Introduction
Lung cancer is the second most common type of cancer
(1) and is the leading cause of
cancer-associated mortality (2),
with >1.38 million mortalities reported worldwide (3). At present, the majority of patients
with lung cancer have a poor prognosis, with a 5-year relative
survival rate of <17% (4). Lung
cancer can be classified into small-cell lung cancer and non-small
cell lung cancer (NSCLC), according to histological characteristics
(5). More than 80% of lung cancer
cases are NSCLC (6), which is
composed of adenocarcinoma, squamous cell carcinoma and large cell
carcinoma (7). Due to a lack of
symptoms associated with early-stage lung cancer, the majority of
lung cancer cases are diagnosed at an advanced stage when there are
limited treatment options (8–10). The
survival rate for patients with early-stage NSCLC is relatively
higher following surgery (11).
However, the majority of patients have a high risk of recurrence.
Systemic chemotherapy is the most mainstream therapeutic option for
the treatment of advanced NSCLC; however, the median survival time
is only slightly >18 months from the time of diagnosis (12,13).
Furthermore, it has been demonstrated that chemotherapy has notable
resistance and insensitivity in the treatment of lung cancer
(14). Therefore, the investigation
of novel anticancer drugs with high efficiency and selectivity
against lung cancer is required.
The well-known tumor suppressor gene p53 has been
identified to be involved in DNA repair, transcription, genomic
stability, cell cycle and apoptosis following cellular exposure to
various types of stress (15–17).
Both cell cycle arrest and apoptosis are important
tumor-suppressive pathways that involve p53 (18,19).
Cell cycle arrest is regulated by the depletion of critical cell
cycle proteins. Upon p53 activation, the cyclin-dependent kinase
(CDK) inhibitor p21WAF1/CIP1 is upregulated, which leads
to the inhibition of numerous cell cycle proteins, including CDK-4,
CDK-6/cyclin D, CDK2/cyclin E and cyclin B1/Cdc2 (20). Cell apoptosis is associated with
complex signaling pathways. p53 mediates cell apoptosis by
upregulating BH3-only proteins, including Bax, and downregulating
Bcl-2 family members, including Bcl-2 (21). As an intracellular suicide program,
cell apoptosis is also mediated by the activation of caspases,
including caspase-3, −8 and −9, and poly(ADP-ribose) polymerase
(22,23). Therefore, triggering pathways
involved with apoptosis and inhibiting cell cycle progression may
present new strategies for the treatment of lung cancer.
Nitric oxide (NO) serves a crucial role in
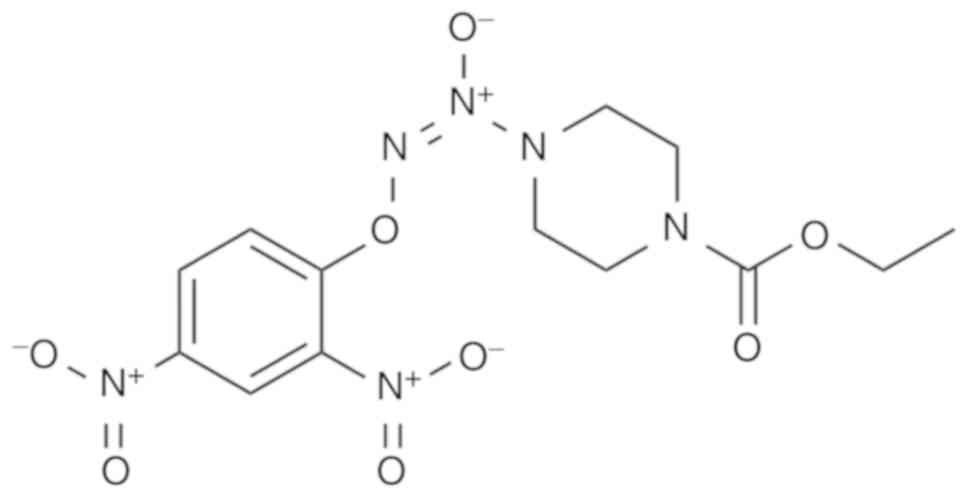
regulating the activity of cancer (24). As an arylated diazeniumdiolate-based
anticancer agent, O2-(2,4-dinitrophenyl)-1-[(4-
ethoxycarbonyl)piperazin- 1-yl]diazen-1-ium-1,2-diolate (JS-K;
Fig. 1) exhibits cytotoxic and
potent antitumor effects in several types of human cancer,
including prostate cancer (25),
hepatoma (26), acute myeloid
leukemia (27), multiple myeloma
(28) and bladder cancer (29). JS-K can also enhance cytotoxicity in
doxorubicin-treated renal carcinoma cells by upregulating the
expression of p53 (30). Notably, a
number of studies have reported that JS-K was selectively cytotoxic
to cancer cells and demonstrated no significant toxicity towards
normal cells (25,28,31). A
previous study demonstrated that a reactive oxygen species (ROS)
activation mechanism contributed to JS-K-induced apoptosis in human
NSCLC cells, including H1944 and H1703 cells (32). However, there is limited
understanding regarding the mechanism of action of JS-K in other
lung cancer cells, including A549 and H460 cells. The present study
aimed to investigate the underlying antitumor mechanism of JS-K in
A549 and H460 human NSCLC cells. To the best of our knowledge, the
present study is the first to demonstrate that
p53/p21WAF1/CIP1 and p27KIP1 proteins are
involved in JS-K-induced apoptosis and cell cycle arrest.
Materials and methods
Chemicals
JS-K was acquired from Sigma-Aldrich; Merck KGA
(Darmstadt, Germany; cat. no. J4137), and stored at −80°C at a
concentration of 20 mM in 100% dimethyl sulfoxide (DMSO). The final
density of DMSO used in the present study was <0.1%. Pifithrin-α
(PFT-α) was purchased from MedChemExpress (New Jersey, USA; cat.
no. HY-15484) and diluted to a final concentration of 30 µM with
100% DMSO. Antibodies against caspase-3 (cat. no. 9665S), caspase-9
(cat. no. 9502S), caspase-8 (cat. no. 4790S), Bcl-2 (cat. no.
4223S), Bax (cat. no. 2772S), p53 (cat. no. 9282S),
p21Waf1/Cip1 (cat. no. 2947S), cyclin B1 (cat. no.
4138P), CDK2 (cat. no. 2546P), cyclin E1 (cat. no. 4129P),
p27Kip1 (cat. no. 3686S), Cdc2 (cat. no. 9112S), p-Cdc2
(cat. no. 4539S) and GAPDH (cat. no. 2118L) were all obtained from
Cell Signaling Technology, Inc. (Danvers, MA, USA). Antibodies
(caspase-3, caspase-9, caspase-8, Bcl-2, Bax, p53,
p21Waf1/Cip1, cyclin B1, CDK2, cyclin E1,
p27Kip1, Cdc2, p-Cdc2 and GAPDH) were diluted at the
1:1,000 with 5% bovine serum albumin (BSA). Goat anti-rabbit
immunoglobulin G (IgG)-horseradish peroxidase antibody was used as
the secondary antibody (Sino Biological, Inc., Beijing, China; cat.
no. SSA004). The secondary antibody was diluted with 5% non-fat
milk in proportion of 1:2,000. The Annexin V-FITC Apoptosis
Detection kit was obtained from Dojindo Molecular Technologies,
Inc. (Kumamoto, Japan; cat. no. AD10). Cell Cycle Analysis kit
(cat. no. C1052) and MTT (cat. no. ST316) were all obtained from
Beyotime Institute of Biotechnology (Shanghai, China).
Cell culture and JS-K treatment
The human NSCLC cell lines A549 and H460 were
obtained from the Cell Bank of the Institute of Biochemistry and
Cell Biology at the China Academy of Sciences (Shanghai, China). In
brief, A549 and H460 cells were respectively cultured in Dulbecco's
modified Eagle's medium (DMEM; cat. no. C11995500BT) and RPMI-1640
medium (cat. no. C11875500BT) (both from Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), which were supplemented with
10% (v/v) fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific,
Inc.; cat. no. 10099141), 100 U/ml penicillin and 100 U/ml
streptomycin. The cells were maintained in a humidified incubator
at 37°C with 5% CO2. Subsequently, the A549 cells were
treated with various concentrations of JS-K (0, 1, 2 or 5 µM) for
48 h. In addition, the H460 cells were treated with different
concentrations of JS-K (0, 5, 10 or 15 µM) for 24 h at which point
the confluency was 60–70%. Another group of A549 cells were
cultured with 5 µM JS-K for 0, 12, 24 or 48 h, and H460 cells were
treated with 15 µM JS-K for 0, 6, 12 and 24 h. Moreover, A549 and
H460 cells were pretreated with PFT-α (40 and 10 µM, respectively)
for 1 h in the presence or absence of JS-K (5 and 15 µM,
respectively).
MTT assay
An
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay was performed to determine the cell toxicity and growth
inhibition following JS-K treatment. Briefly, A549 and H460 cells
were seeded in 96-well plates at a density of 5,000 cells/well
overnight for attachment and recovery. Subsequently, the cells were
pretreated with 0, 1, 2, 5, 10, 15 and 20 µM JS-K for 24 or 48 h.
After the indicated time, 20 µl MTT solution (5 mg/ml) was added to
each well and the cells were maintained in a humidified incubator
at 37°C with 5% CO2 for 4 h. The supernatants were
removed and 150 µl DMSO was added to each well to completely
dissolve the formazan crystals. Subsequently, the plates were
placed on an orbital shaker for 15 min and the absorbance was then
assessed at 490 nm with an automated spectrophotometric plate
reader (PerkinElmer, Inc., Waltham, MA, USA). All experiments were
repeated independently in triplicate. The inhibition ratio was
calculated using the following equation: Inhibition ratio (%) =
(OD490 control-OD490
JS-K-treated)/OD490 control × 100%, where OD is the
optical density.
Cell cycle analysis
Cells were quantified using a Cell Cycle Analysis
kit (Beyotime Institute of Biotechnology, Haimen, China). A549
cells were pretreated with 0, 1, 2 or 5 µM JS-K for 48 h, and H460
cells were pretreated with 0, 5, 10 or 15 µM for 24 h. The cells
were then washed with cold phosphate-buffered saline (PBS) and
fixed with 70% ethanol overnight at 4°C. Subsequently, the ethanol
was removed and the cells were incubated with propyl iodide
organism dye for 30 min at 37°C. The dyed cells were detected with
a BD Biosciences FACSCanto II flow cytometer (BD Biosciences,
Franklin Lakes, NJ, USA) and then analyzed using ModFit and
CellQuest software 6.1 (BD Biosciences).
Analysis of cell apoptosis
To detect JS-K-induced apoptosis of the lung cancer
cells, an Annexin V-fluorescein isothiocyanate (FITC)/propidium
iodide (PI) double staining assay was performed with the Annexin
V-FITC/PI kit (Sigma-Aldrich; Merck KGAA). Briefly, A549 cells were
treated with 0, 1, 2 or 5 µM JS-K for 48 h, and H460 cells were
treated with 0, 5, 10 or 15 µM JS-K for 24 h. The cells were then
collected and resuspended in 1X binding buffer, which contained 10
mM 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid (pH 7.5),
2.5 mM CaCl2 and 140 nM NaCl. Cells were stained in the
dark with Annexin V-FITC and PI for 15 min, according to the
manufacturer's protocol, prior to flow cytometric analysis. Annexin
V-positive cells were regarded to be in the early stage of
apoptosis, whereas Annexin V and PI-positive cells were considered
to be in the late phase of apoptosis.
Western blot analysis of
JS-K-regulated apoptotic proteins and cell cycle proteins
The JS-K-treated cells were collected and lysed in
lysis buffer, containing 1 ml RIPA buffer and 10 µl PMSF (Beyotime
Institute of Biotechnology). The samples were then centrifuged for
20 min at 16,900 × g at 4°C to acquire the supernatants. The
protein levels were quantified using a bicinchoninic acid assay
(Beyotime Institute of Biotechnology) and equal amounts of protein
were loaded to a 10% sodium dodecyl sulfate-polyacrylamide gel. The
amount of p-Cdc2 loaded per lane was 40 µg, and for the other
proteins 20 µg was loaded. Following electrophoresis, the separated
proteins were transferred to polyvinylidene difluoride membranes,
which were then blocked with 5% non-fat milk for 40 min at room
temperature. The primary antibodies against cleaved-caspase-3,
cleaved-caspase-9, cleaved-caspase-8, Bcl-2, Bax, p53, p21, p27,
cyclin B1, CDK2, cyclin E1, Cdc2, cleaved-Cdc2 and GAPDH were
incubated with the membranes at 4°C overnight. Subsequently,
anti-rabbit IgG monoclonal antibody conjugated with horseradish
peroxidase was added for 1 h at room temperature. Proteins were
visualized with chemiluminescent reagent and a Tanon 5200 system
(Tanon Science & Technology Co., Ltd., Shanghai, China). The
data were analyzed using ImageJ (v1.8.0) analysis software
(National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
All data were acquired from a minimum of three
independent repetitions. The results are presented as the mean ±
standard deviation (SD) and the SD is indicated with an error bar
in all figures. Statistical analysis was performed using SPSS
software (version 17.0; IBM Corp., Armonk, NY, USA). Comparisons
between groups were evaluated using one-way analysis of variance
(ANOVA) (followed by Tukey's post hoc test). P<0.05 and
P<0.01 were considered to indicate statistically significant
differences.
Results
JS-K inhibits proliferation and
induces apoptosis of A549 and H460 cells
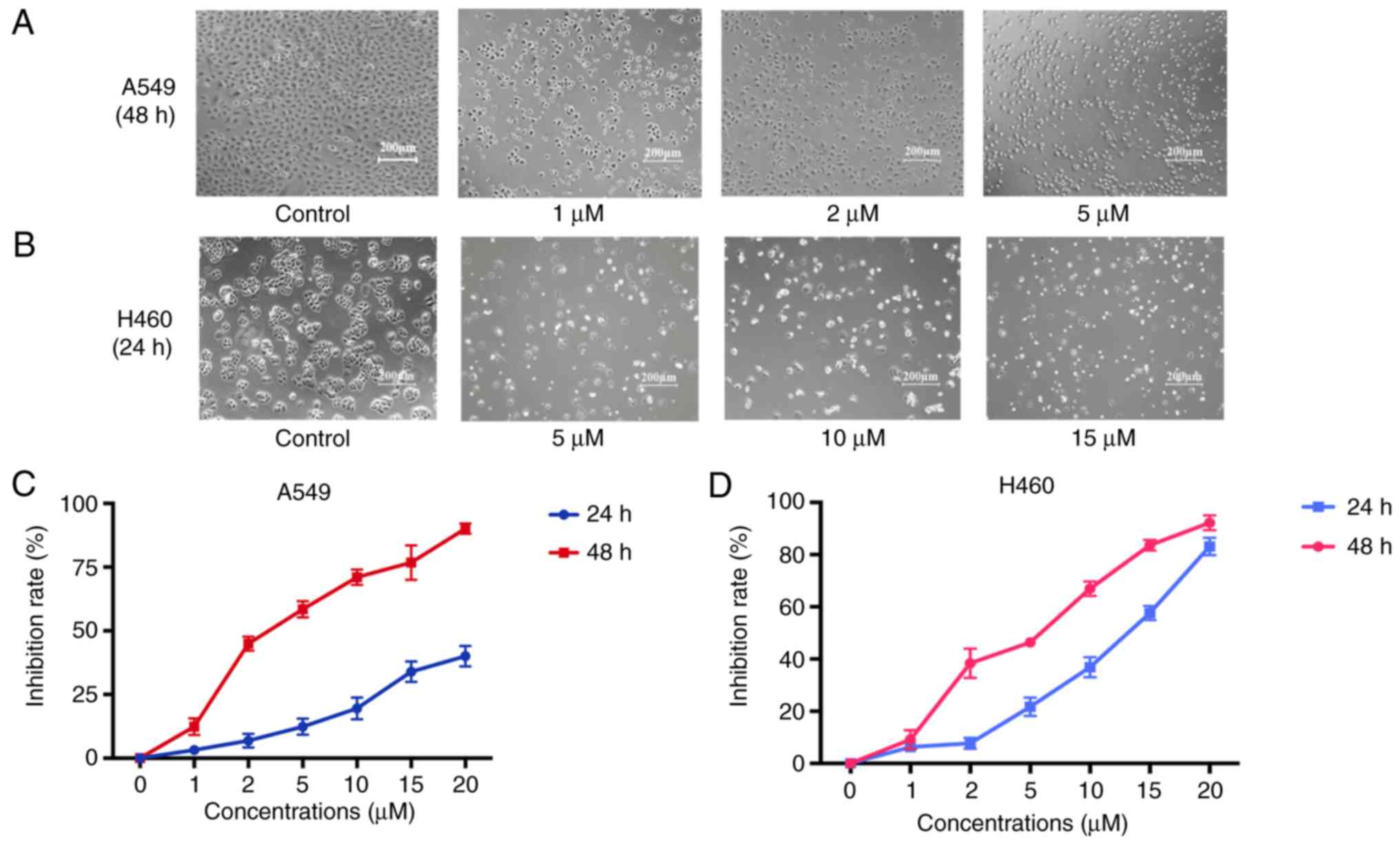
The MTT assay with different concentrations of JS-K
for 24 and 48 h demonstrated that JS-K exerted a significant
inhibitory effect on the proliferation of A549 and H460 cells in a
concentration- and time-dependent manner (Fig. 2). The IC50 value of JS-K
at 48 h for A549 cells was 3.48±0.02 µM. The IC50 value
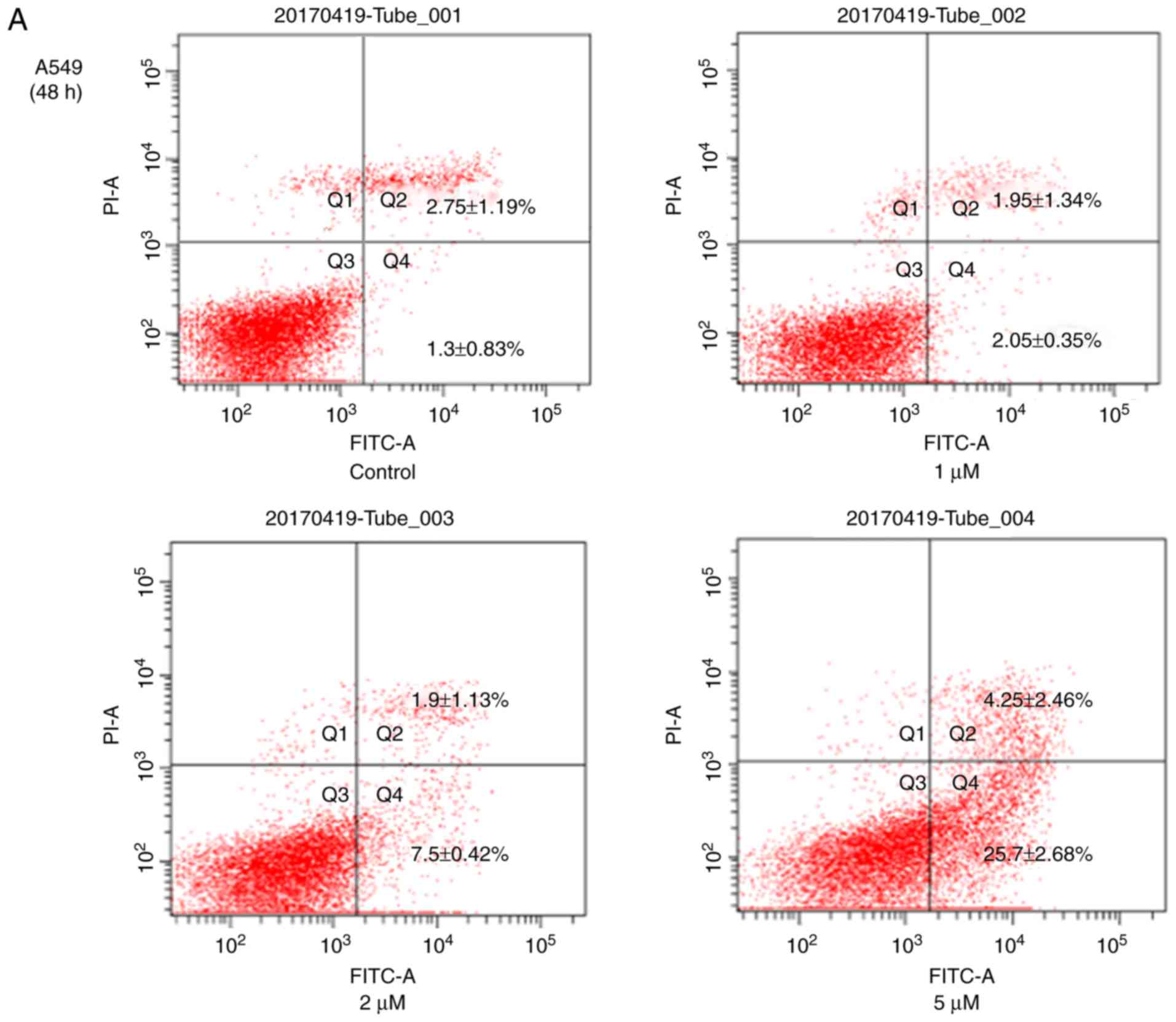
of JS-K at 24 h for H460 cells was 11.17±0.03 µM. The Annexin
V-FITC/PI cell apoptosis detection kit was also used to detect
apoptosis of H460 and A549 cells (Fig.
3). The results revealed a significant increase in the
apoptosis rates of these cells with increasing concentrations of
JS-K. In addition, the Q2 and Q4 cell
population in A549 (4.05–29.95%) and H460 (5.83–21.0%) cells was
increased compared with the control group. As demonstrated in
Figs. 2 and 3, JS-K inhibited proliferation and
promoted apoptosis of H460 and A549 cells in a
concentration-dependent manner.
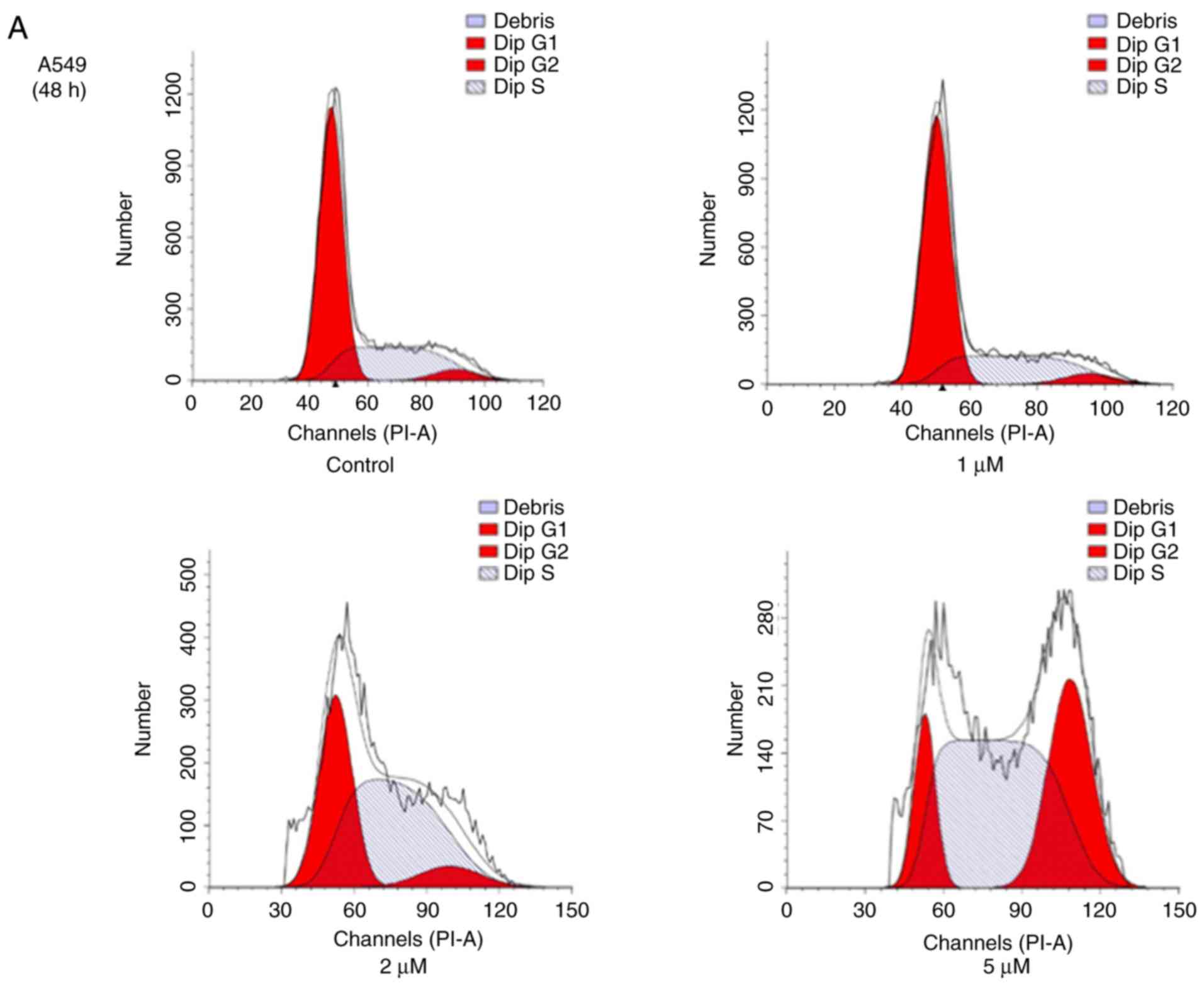
JS-K promotes cell cycle arrest
To reveal whether JS-K exhibits an effect on cell
cycle arrest in human lung carcinoma cells, A549 and H460 cells
were treated with various concentrations of JS-K for 48 and 24 h,
respectively, and were then subjected to flow cytometric analysis
following DNA staining. The population of cells in the
G2/M phase increased in A549 and H460 cells, coinciding
with a reduction in cells in the G1 phase, when compared
with the control group. The results revealed that the proportion of
cells in the G2/M phase increased with increasing
concentrations of JS-K following the indicated time-points
(Fig. 4).
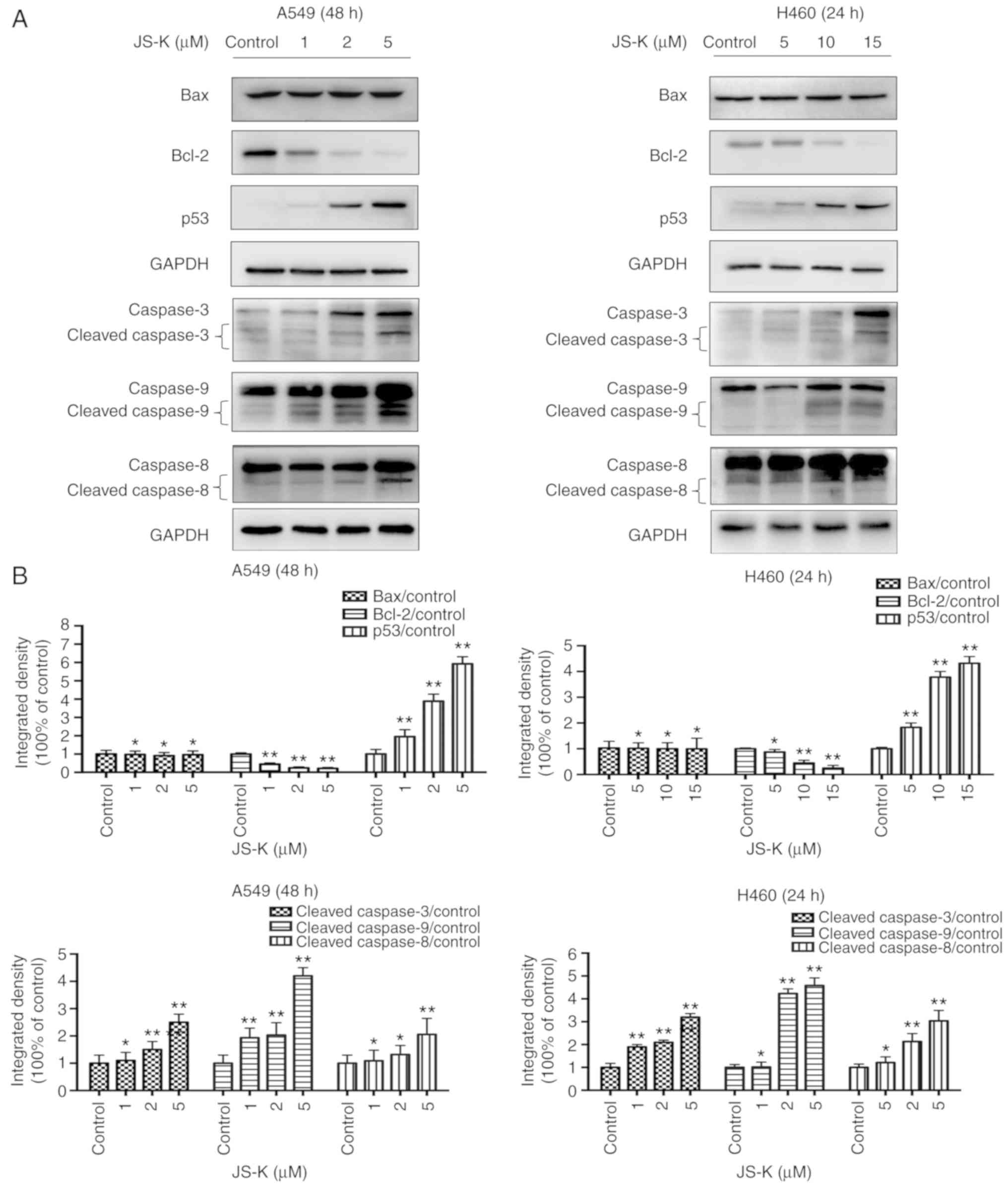
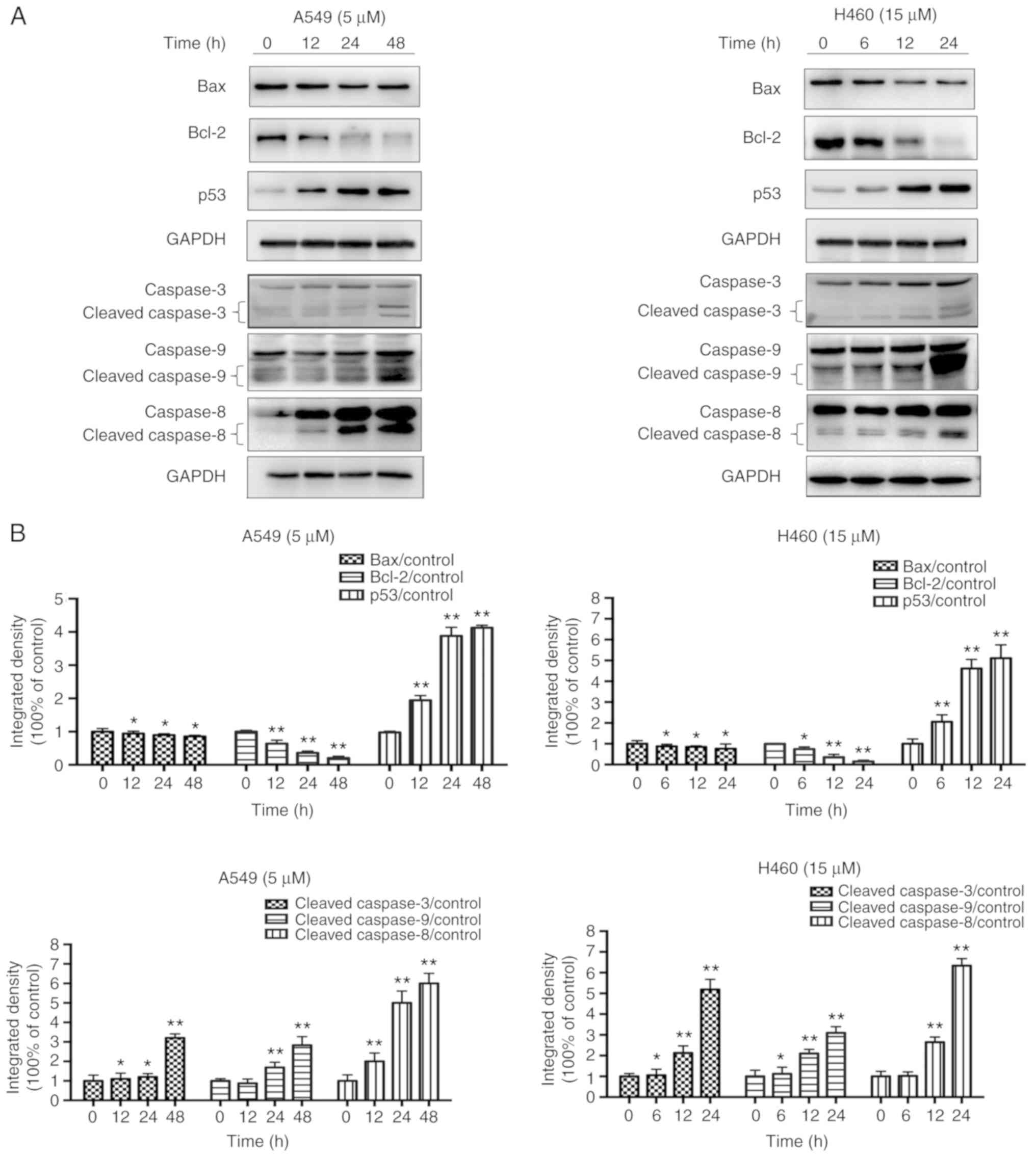
JS-K activates proteins associated
with apoptosis and cell cycle arrest
The expression levels of apoptosis-associated
proteins, including cleaved-caspase-9, cleaved-caspase-8,
cleaved-caspase-3 and p53, and the ratio of Bax/Bcl-2 were
identified to increase in a concentration- and time-dependent
manner following treatment with JS-K compared with the control
cells (Figs. 5 and 6). The amount of p-Cdc2 loaded per lane
was 40 µg, and for the others 20 µg was loaded. In addition, the
expression levels of proteins associated with cycle arrest,
including cyclin E1, cyclin B1, p21, p27 and p-Cdc2, were revealed
to increase in a concentration- and time-dependent manner following
treatment with JS-K compared with the control cells. Conversely,
the expression levels of CDK2, cyclin B1 and Cdc2 were revealed to
decrease in a concentration- and time-dependent manner following
treatment with JS-K (Figs. 7 and
8).
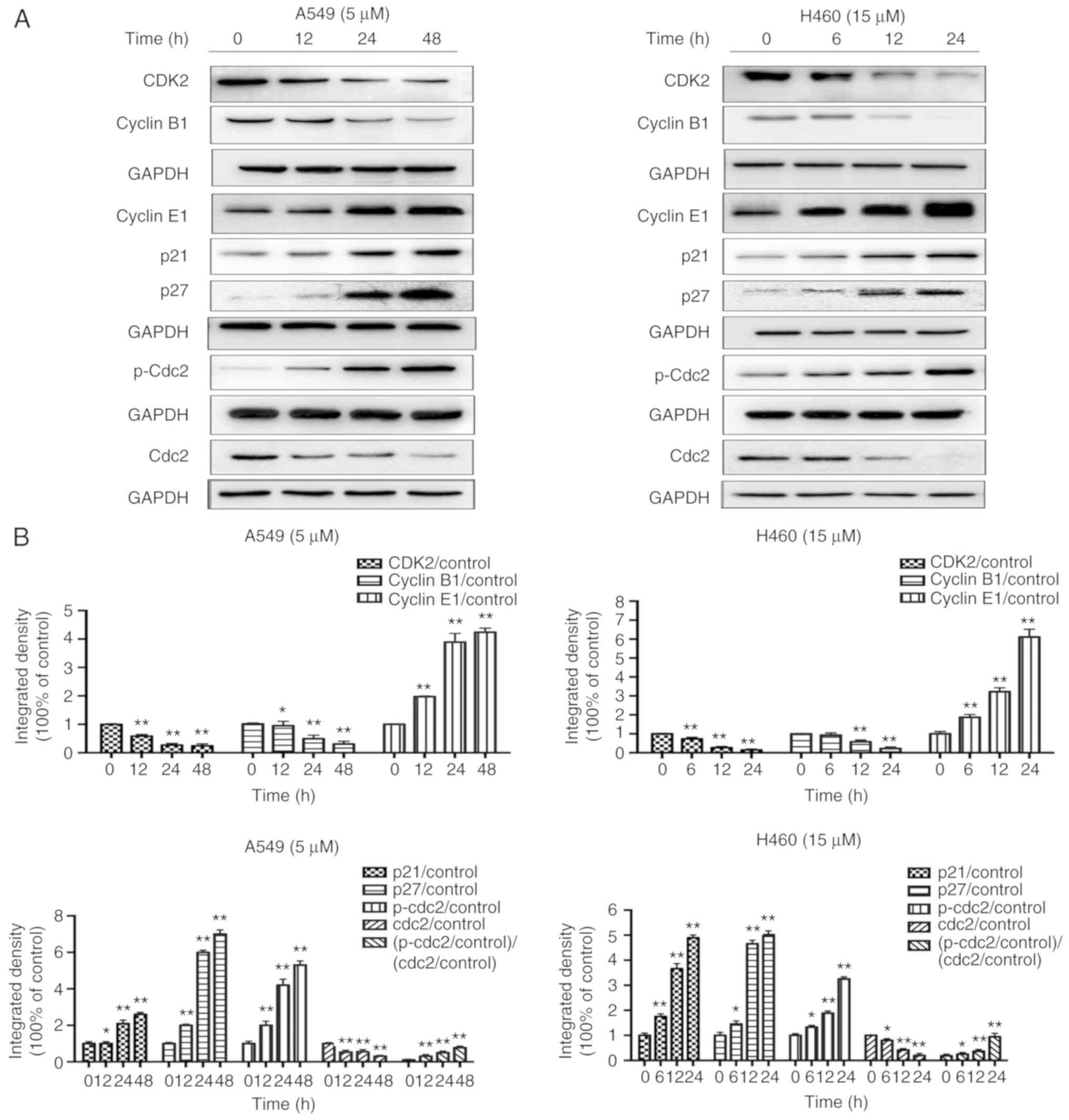
 | Figure 7.JS-K influences the
p21WAF1/CIP1 and p27KIP1 pathways. JS-K
altered the expression levels of cell cycle-associated proteins in
A549 and H460 cells following treatment for the indicated
time-points. (A) The protein expression levels of CDK2, cyclin B1,
cyclin E1, p21, p27, p-Cdc2 and Cdc2 were detected by western blot
analysis. JS-K downregulated the expression of CDK2, cyclin B1 and
Cdc2, and upregulated cyclin E1, p21, p27 and p-Cdc2 in a
concentration-dependent manner. (B) Quantification of the western
blot analysis results for A549 and H460 cells. The amount of p-Cdc2
loaded per lane was 40 µg, while for the other proteins 20 µg was
loaded. Data are representative of three independent experiments.
Values are presented as the mean ± standard deviation (n=3).
*P<0.05, **P<0.01 vs. the control group. JS-K,
O2-(2,4-dinitrophenyl)-1-[(4-ethoxycarbonyl)piperazin-1-yl]diazen-1-ium-1,2-diolate. |
p53 serves a crucial role in
JS-K-induced apoptosis and cell cycle arrest in A549 and H460
cells
As demonstrated in Figs.
9 and 10 the expression levels
of proteins associated with apoptosis and cell cycle arrest were
altered following JS-K treatment. However, it was identified that
PFT-α could alleviate JS-K-induced cell apoptosis and cell cycle
arrest. The results revealed that PFT-α significantly reduced the
expression levels of p53, Bax, cleaved caspase-3, cleaved caspase-8
and cleaved caspase-9, and upregulated the expression level of
Bcl-2 in comparison with cells treated with JS-K alone. In
addition, the expression levels of proteins associated with cell
cycle arrest were altered following treatment with PFT-α. The
results indicated that PFT-α significantly reduced the expression
level of cyclin E1, p21, p27 and p-Cdc2. and upregulated the
expression level of CDK2, cyclin B1 and Cdc2 in comparison with
cells treated with JS-K alone. In summary, the results indicated a
crucial role of p53 in JS-K-induced cellular apoptosis and cell
cycle arrest in A549 and H460 cells.
Discussion
Despite advancements in therapeutic strategies and
the development of new anticancer drugs, lung cancer is usually
incurable. Therefore, investigations of novel therapeutic
strategies and anticancer agents are urgently required. The
antitumor effect of JS-K has attracted attention in recent years.
JS-K is a diazeniumdiolate-based NO-donor prodrug, which has been
widely studied due to its potential in increasing ROS levels, and
the mechanisms of JS-K are largely understood. For example, JS-K
has been reported to induce apoptosis of bladder cancer cells by
increasing the levels of ROS (29).
Although JS-K has been revealed to induce apoptosis and ROS
accumulation in several types of human cancer cells, its effects on
cell cycle signaling and apoptosis are poorly understood. In the
present study, a potent antiproliferative effect of JS-K was
observed in A549 and H460 cells. JS-K was identified to exert a
marked effect on G2/M phase arrest and evidently
triggered apoptosis in concentration- and time-dependent manners in
both cell lines via the p53/p21WAF1/CIP1 and
p27KIP1 pathways.
The proliferation of tumor cells is closely
associated with cell cycle arrest, which can serve as a marker for
the chemopreventative or antitumor activity of drugs (33). Three checkpoints in the
G1, S and G2/M phases are mediated by various
cell cycle regulatory proteins, including cyclins, CDKs and tumor
suppressor proteins, including p53, p21WAF1/CIP1 and
p27KIP1 (34).
CDK-cyclin complexes are drivers of the cell cycle and are mediated
by two families of CKIs, the CIP/KIP family, which includes
p21WAF1/CIP1 and p27KIP1, and the INK4
family, which includes p15INK4b and p19INK4d
(35). As the only CDK2-associated
cyclin, cyclin E is a strong independent prognostic marker in
patients with early-stage NSCLC (36). Results of the present study
demonstrated that JS-K upregulated the expression level of cyclin E
and downregulated the expression level of CDK2 in a concentration
and time-dependent manner in A549 and H460 cells. Cdc2, also termed
CDK-1, is associated with cyclin B1. The G2/M transition
is often regulated by Cdc2 in mammalian cells (23). Cdc2 serves a crucial role in the
induction of mitosis. Suppressing the activity of Cdc2 resulted in
cell cycle arrest at the G2/M phase in several types of
cell lines (37,38). In addition, it has been demonstrated
that activation of the Cdc2/cyclin B1 complex was indispensable for
the transition from the G2 to the M phase (39). In the present study, JS-K was
identified to induce cell cycle arrest at the G2/M phase
(Fig. 4). This effect was
associated with a downregulation of cyclin B1 and Cdc2, an
upregulation of p21WAF1/CIP1, and phosphorylation of
Cdc2 in a concentration and time-dependent manner in A549 and H460
cells (Fig. 7). For the western
blot analysis, the amount of p-Cdc2 loaded per lane was 40 µg,
while for the other 20 µg was loaded. We consider that different
amounts of samples may lead to some errors in the results. After
adjusting the amounts of samples, we determined that the amount of
sample of each analyzed lane was uniform by exposing the internal
reference protein (GAPDH). The results revealed that the expression
of the reference protein (GAPDH) and the target protein (p-Cdc2)
was increased after adjusting the amounts of samples. Satisfactory
observation results were obtained. The trend of GAPDH expression of
each analyzed lane was consistent with that before samples
adjustment. Although the amount of p-Cdc2 was different from that
of Cdc2, the batches of samples on the analyzed lanes were the
same. In addition, three batches of samples were repeated to obtain
the means and relative errors. Then the relative errors were
greatly reduced. In order to determine the phosphorylation, the
following was assessed in the quantification section:
‘(p-Cdc2/control)/(Cdc2/control)’. Therefore, the errors caused by
adjusting the amounts of samples had little effect on the results
of the quantification analysis.
Following binding to cyclin-CDK complexes, members
of the Cip/Kip family, including p21WAF1/CIP1 and
p27KIP1, can prevent kinase activation and subsequently
inhibit the progression of the cell cycle at the G2/M
phase (40). p27KIP1
acts as a tumor suppressor by binding to complexes of cyclin E/CDK2
to prevent cell cycle progression from the G1 phase to
the S phase (41). A previous study
revealed that a low expression level of p27KIP1 was
associated with tumor aggressiveness and poor patient survival
(42). One of the functions of
p21WAF1/CIP1 is to maintain cells at G2/M
phases following DNA damage (18).
In addition, p21WAF1/CIP1 can interact with cyclin B1,
which prevents dephosphorylation of Cdc2 by Cdc25 and inhibit the
activity of CDK-1 (43). Activated
p53 has been revealed to serve a role in the regulation of cell
cycle progression following DNA damage. The mechanism of p53 in
G2/M phase cell cycle arrest involves the
transactivation of the CDK inhibitor p21 (44). The results of the present study
revealed that the expression levels of p53, p21WAF1/CIP1
and p27KIP1 increased in a concentration- and
time-dependent manner in A549 and H460 cells following treatment
with JS-K. These findings indicated that JS-K-induced
G2/M phase cell cycle arrest may be mediated by the
upregulation of p53, p21WAF1 and p27KIP1.
To maintain tissue homeostasis, apoptosis serves as
a physiological pathway to eliminate damaged or infected cells
(45). Apoptosis induction has been
considered to be a useful strategy for cancer therapy. In the
present study, the results of MTT assay suggested that JS-K acted
as an anti-proliferation agent against A549 and H460 cells
(Fig. 2). Therefore, in addition to
cell cycle arrest, the present study investigated whether
JS-K-induced cell growth arrest could be due to the induction of
apoptosis. In the apoptosis assay, a significant increase in the
apoptosis rates of A549 and H460 cells were revealed by flow
cytometry following treatment with JS-K (Fig. 3).
Apoptosis can be activated via the extrinsic- or
death receptor-associated pathways, as well the intrinsic- or
organelle-mediated pathways (45).
It is well known that p53 acts upstream of other pro-apoptotic
proteins and mediates both the intrinsic and the extrinsic
apoptotic pathways (46). Cysteine
proteases, particularly caspases, can be classified as initiator
caspases, including caspases-8 and −9, or as executioner caspases,
including caspases-3, −6 and −7, based on their mechanism of action
(47). Caspase-3, an executioner
caspase, can be activated by the extrinsic pathway involving the
activation of caspase-8 and the intrinsic pathway involving the
activation of caspase-9 (48,49).
The present study revealed that JS-K treatment could trigger both
the mitochondria-initiated intrinsic pathway and extrinsic pathway
in A549 and H460 cells. The results demonstrated that cleaved
caspase-3, −8 and −9 were significantly upregulated following
treatment with JS-K, which indicated that JS-K induced apoptosis in
A549 and H460 cells via the mitochondria-initiated intrinsic
pathway and extrinsic pathway by the upregulation of p53 (Figs. 5 and 6).
p53 is a critical transcription factor that
regulates the transcription and expression of various target genes,
including Bcl-2 and Bax, which can result in cell cycle arrest and
apoptosis (50,51). The anti-apoptotic ability of Bcl-2
is suppressed following binding with Bax protein (52). Overall, p53 can positively regulate
the expression of the proapoptotic proteins Bax, Bad and Bak, which
prevents inhibition of Bcl-2 (49).
Compared with the expression level of Bcl-2 alone, the ratio of
Bcl-2 to Bax has been reported to a serve a larger role in cell
survival or death following an apoptotic stimulus (53,54).
The present study demonstrated that JS-K markedly upregulated the
expression levels of cell cycle-associated proteins, including
p21WAF1/CIP1 and p53, and also enhanced the ratio of
Bax/Bcl-2 by increasing the expression level of Bax and reducing
the expression level of the anti-apoptotic protein Bcl-2 in A549
and H460 cells (Fig. 5). These
results indicated that p53 and Bax/Bcl-2 served an important role
in JS-K-induced cell apoptosis. In addition, the results
demonstrated that JS-K can be inhibited by PFT-α (Figs. 9 and 10). Following inhibition of JS-K with
PFT-α, Bcl-2 suppression was markedly reversed, and
p21WAF1/CIP1 and p27KIP1 were significantly
downregulated with a decrease in the expression level of p53.
Therefore, targeting the p53/p21WAF1/CIP1 and
p27KIP1 signaling pathway may be considered as a
potential strategy to inhibit the proliferation of A549 and H460
cells.
This research was aimed at evaluating the anticancer
effect of JS-K and exploring its mechanism in A549 and H460 human
NSCLC cells. It is unclear whether JS-K acts on other NSCLC cells
via the p53/p21WAF1/CIP1 and p27KIP1
pathways. In addition, in vivo experiments were not carried
out. In order to further elucidate the mechanism of JS-K, we will
use JS-K to screen NSCLC cells comprehensively, and conduct in
vivo studies in the future. In conclusion, the present study
revealed that JS-K could induce cell cycle arrest and apoptosis in
A549 and H460 cells via the p53/p21WAF1/CIP1 and
p27KIP1 pathways. These findings promote understanding
regarding the antitumor effect of JS-K in NSCLC and suggest that
JS-K may be a novel candidate for lung cancer therapy.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Yangfan Plan
of Talents Recruitment Grant, Guangdong, China (Yue Ren Cai Ban
[2016] 6); the Scientific Research Fund of Guangdong Medical
College, China (M2016031, M2016032).
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
DC, WL and RZ conceived and designed the study. DC
and WL provided administrative support. ZS, RZ, XH and YY provided
the materials of the study. BL, JW, ZS, CG and XH collected and
assembled the data. SH, BL, JW, YY and WL performed the data
analysis and interpretation. SH, CG and WL wrote the manuscript.
All authors read and approved the manuscript and agree to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Daniels MG, Bowman RV, Yang IA, Govindan R
and Fong KM: An emerging place for lung cancer genomics in 2013. J
Thorac Dis. 5 (Suppl 5):S491–S497. 2013.PubMed/NCBI
|
|
3
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J and
Thun MJ: Cancer statistics, 2009. CA Cancer J Clin. 59:225–249.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Askoxylakis V, Thieke C, Pleger ST, Most
P, Tanner J, Lindel K, Katus HA, Debus J and Bischof M: Long-term
survival of cancer patients compared to heart failure and stroke: A
systematic review. BMC Cancer. 10:1052010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Saito S, Espinoza-Mercado F, Liu H, Sata
N, Cui X and Soukiasian HJ: Current status of research and
treatment for non-small cell lung cancer in never-smoking females.
Cancer Biol Ther. 18:359–368. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zarogoulidis K, Zarogoulidis P, Darwiche
K, Boutsikou E, Machairiotis N, Tsakiridis K, Katsikogiannis N,
Kougioumtzi I, Karapantzos I, Huang H and Spyratos D: Treatment of
non-small cell lung cancer (NSCLC). J Thorac Dis. 4 (Suppl
5):S389–S396. 2013.
|
|
7
|
Farhat FS and Houhou W: Targeted therapies
in non-small cell lung carcinoma: What have we achieved so far?
Ther Adv Med Oncol. 5:249–270. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pirozynski M: 100 years of lung cancer.
Respir Med. 100:2073–2084. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schwartz AG, Prysak GM, Bock CH and Cote
ML: The molecular epidemiology of lung cancer. Carcinogenesis.
28:507–518. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Spiro SG and Silvestri GA: One hundred
years of lung cancer. Am J Respir Crit Care Med. 172:523–529. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Blandin Knight S, Crosbie PA, Balata H,
Chudziak J, Hussell T and Dive C: Progress and prospects of early
detection in lung cancer. Open Boil. 7:1700702017. View Article : Google Scholar
|
|
12
|
Vilmar AC and Sorensen JB: Customising
chemotherapy in advanced nonsmall cell lung cancer: Daily practice
and perspectives. Eur Respir Rev. 20:45–52. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang T, Nelson RA, Bogardus A and Grannis
FW Jr: Five-year lung cancer survival: Which advanced stage
nonsmall cell lung cancer patients attain long-term survival?
Cancer. 116:1518–1525. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang H, Feng QQ, Gong JH and Ma JP:
Anticancer effects of isofraxidin against A549 human lung cancer
cells via the EGFR signaling pathway. Mol Med Rep. 18:407–414.
2018.PubMed/NCBI
|
|
15
|
Bailey SM, Meyne J, Chen DJ, Kurimasa A,
Li GC, Lehnert BE and Goodwin EH: DNA double-strand break repair
proteins are required to cap the ends of mammalian chromosomes.
Proc Natl Acad Sci USA. 96:14899–14904. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vazquez A, Bond EE, Levine AJ and Bond GL:
The genetics of the p53 pathway, apoptosis and cancer therapy. Nat
Rev Drug Discov. 7:979–987. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Choisy-Rossi C, Reisdorf P and
Yonish-Rouach E: Mechanisms of p53-induced apoptosis: In search of
genes which are regulated during p53-mediated cell death. Toxicol
Lett. 102-103:491–496. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bunz F, Dutriaux A, Lengauer C, Waldman T,
Zhou S, Brown JP, Sedivy JM, Kinzler KW and Vogelstein B:
Requirement for p53 and p21 to sustain G2 arrest after
DNA damage. Science. 282:1497–1501. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vogelstein B, Lane D and Levine AJ:
Surfing the p53 network. Nature. 408:307–310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pietenpol JA and Stewart ZA: Cell cycle
checkpoint signaling: Cell cycle arrest versus apoptosis.
Toxicology. 181-182:475–481. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pflaum J, Schlosser S and Muller M: p53
family and cellular stress responses in cancer. Front Oncol.
4:2852014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shi Y: Mechanisms of caspase activation
and inhibition during apoptosis. Mol Cell. 9:459–470. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yunlan L, Juan Z and Qingshan L: Antitumor
activity of di-n-
butyl-(2,6-difluorobenzohydroxamato)tin(IV) against human gastric
carcinoma SGC-7901 cells via G2/M cell cycle arrest and cell
apoptosis. PLoS One. 9:e907932014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang Z, Fu J and Zhang Y: Nitric oxide
donor-based cancer therapy: Advances and prospects. J Med Chem.
60:7617–7635. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Laschak M, Spindler KD, Schrader AJ,
Hessenauer A, Streicher W, Schrader M and Cronauer MV: JS-K, a
glutathione/glutathione S-transferase-activated nitric oxide
releasing prodrug inhibits androgen receptor and WNT-signaling in
prostate cancer cells. BMC Cancer. 12:1302012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu L, Huang Z, Chen J, Wang J and Wang S:
Protein phosphatase 2A mediates JS-K-induced apoptosis by affecting
Bcl-2 family proteins in human hepatocellular carcinoma HepG2
cells. J Cell Biochem. 119:6633–6643. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shami PJ, Saavedra JE, Wang LY, Bonifant
CL, Diwan BA, Singh SV, Gu Y, Fox SD, Buzard GS, Citro ML, et al:
JS-K, a glutathione/glutathione S-transferase-activated nitric
oxide donor of the diazeniumdiolate class with potent
antineoplastic activity. Mol Cancer Ther. 2:409–417.
2003.PubMed/NCBI
|
|
28
|
Kiziltepe T, Hideshima T, Ishitsuka K,
Ocio EM, Raje N, Catley L, Li CQ, Trudel LJ, Yasui H, Vallet S, et
al: JS-K, a GST-activated nitric oxide generator, induces DNA
double-strand breaks, activates DNA damage response pathways, and
induces apoptosis in vitro and in vivo in human multiple myeloma
cells. Blood. 110:709–718. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Qiu M, Chen L, Tan G, Ke L, Zhang S, Chen
H and Liu J: A reactive oxygen species activation mechanism
contributes to JS-K-induced apoptosis in human bladder cancer
cells. Sci Rep. 5:151042015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Qiu M, Ke L, Zhang S, Zeng X, Fang Z and
Liu J: JS-K, a GST-activated nitric oxide donor prodrug, enhances
chemo- sensitivity in renal carcinoma cells and prevents cardiac
myocytes toxicity induced by Doxorubicin. Cancer Chemother
Pharmacol. 80:275–286. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Simeone AM, McMurtry V, Nieves-Alicea R,
Saavedra JE, Keefer LK, Johnson MM and Tari AM: TIMP-2 mediates the
anti-invasive effects of the nitric oxide-releasing prodrug JS-K in
breast cancer cells. Breast Cancer Res. 10:R442008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Maciag AE, Chakrapani H, Saavedra JE,
Morris NL, Holland RJ, Kosak KM, Shami PJ, Anderson LM and Keefer
LK: The nitric oxide prodrug JS-K is effective against
non-small-cell lung cancer cells in vitro and in vivo: Involvement
of reactive oxygen species. J Pharmacol Exp Ther. 336:313–320.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ou X, Lu Y, Liao L, Li D, Liu L, Liu H and
Xu H: Nitidine chloride induces apoptosis in human hepatocellular
carcinoma cells through a pathway involving p53, p21, Bax and
Bcl-2. Oncol Rep. 33:1264–1274. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shin SY, Yong Y, Kim CG, Lee YH and Lim Y:
Deoxypodophyllotoxin induces G2/M cell cycle arrest and
apoptosis in HeLa cells. Cancer Lett. 287:231–239. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hwang HJ, Kang YJ, Hossain MA, Kim DH,
Jang JY, Lee SH, Yoon JH, Moon HR, Kim HS, Chung HY, et al: Novel
dihydrobenzofuro[4,5-b][1,8]naphthyridin-6-one derivative, MHY-449,
induces apoptosis and cell cycle arrest in HCT116 human colon
cancer cells. Int J Oncol. 41:2057–2064. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Muller-Tidow C, Metzger R, Kugler K,
Diederichs S, Idos G, Thomas M, Dockhorn-Dworniczak B, Schneider
PM, Koeffler HP, Berdel WE, et al: Cyclin E is the only
cyclin-dependent kinase 2-associated cyclin that predicts
metastasis and survival in early stage non-small cell lung cancer.
Cancer Res. 61:647–653. 2001.PubMed/NCBI
|
|
37
|
Bulavin DV, Higashimoto Y, Popoff IJ,
Gaarde WA, Basrur V, Potapova O, Appella E and Fornace AJ Jr:
Initiation of a G2/M checkpoint after ultraviolet radiation
requires p38 kinase. Nature. 411:102–107. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen YL, Lin SZ, Chang JY, Cheng YL, Tsai
NM, Chen SP, Chang WL and Harn HJ: In vitro and in vivo studies of
a novel potential anticancer agent of isochaihulactone on human
lung cancer A549 cells. Biochem Pharmacol. 72:308–319. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lew DJ and Kornbluth S: Regulatory roles
of cyclin dependent kinase phosphorylation in cell cycle control.
Curr Opin Cell Biol. 8:795–804. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K
and Linn S: Molecular mechanisms of mammalian DNA repair and the
DNA damage checkpoints. Annu Rev Biochem. 73:39–85. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Srivastava RK, Chen Q, Siddiqui I, Sarva K
and Shankar S: Linkage of curcumin-induced cell cycle arrest and
apoptosis by cyclin-dependent kinase inhibitor p21(/WAF1/CIP1).
Cell Cycle. 6:2953–2961. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Migita T, Oda Y, Naito S and Tsuneyoshi M:
Low expression of p27Kip1 is associated with tumor size
and poor prognosis in patients with renal cell carcinoma. Cancer.
94:973–979. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
O'Connell MJ, Walworth NC and Carr AM: The
G2-phase DNA-damage checkpoint. Trends Cell Biol. 10:296–303. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chaudhary P, Sharma R, Sahu M, Vishwanatha
JK, Awasthi S and Awasthi YC: 4-Hydroxynonenal induces
G2/M phase cell cycle arrest by activation of the ataxia
telangiectasia mutated and Rad3-related protein (ATR)/checkpoint
kinase 1 (Chk1) signaling pathway. J Biol Chem. 288:20532–20546.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Guicciardi ME, Malhi H, Mott JL and Gores
GJ: Apoptosis and necrosis in the liver. Compr Physiol. 3:977–1010.
2013.PubMed/NCBI
|
|
46
|
Zhu X, Wang K, Zhang K, Zhang T, Yin Y and
Xu F: Ziyuglycoside I inhibits the proliferation of MDA-MB-231
breast carcinoma cells through inducing p53-mediated G2/M cell
cycle arrest and intrinsic/extrinsic apoptosis. Int J Mol Sci.
17:E19032016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
McIlwain DR, Berger T and Mak TW: Caspase
functions in cell death and disease. Cold Spring Harb Perspect
Biol. 5:a0086562013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT,
Liu B and Bao JK: Programmed cell death pathways in cancer: A
review of apoptosis, autophagy and programmed necrosis. Cell
Prolif. 45:487–498. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhang Q, Ma S, Liu B, Liu J, Zhu R and Li
M: Chrysin induces cell apoptosis via activation of the
p53/Bcl-2/caspase-9 pathway in hepatocellular carcinoma cells. Exp
Ther Med. 12:469–474. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Brady CA, Jiang D, Mello SS, Johnson TM,
Jarvis LA, Kozak MM, Kenzelmann Broz D, Basak S, Park EJ,
McLaughlin ME, et al: Distinct p53 transcriptional programs dictate
acute DNA-damage responses and tumor suppression. Cell.
145:571–583. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Burmakin M, Shi Y, Hedstrom E, Kogner P
and Selivanova G: Dual targeting of wild-type and mutant p53 by
small molecule RITA results in the inhibition of N-Myc and key
survival oncogenes and kills neuroblastoma cells in vivo and in
vitro. Clin Cancer Res. 19:5092–5103. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zeng G, Liu J, Chen H, Liu B, Zhang Q, Li
M and Zhu R: Dihydromyricetin induces cell cycle arrest and
apoptosis in melanoma SK-MEL-28 cells. Oncol Rep. 31:2713–2719.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Reed JC: Bcl-2 family proteins: Regulators
of apoptosis and chemoresistance in hematologic malignancies. Semin
Hematol 34 (4 Suppl 5). S9–S19. 1997.
|
|
54
|
Pettersson F, Dalgleish AG, Bissonnette RP
and Colston KW: Retinoids cause apoptosis in pancreatic cancer
cells via activation of RAR-gamma and altered expression of
Bcl-2/Bax. Br J Cancer. 87:555–561. 2002. View Article : Google Scholar : PubMed/NCBI
|