2 lentivirus (cat. no. LVP082354) and
pLenti-GIII-CMV PLK4 lentivirus were purchased from Applied
Biological Materials, Inc. (Richmond, BC, Canada). U87 cells
(2×105) were seeded in 6-well plates with 5 ml medium.
Next, 10 µl lentivirus was added to the medium and incubated at
37°C for 24 h. Reverse transcription-quantitative polymerase chain
reaction (RT-qPCR) and western blotting were performed to confirm
transfection efficiency.
RNA isolation and RT-qPCR
RNA isolation and RT-qPCR were performed as
previously described (16). The
following primers were used: PLK4 forward, CCTTCTGCAAATCTGGATGG and
reverse, ACAGTGGTTTGGGAATCTGC; ATAD2 forward,
AAGGAAGTTGAAACCTACCACCG and reverse, GCAAGTTGCTCCGTTATTTCCA; 18S
forward, GGCCCTGTAATTGGAATGAGTC and reverse, CCAAGATCCAACTACGAGCTT
reverse.
Western blotting
Western blotting was performed as previously
described (16). An anti-PLK4
primary antibody was purchased from Abcam (Cambridge, UK; cat. no.
ab137398; 1:1,000; rabbit). Anti-rabbit IgG (cat. no. ab171870;
1:1,000; Abcam) was used as a negative control. Horseradish
peroxidase-conjugated goat anti-rabbit IgG (cat. no. ab97051;
1:2,000; Abcam) and goat anti-mouse IgG (cat. no. ab205719;
1:2,000; Abcam) were used as secondary antibodies.
Luciferase assays
PLK4 3′ untranslated region (UTR)
Lenti-reporter-Luciferase virus was purchased from Applied
Biological Materials, Inc. (cat. no. MV-m16562). U87 cells were
infected with 1 µg of either empty vector or PLK4 promoter
luciferase reporter lentivirus and cultured for 3–5 days at 37°C
with 5% CO2, and then infected with either control or
ATAD2 overexpression lentivirus. Cells were cultured for 7 days at
37°C with 5% CO2. Luciferase assays were performed using
the Bright-Glo™ Luciferase Assay system (Promega Corporation,
Madison, WI, USA) on the Victor3 plate counter (PerkinElmer, Inc.,
Waltham, MA, USA). The luciferase activity of each sample was
normalized to Renilla luciferase activity.
Flow cytometry
Flow cytometry was performed as previously described
(16). The Alexa Fluor®
488 Annexin V/Dead Cell Apoptosis kit (Thermo Fisher Scientific,
Inc.; V13241) was used to measure U87 cell apoptosis according to
the manufacturer's protocol.
Immunohistochemistry (IHC)
IHC was performed as previously described (16). Glioma samples were collected from 41
patients (aged 22–68; 14 males and 27 females). These patients had
undergone surgical resection from 2006 to 2015 at the Department of
Neurosurgery (First Affiliated Hospital of Xi'an Jiaotong
University, Xi'an, China). All patients had been pathologically
diagnosed and had died due to tumor recurrence, which was confirmed
by computed tomography or magnetic resonance imaging. Three normal
human brain tissue samples collected from patients with epilepsy
(n=3; aged 32–41; male) were used as the negative controls. An
anti-PLK4 primary antibody (cat. no. ab137398; 1:200; Abcam;
rabbit) was used for PLK4 staining, and nuclei were counterstained
with hematoxylin or Hoechst, respectively. Anti-rabbit IgG (cat.
no. ab171870; 1:200; Abcam) was used as a negative control. Goat
anti-rabbit IgG (cat. no. ab97051; 1:5,000; Abcam) and goat
anti-mouse IgG (cat. no. ab205719; 1:5,000; Abcam) were used as
secondary German immunohistochemical scoring (GIS) was used to
measure the expression of PLK4 (17), in which the final immunoreactive
score = % positive cells × average staining intensity. The
percentage of positive cells was graded as follows: 0, negative; 1,
<10% positive; 2, 11–50%; 3, 51–80%; 4, >80%. Staining
intensity was graded as: 0, negative; 1, weakly positive; 2,
moderately positive; 3, strongly positive. A combination of >3
was considered positive. Additionally, survival data from the
Rembrandt database (Affymetrix HG U133 v.20 plus) was extracted and
analyzed with G-doc (gdoc.georgetown.edu/gdoc/workflows/index) to compare
the outcomes and PLK4 expression in glioma patients.
In vivo intracranial xenograft tumor
models
Female nude mice (6 weeks; ~15 g; n=5 in each group)
aged 6 weeks were used for in vivo experiments. The nude
mice were purchased from Laboratory Animal Center, Xi'an Jiaotong
University (Xi'an, China). Briefly, 1×105 U87 cells in 5
µl PBS transduced with non-target or shPLK4 lentivirus were
implanted into the brains of nude mice following anesthesia. Mice
were monitored once a day until at least one of the symptoms
associated with tumor growth appeared, including an arched back,
unsteady gait, leg paralysis and weight loss of 15%; at which
point, the mice were sacrificed and brains were harvested following
a ketamine/xylazine anesthesia overdose.
Gene expression analysis
Expression data of 669 kinase-encoding genes was
extracted from GSE67089 dataset (18), which included 30 primary glioma
sphere cultures (Glioma group) and three human fetal brain-derived
sphere cultures (Control group). Hierarchical biclustering was
performed to compare the expression of those genes, using Cluster
3.0 (www.geo.vu.nl/~huik/cluster.htm). Euclidean distance
and average linkage were used as similarity metric and clustering
method, respectively. The expression comparison was presented as
fold-changes.
Pearson r correlation analysis
Expression data was extracted from TCGA database
(cancergenome.nih.gov/). All data were
converted into Log2 form and the Pearson correlation
coefficient was calculated with the following formula:
rx,y=∑(x-x¯)(y-y¯)∑i=1n(xi-x¯)2∑i=1n(yi-y¯)2
In terms of the strength of relationship, the value
of the r coefficient varies between 1 and −1. When the value of the
r coefficient is close to 1 or −1, there is a strong positive or
negative association between the 2 variables, respectively. The
statistical significance was calculated using F-test.
Statistical analysis
Results are presented as the mean ± standard
deviation of three replicates. A two tailed t-test was used to
evaluate the statistical differences between two groups. One-way
analysis of variance followed by Dunnett's post hoc test was used
to evaluate the statistical differences among multiple groups. The
statistical significance of Kaplan-Meier survival plots was
analyzed by the log-rank test. Statistical analysis was performed
using SPSS 19.0 (IBM Corp., Armonk, NY, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
PLK4 is overexpressed in GBM
It is well-known that patients with GBM have a
considerably poor prognosis, even following maximal treatment
(3). To identify the molecular
mechanism for GBM tumor recurrence and treatment resistance, the
expression of 669 kinase-encoding genes was first compared using
DNA microarray data (GSE67089) (18) from 30 primary glioma sphere cultures
and three human fetal brain-derived sphere cultures (control).
Based on this analysis, 28 kinase-encoding genes were identified to
be significantly enriched in GBM samples, compared with astrocytes.
Next, a total of 669 kinase-encoding genes were picked from the
transcriptome microarray data of the glioma sphere samples-the
results demonstrated a wide range of upregulated genes following
radiation at 12 Gy, compared to naïve GBM cells. In total, seven
kinase-encoding genes were found to overlap in these two sets of
comparisons, including NIMA-related kinase 2, BUB1 mitotic
checkpoint serine/threonine kinase B, cell division cycle 7, BUB1,
PLK4, CDKN3 and CHEK2 (Fig.
1A).
With PLK4 identified as a functional regulator of
tumor proliferation and treatment resistance in a variety of human
tumors, including gastric and breast cancer (6,11), the
present study focused on the physiological function of PLK4 in GBM.
Expression data from the TCGA database were analyzed and PLK4 was
found to be upregulated in all four subtypes of GBM, including
classical, mesenchymal, neural and proneural, as compared with the
normal brain tissues (Fig. 1B).
This expression profile by microarray was validated by RT-qPCR in
three GBM cell lines (U87, U138 and U251) and a normal astrocyte
cell line (Fig. 1C). Western
blotting showed a higher PLK4 expression in GBM cells, compared
with normal astrocyte cells (Fig.
1D). Taken together, these results demonstrated that PLK4
expression was significantly elevated in GBM.
Overexpression of PLK4 implies poor
prognosis in GBM
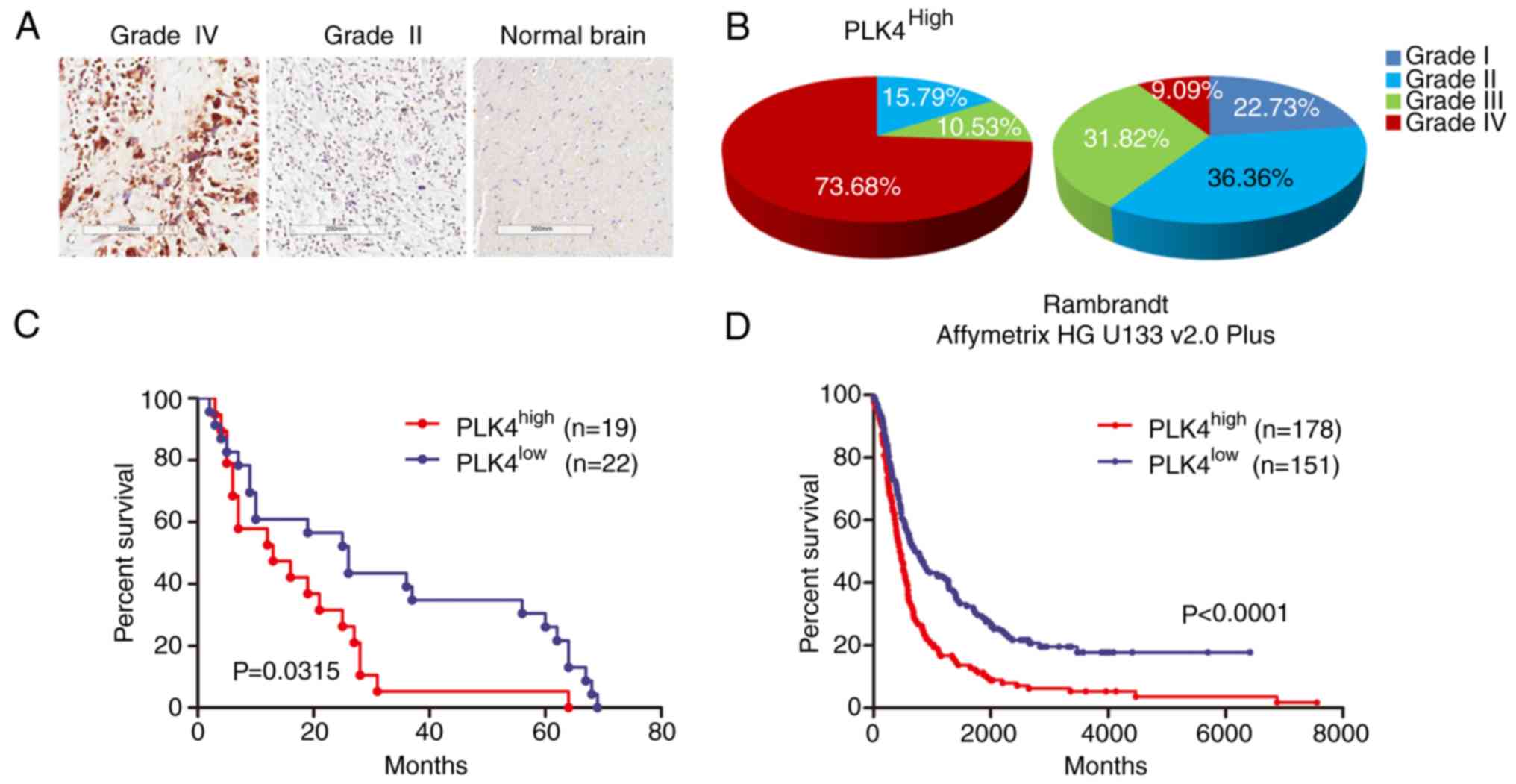
To further verify the expression of PLK4 in GBM
tumors, IHC staining was performed in glioma samples from 41
patients that had undergone surgical resection from 2006 to 2015 at
the Department of Neurosurgery of the First Affiliated Hospital of
Xi'an Jiaotong University, Xi'an, Shaanxi Province, China GIS was
used to quantify the expression levels of PLK4. In sharp contrast
to the low PLK4 expression in normal brain tissues and low-grade
glioma samples, PLK4 expression was highly expressed in GBM
(Fig. 2A and B). Furthermore, a
longer overall survival was observed in samples with a lower PLK4
expression, compared to those with a higher expression (median
survival, 26 vs. 13 months; Fig.
2C). Similarly, data from the Rembrandt database (Affymetrix HG
U133 v.20 plus) demonstrated that enriched PLK4 expression could be
associated with poorer survival, when compared to samples with an
intermediate or low PLK4 expression (Fig. 2D). In combination, this demonstrated
that increased PLK4 expression was associated with a poor prognosis
in patients with GBM, and could potentially serve as a clinically
relevant molecular marker for GBM.
PLK4 promotes GBM proliferation and
tumorigenesis
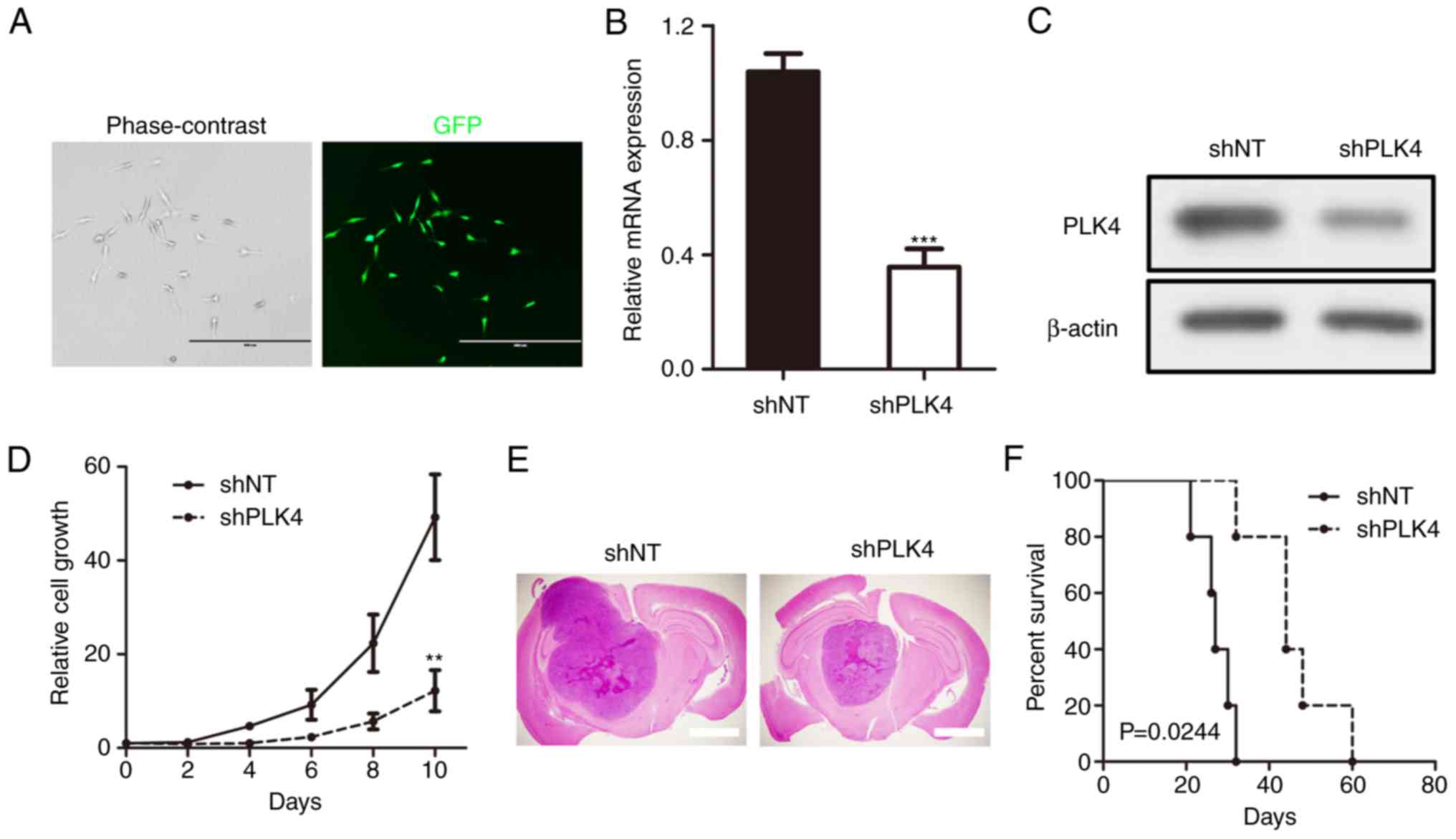
To further investigate the physiological functions
of PLK4 in GBM tumorigenesis, U87 GBM cells were used as an in
vitro cell model and were infected with either non-targeting
control (shNT) or pGFP-shPLK4 lentiviruses. The efficiency of the
lentivirus infection was confirmed by both GFP fluorescence
(Fig. 3A) and RT-qPCR (Fig. 3B). The results indicated that the
mRNA expression of PLK4 was significantly reduced in shPLK4 U87
cells. Western blotting yielded the same results (Fig. 3C). In addition, in vitro
growth kinetics of shPLK4 lentivirus-infected U87 cells were
inhibited proportionally to PLK4 reduction (Fig. 3D).
A mouse intracranial tumor model was used to
investigate the functional role of PLK4 on GBM tumorigenesis in
vivo. The results showed that shNT-transduced U87 cells formed
GBM-like tumors within 30 days in mice (median survival, 27.2±4.21
days). However, a longer survival was observed in shPLK4-transduced
U87 ×enografted mice (45.6±10.04 days), highlighting a potential
anti-tumorigenesis effect of PLK4 knockdown (Fig. 3E and F). In combination, these
findings implied that PLK4 promoted GBM proliferation and
tumorigenesis in vitro and in vivo.
PLK4 induces radioresistance in
GBM
As PLK4 was found to be one of the most upregulated
kinase-encoding genes for GBM following radiotherapy, it was
assumed that PLK4-dependent radioresistance was essential for GBM
cells. U87 cells were therefore treated with or without 12 Gy
radiotherapy and analyzed by RT-qPCR. The results indicated that
the PLK4 mRNA expression was significantly elevated following
radiotherapy (Fig. 4A), while
western blotting yielded the same results (Fig. 4B). To further clarify whether PLK4
induces radioresistance in GBM, PLK4 was knocked down via shPLK4
lentivirus infection, followed by 12 Gy radiotherapy. The RT-qPCR
results showed that PLK4 mRNA expression was markedly increased
following radiation and was partially eliminated by shPLK4
(Fig. 4C), as well as the in
vitro cell proliferation (Fig.
4D). In addition, flow cytometry indicated that the percentage
of U87 cells undergoing both early (AV+; PI−)
and late (AV+; PI+) apoptosis were markedly
increased following PLK4 knockdown followed by radiation, compared
with radiotherapy alone (Fig. 4E).
PLK4 was therefore essential for radioresistance, and the knockdown
of PLK4 could increase radiosensitivity in GBM cells.
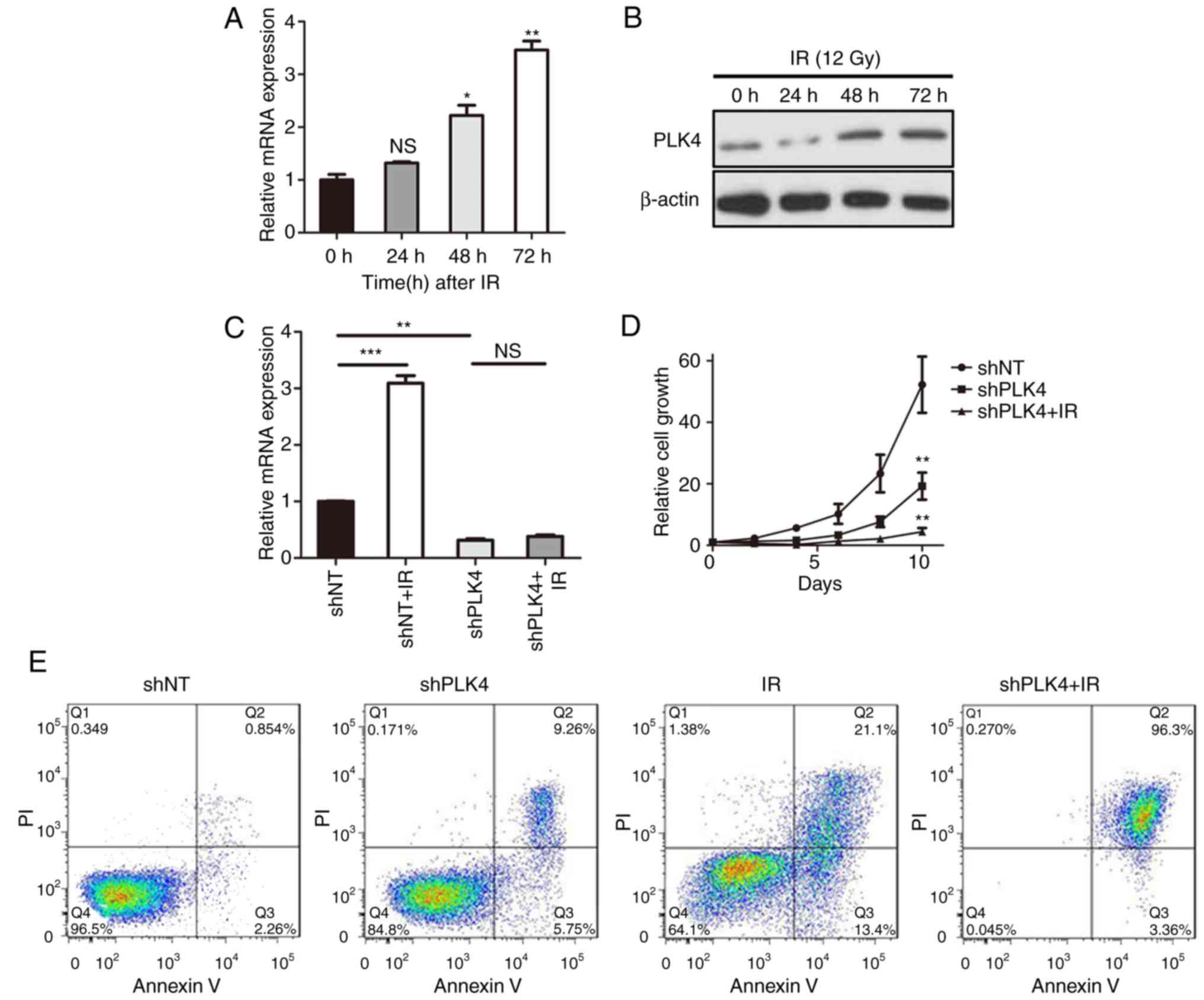 | Figure 4.PLK4 induces radioresistance in GBM.
(A) RT-qPCR and (B) western blot analysis for PLK4 expression in
U87 cells treated with or without radiation (12 Gy; *P<0.05,
**P<0.01 vs. 0 h; one-way ANOVA followed by Dunnett's post hoc
test. β-actin served as the control. (C) RT-qPCR results showed
that PLK4 mRNA expression was markedly increased following
radiation (12 Gy), and could be partially eliminated by shPLK4.
**P<0.01, ***P<0.001; one-way ANOVA followed by Dunnett's
post hoc test. (D) In vitro cell proliferation assay for U87
cells transduced with either shNT or shPLK4 lentiviruses, followed
(or not) by radiotherapy (12 Gy). **P<0.01 vs. shNT; one-way
ANOVA followed by Dunnett's post hoc test. (E) Flow cytometry
(Annexin V and propidium iodide) determined the apoptosis of U87
cells transduced with shNT or shPLK4, followed (or not) by
radiotherapy (12 Gy). (F) RT-qPCR analysis of U87 cells transduced
with PLK4 overexpression or control lentivirus. **P<0.01;
t-test. (G) Flow cytometry determined the apoptosis of U87 cells
transduced with PLK4 overexpression or control lentivirus, followed
(or not) by radiotherapy (12 Gy). GBM, glioblastoma; ANOVA,
analysis of variance; RT-qPCR, reverse transcription quantitative
polymerase chain reaction; ns, not significant; PLK4, polo-like
kinase 4; sh, small hairpin RNA; IR, radiation. |
As it is well known that PLK4 predominantly
functions as a mitosis regulating kinase (5), knockdown of PLK4 will increase cell
cycle arrest in GBM. To eliminate the effects of PLK4 knock down on
mitosis, PLK4 was overexpressed in U87 cells (Fig. 4F) then combined with radiation. The
results indicated that exogenous overexpression markedly reduced
cell apoptosis in U87 cells following radiotherapy (Fig. 4G), demonstrating that PLK4-induced
radioresistance in GBM was independent, at least partially, from
its functions in mitosis regulation.
PLK4 is transcriptionally regulated by
ATAD2 in GBM
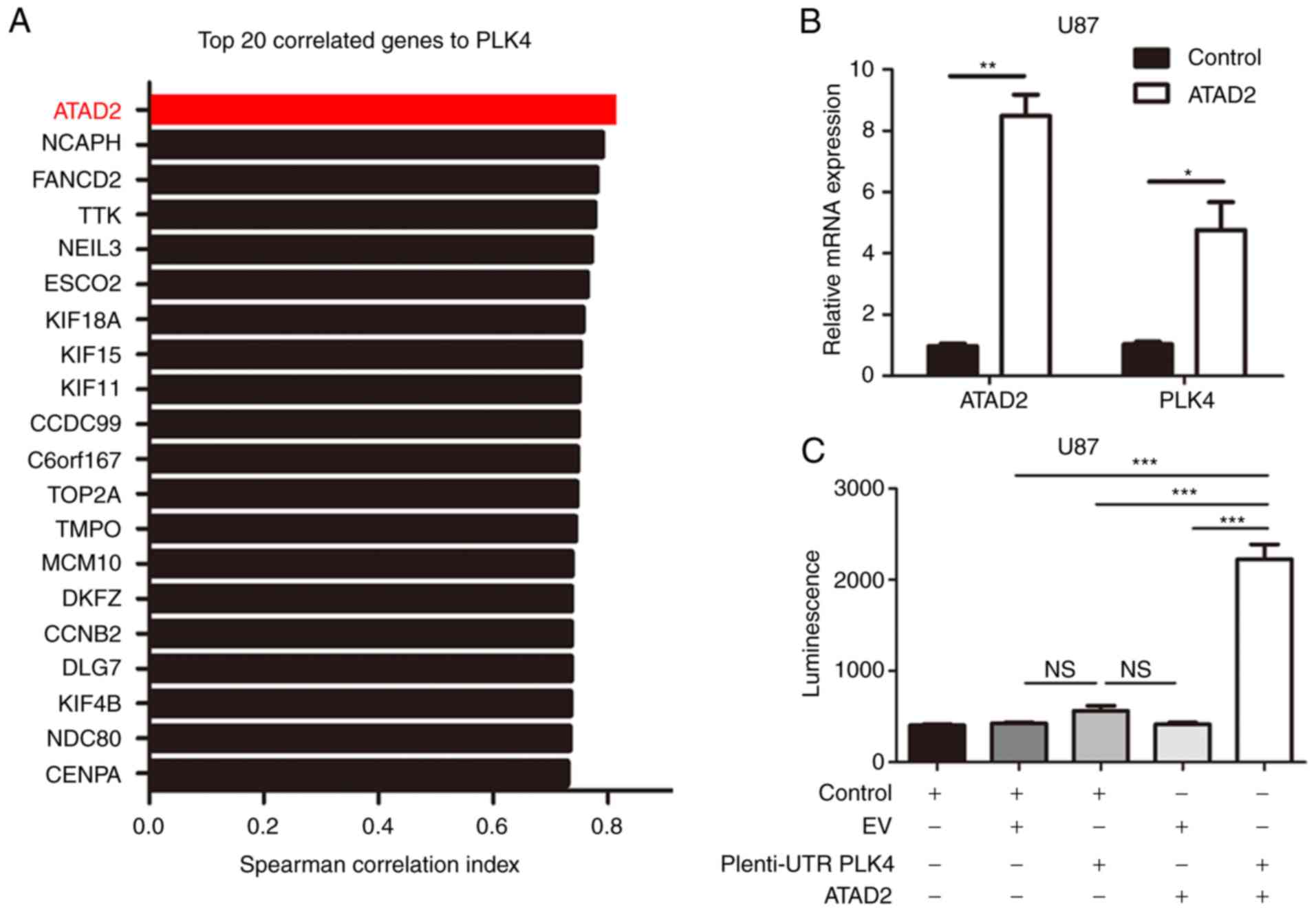
To further assess the regulatory mechanism of PLK4
expression in GBM, expression data from the TCGA dataset were
analyzed. The results indicated that ATAD2 was one of the top genes
correlated to PLK4 mRNA (Fig. 5A).
Furthermore, the shRNA-mediated overexpression of ATAD2 increased
the expression of PLK4 mRNA in U87 cells (Fig. 5B). To further study the mechanism of
the ATAD2-dependent regulation of PLK4, luciferase reporter assays
were performed on U87 cells transfected with human PLK4 promoter.
According to the findings, the lentivirus-mediated overexpression
of ATAD2 elevated the transcription activity of the PLK4 promoter
in U87 cells, compared with the control cells (Fig. 5C). Collectively, these data suggest
that ATAD2 may be a key regulator of PLK4 transcription in GBM
cells.
Discussion
Accumulating data has demonstrated that
kinase-dependent tumorigenesis and treatment resistance is
essential for the recurrence of multiple types of tumors, and
targeting kinase activity has been proven to be an effective way to
reduce tumor growth (19–25). PLK4 is a serine/threonine
centrosomal protein kinase, whose main function is to regulate the
number of centrosomes in cells (26). Previous studies have indicated that
PLK4 serves an essential role in cell proliferation, tumorigenesis,
invasion and viability, as well as treatment resistance in a wide
range of cancers, including gastric adenocarcinoma, colon, liver
and breast cancer (6,9,12,27).
However, the function of PLK4 in GBM remains ambiguous. In the
present study, PLK4 was identified as one of the most enriched
kinase-encoding genes in GBM, and was proven to be an essential
regulator of GBM proliferation. PLK4 was shown to be functionally
indispensable for both in vitro cell proliferation and in
vivo tumorigenesis in GBM. PLK4 expression was highly enriched
in GBM and implied poor prognosis.
Mechanically, PLK4 overexpression has been shown to
regulate tumor proliferation and cell migration by inducing
centrosome amplification and CIN, resulting in the suppression of
cilia formation (6). Dzhindzhev
et al (28) reported that
PLK4 regulates centrosome duplication by interacting with
centrosomal protein of 152 kDa (CEP152). These findings suggest
that PLK4 may induce proliferation in tumor cells by inducing
centrosome amplification and CIN. The inhibition of PLK4 in a lung
cancer model induced apoptosis through a temperature-sensitive p53
mutant, while PLK4 overexpression diminished p53-dependent
apoptosis (29). In addition, the
Rho-GTPase signaling pathway could be disrupted by haploid
expression of PLK4 during cytokinesis in liver cancer, leading to
aneuploidy and tumorigenesis (12).
Additional research has revealed that the artificial silencing of
PLK4 inhibits stress-induced Akt activation, thus promoting
apoptosis in lung cancer cells, and that the gradual activation of
p53 downregulates PLK4 to promote apoptosis (30). In accordance with these findings, it
was found in the present study that PLK4 induced tumorigenesis and
radioresistance in GBM, with this mechanism potentially dependent
on the induction of centrosome amplification and CIN. However, the
mechanism and downstream target of PLK4 in GBM remains unclear and
further study is required.
Since PLK4 was identified as an essential kinase for
tumorigenesis and radioresistance in GBM, it was useful to
investigate the detailed mechanism of PLK4 regulation in GBM.
Spearman correlation analysis was performed between PLK4 and 14,731
genes using the TCGA dataset. Among these genes, ATAD2 was the gene
most significantly correlated with PLK4 in GBM. ATAD2 was
identified as a significantly conserved gene predominantly
expressed in germ cells, but was remarkably elevated in a wide
range of different subtypes of tumors, including thyroid, breast,
cervical and gastric cancer (31–34).
As a transcriptional coactivator of a wide subset of estradiol
target genes, ATAD2 is associated with a variety of key regulatory
mechanisms in human cancer cells, including the regulation of cell
proliferation and tumor metastasis, via the transcriptional
regulation of cyclin D1, c-myc and E2F (32,33).
However, the potential downstream targets of ATAD2 in the DNA
repair response of GBM remains unclear. In the present study, it
was found that the PLK4 expression was increased by the exogenous
overexpression of ATAD2, which suggested that the ATAD2-dependent
transcriptional regulation of PLK4 was essential for tumor growth
and treatment resistance in GBM cells. However, the transcription
factor which directly binds to the PLK4 promoter area is unknown.
Further experiments, including chromatin immunoprecipitation
sequencing for PLK4, should be performed on co-immunoprecipitation
of ATAD2-binding protein in order to clarify the interaction and
exact pathway between ATAD2 and PLK4.
In the present study, bioinformatics analysis
results identified PLK4 as one of the most upregulated
kinase-encoding genes in GBM which was found to be functionally
required for both in vitro cell proliferation and in
vivo tumorigenesis. Clinically, elevated PLK4 expression was
observed in high grade glioma patients and was linked to poor
prognosis in GBM. In addition, PLK4 enhanced radioresistance in GBM
cells, whereas PLK4 knockdown significantly increased the
radiosensitivity of GBM cells. Mechanically, PLK4 expression was
markedly elevated by the exogenous overexpression of ATAD2 in GBM
cells. Collectively, the data showed that the ATAD2-dependent
transcriptional regulation of PLK4 promoted cell proliferation and
tumorigenesis, as well as GBM radioresistance, thus potentially
inducing tumor recurrence. PLK4 could therefore serve as a
potential therapeutic target for GBM treatment.
Acknowledgements
The authors would like to thank Dr Ruichun Li, Dr
Ping Mao and all other members of the Department of Neurosurgery
and Center of Brain Science (First Affiliated Hospital of Xi'an
Jiaotong University) for their technical assistance.
Funding
Project was supported by National Natural Science
Foundation of China (grant no. 81802502).
Availability of data and materials
The data used or analyzed during the current study
are available from the corresponding author on reasonable
request.
Authors' contributions
JW performed the experiments. JW, MW, XM and KG
analyzed the data. DNA microarray database analysis was performed
by JW, JZ and XB. JW, NW and JZ collected the patient samples and
performed follow-up surveys. Bioinformatics analysis was performed
by JW, MW and XM. Mice intracranial xenograft experiments were
performed by JW, KG, XB and NW. Pearson's correlation analysis was
performed by JW, WX and HL. The primers were designed by JW and HL.
JW and JZ wrote the manuscript. All authors read and approved the
final manuscript and agree to be accountable for all aspects of the
work in ensuring that questions related to the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
The use of experimental animals in this study was
approved by the Ethics Committee of the School of Medicine, Xi'an
Jiaotong University, Xi'an, Shaanxi Province, China (approval no.
2016-085). The collection and use of the tumor samples and patient
information was approved by the patients and the Scientific Ethics
Committee of the First Affiliated Hospital of Xi'an Jiaotong
University, Xi'an, Shaanxi Province, China (approval no.
2016-18).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ho VK, Reijneveld JC, Enting RH, Bienfait
HP, Robe P, Baumert BG and Visser O; Dutch Society for
Neuro-Oncology (LWNO), : Changing incidence and improved survival
of gliomas. Eur J Cancer. 50:2309–2318. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al: Radiotherapy plus concomitant and adjuvant temozolomide
for glioblastoma. N Engl J Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Signorovitch J, Li N, Ohashi E, Dastani H,
Shaw J and Orsini L: Overall survival (Os), quality of life (Qol),
and neurocognitive function (Nf) in recurrent glioblastoma
multiforme (Gbm): A systematic literature review. Value Health.
18:A4332015. View Article : Google Scholar
|
|
4
|
A PLK4 inhibitor has single-agent activity
in preclinical tumor models. Cancer Discov. 4:OF112014. View Article : Google Scholar
|
|
5
|
Korzeniewski N, Hohenfellner M and
Duensing S: CAND1 promotes PLK4-mediated centriole overduplication
and is frequently disrupted in prostate cancer. Neoplasia.
14:799–806. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shinmura K, Kurabe N, Goto M, Yamada H,
Natsume H, Konno H and Sugimura H: PLK4 overexpression and its
effect on centrosome regulation and chromosome stability in human
gastric cancer. Mol Biol Rep. 41:6635–6644. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cunha-Ferreira I, Rodrigues-Martins A,
Bento I, Riparbelli M, Zhang W, Laue E, Callaini G, Glover DM and
Bettencourt-Dias M: The SCF/Slimb ubiquitin ligase limits
centrosome amplification through degradation of SAK/PLK4. Curr
Biol. 19:43–49. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rogers GC, Rusan NM, Roberts DM, Peifer M
and Rogers SL: The SCF Slimb ubiquitin ligase regulates Plk4/Sak
levels to block centriole reduplication. J Cell Biol. 184:225–239.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Guderian G, Westendorf J, Uldschmid A and
Nigg EA: Plk4 trans-autophosphorylation regulates centriole number
by controlling betaTrCP-mediated degradation. J Cell Sci.
123:2163–2169. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Holland AJ, Lan W, Niessen S, Hoover H and
Cleveland DW: Polo-like kinase 4 kinase activity limits centrosome
overduplication by autoregulating its own stability. J Cell Biol.
188:191–198. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Marina M and Saavedra HI: Nek2 and Plk4:
Prognostic markers, drivers of breast tumorigenesis and drug
resistance. Front Biosci. 19:352–365. 2014. View Article : Google Scholar
|
|
12
|
Rosario CO, Ko MA, Haffani YZ, Gladdy RA,
Paderova J, Pollett A, Squire JA, Dennis JW and Swallow CJ: Plk4 is
required for cytokinesis and maintenance of chromosomal stability.
Proc Natl Acad Sci USA. 107:6888–6893. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mason JM, Lin DC, Wei X, Che Y, Yao Y,
Kiarash R, Cescon DW, Fletcher GC, Awrey DE, Bray MR, et al:
Functional characterization of CFI-400945, a Polo-like kinase 4
inhibitor, as a potential anticancer agent. Cancer Cell.
26:163–176. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sampson PB, Liu Y, Forrest B, Cumming G,
Li SW, Patel NK, Edwards L, Laufer R, Feher M, Ban F, et al: The
discovery of Polo-like kinase 4 inhibitors: Identification of
(1R,2S).2-(3-((E).4-(((cis).2,6-dimethylmorpholino)methyl)styryl)-1H.indazol-6-yl)-5′-methoxyspiro[cyclopropane-1,3′-indolin]-2′-one
(CFI-400945) as a potent, orally active antitumor agent. J Med
Chem. 58:147–169. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yu B, Yu Z, Qi PP, Yu DQ and Liu HM:
Discovery of orally active anticancer candidate CFI-400945 derived
from biologically promising spirooxindoles: Success and challenges.
Eur J Med Chem. 95:35–40. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang J, Cheng P, Pavlyukov MS, Yu H, Zhang
Z, Kim SH, Minata M, Mohyeldin A, Xie W, Chen D, et al: Targeting
NEK2 attenuates glioblastoma growth and radioresistance by
destabilizing histone methyltransferase EZH2. J Clin Invest.
127:3075–3089. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
van Diest PJ, van Dam P, Henzen-Logmans
SC, Berns E, van der Burg ME, Green J and Vergote I: A scoring
system for immunohistochemical staining: consensus report of the
task force for basic research of the EORTC-GCCG. European
organization for research and treatment of cancer-gynaecological
cancer cooperative group. J Clin Pathol. 50:801–804. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mao P, Joshi K, Li J, Kim SH, Li P,
Santana-Santos L, Luthra S, Chandran UR, Benos PV, Smith L, et al:
Mesenchymal glioma stem cells are maintained by activated
glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc
Natl Acad Sci USA. 110:8644–8649. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen C, Wang X, Xiong X, Liu Q, Huang Y,
Xu Q, Hu J, Ge G and Ling K: Targeting type Igamma
phosphatidylinositol phosphate kinase inhibits breast cancer
metastasis. Oncogene. 34:4635–4646. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hu J, Ahuja LG, Meharena HS, Kannan N,
Kornev AP, Taylor SS and Shaw AS: Kinase regulation by hydrophobic
spine assembly in cancer. Mol Cell Biol. 35:264–276. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stransky N, Cerami E, Schalm S, Kim JL and
Lengauer C: The landscape of kinase fusions in cancer. Nat Commun.
5:48462014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim SH, Joshi K, Ezhilarasan R, Myers TR,
Siu J, Gu C, Nakano-Okuno M, Taylor D, Minata M, Sulman EP, et al:
EZH2 protects glioma stem cells from radiation-induced cell death
in a MELK/FOXM1-dependent manner. Stem Cell Reports. 4:226–238.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Minata M, Gu C, Joshi K, Nakano-Okuno M,
Hong C, Nguyen CH, Kornblum HI, Molla A and Nakano I: Multi-kinase
inhibitor C1 triggers mitotic catastrophe of glioma stem cells
mainly through MELK kinase inhibition. PLoS One. 9:e925462014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Joshi K, Banasavadi-Siddegowda Y, Mo X,
Kim SH, Mao P, Kig C, Nardini D, Sobol RW, Chow LM, Kornblum HI, et
al: MELK-dependent FOXM1 phosphorylation is essential for
proliferation of glioma stem cells. Stem Cells. 31:1051–1063. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gu C, Banasavadi-Siddegowda YK, Joshi K,
Nakamura Y, Kurt H, Gupta S and Nakano I: Tumor-specific activation
of the C-JUN/MELK pathway regulates glioma stem cell growth in a
p53-dependent manner. Stem Cells. 31:870–881. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Habedanck R, Stierhof YD, Wilkinson CJ and
Nigg EA: The Polo kinase Plk4 functions in centriole duplication.
Nat Cell Biol. 7:1140–1146. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Macmillan JC, Hudson JW, Bull S, Dennis JW
and Swallow CJ: Comparative expression of the mitotic regulators
SAK and PLK in colorectal cancer. Ann Surg Oncol. 8:729–740. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dzhindzhev NS, Yu QD, Weiskopf K,
Tzolovsky G, Cunha-Ferreira I, Riparbelli M, Rodrigues-Martins A,
Bettencourt-Dias M, Callaini G and Glover DM: Asterless is a
scaffold for the onset of centriole assembly. Nature. 467:714–718.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li J, Tan M, Li L, Pamarthy D, Lawrence TS
and Sun Y: SAK, a new polo-like kinase, is transcriptionally
repressed by p53 and induces apoptosis upon RNAi silencing.
Neoplasia. 7:312–323. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nakamura T, Saito H and Takekawa M: SAPK
pathways and p53 cooperatively regulate PLK4 activity and
centrosome integrity under stress. Nat Commun. 4:17752013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sun W, Lan X, Zhang H, Wang Z, Dong W, He
L, Zhang T, Zhang P, Liu J and Qin Y: NEAT1_2 functions as a
competing endogenous RNA to regulate ATAD2 expression by sponging
microRNA-106b-5p in papillary thyroid cancer. Cell Death Dis.
9:3802018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kalashnikova EV, Revenko AS, Gemo AT,
Andrews NP, Tepper CG, Zou JX, Cardiff RD, Borowsky AD and Chen HW:
ANCCA/ATAD2 overexpression identifies breast cancer patients with
poor prognosis, acting to drive proliferation and survival of
triple-negative cells through control of B-Myb and EZH2. Cancer
Res. 70:9402–9412. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zheng L, Li T, Zhang Y, Guo Y, Yao J, Dou
L and Guo K: Oncogene ATAD2 promotes cell proliferation, invasion
and migration in cervical cancer. Oncol Rep. 33:2337–2344. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang M, Zhang C, Du W, Yang X and Chen Z:
ATAD2 is overexpressed in gastric cancer and serves as an
independent poor prognostic biomarker. Clin Transl Oncol.
18:776–781. 2016. View Article : Google Scholar : PubMed/NCBI
|