Introduction
Acute lymphoblastic leukemia (ALL) is the most
commonly diagnosed malignancy in children, accounting for
approximately 30% of all pediatric cancers worldwide (1). Advances in the treatment of children
with ALL have led to 5-year disease-free survival rates exceeding
85% (2). However, children with ALL
cells that show resistance to glucocorticoids (GCs) have a
significantly poorer treatment outcome when compared with patients
with ALL cells that are sensitive to GCs (3–6). The
underlying mechanisms of this phenomenon are not yet clear.
Therefore, there is an immediate need to elucidate the mechanisms
of GC resistance and thereby explore novel therapeutic strategies
to reverse GC resistance, which will help achieve complete
treatment potential and significantly improve prognosis.
GCs are widely used in antileukemia therapy, due to
their extreme pro-apoptotic effects on lymphoblasts. The
physiologic and pharmacologic actions of GCs are mediated by the GC
receptor (GR), which consists of three isoforms: GRα, GRβ and GRγ
(7). In spite of GC binding to all
three isoforms, only GRα mediates appropriate GC signaling, whereas
GRβ and GRγ prevent the GC/GR complex from binding to DNA (8). Thus, these two latter forms of GRs
inhibit GC activity and play an important role in GC resistance
(9).
Bcl-2-interacting mediator of cell death (BCL2L11,
BIM) is a pro-apoptotic BH3-only member of the B-cell
leukemia/lymphoma (BCL2) family, and plays a critical role in the
development of the lymphoid system. Three major alternative
transcription variants, BIM-EL, BIM-L and BIM-S, which are formed
by alternative splicing within exon 2, induce apoptosis through a
pro-apoptotic BH3 domain that is encoded exclusively by exon 4 of
the BIM gene (10). BIM is a
critical mediator of GC-induced cell death of normal lymphocytes
and ALL cells and is upregulated upon GC stimulation (11).
MicroRNAs (miRNAs) belong to a class of endogenously
expressed non-coding single-stranded RNAs of 18–24 nucleotides and
induce gene silencing by binding to target mRNAs with partial
complementarity (12,13). miRNAs play a significant regulatory
role in GC resistance by several mechanisms such as: i)
Post-transcriptionally modulating GR mRNA translation thereby
affecting GR levels; ii) altering the receptor-isoform ratio; or
iii) controlling activity of other transcriptional factors.
Involvement of miRNAs in the GR transcriptional pathways have been
studied. High levels of miR-142-3p resulted in low levels of GRα
protein expression, thus leading to resistance of T-leukemic cells
to GCs (14). It has also been
reported that miR-124 induces resistance to GC treatment by
targeting GR in ALL (15). Aberrantly
expressed miR-130b (16) and miR-21
(17) exhibited similar expression
pattern with respect to GRα transcripts inhibiting GC-induced
apoptosis in multiple myeloma.
On the contrary, several studies have investigated
the mechanisms that contribute to the upregulation of BIM and
induction of apoptosis in lymphocytes. A recent report demonstrated
that GC treatment induced the expression of pro-apoptotic protein
BIM via downregulation of the miR-17~92 cluster (18). The loss of miR17 family expression and
concomitant increases in the miR17 target BIM were found to occur
in GC-sensitive ALL cells but not in GC-resistant ALL cells
(19). Furthermore, two miRNAs,
miR-142-3p and miR-17-5p, are computationally predicted to be
closely related to GC resistance in pediatric ALL (20).
In the present study, we demonstrated that GC
treatment upregulated GR protein and BIM protein expression, and
induced apoptosis in a GC-sensitive B-cell precursor ALL (BCP-ALL)
cell line. However, GR protein and BIM protein levels were not
upregulated in a GC-resistant BCP-ALL cell line. Expression levels
of the miR-17~92 cluster and miR-142-3p were upregulated in a
GC-resistant BCP-ALL cell line which was induced by increasing
concentrations of GC treatment. Our data suggest that continuous GC
treatment leads to elevated expression of the miR-17~92 cluster and
miR-142-3p, with concomitant downregulation of GR.
Materials and methods
Cell lines, culture conditions and
reagents
BCP-ALL cell lines, 697 (cat. no. CRL-7433; ATCC,
Manassas, VA, USA), MB-YU (21) and
REH (cat. no. ACC22; DSMZ, Braunschweig, Germany) were used in the
present study. Cells were maintained in Roswell Park Memorial
Institute (RPMI)-1640 medium (Wako Pure Chemical Industries, Osaka,
Japan) containing 10% fetal bovine serum (FBS; Gibco, Thermo Fisher
Scientific, Inc., Waltham, MA, USA) at 37°C in humidified air with
5% CO2. Dexamethasone (DEX) was obtained from
Sigma-Aldrich/Merck KGaA (Darmstadt, Germany) and dissolved in PBS.
The following antibodies were used in this study: Glucocorticoid
receptor (GR; cat. no. sc-899; Santa Cruz Biotechnology,
Heidelberg, Germany), polyclonal rabbit anti-BIM (cat. no.
ADI-AAP-330; Stresgen, Farmingdale, NY, USA), anti-cleaved PARP
(cat. no. 5625; Cell Signaling Technology, Danvers, MA, USA),
anti-caspase-3 (M097-3; MBL Int Corp., Woburn MA, USA) and
anti-actin (cat. no. A5316; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany). The secondary antibodies conjugated with horseradish
peroxidase, goat anti-mouse IgG (H+L) (cat. no. 31430) and goat
anti-rabbit IgG (cat. no. 31460) were obtained from Invitrogen
(Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Induction of DEX-resistant BCP-ALL
cells
To induce acquired DEX-resistance, aliquots of
parental cells were seeded into 25 cm2 culture bottles
and cultured in RPMI-1640 medium supplemented with 10% FBS and
increasing concentrations of DEX (from 0.1 nM to 1 µM). Fresh
medium with DEX was changed every 48 h. Cells were transferred into
new culture bottles every 7 days. We continued this process while
observing cell death every day, and performing cell counting
regularly by using an Invitrogen Cell Counter (Thermo Fisher
Scientific, Inc.). Cells were treated with increasing
concentrations of DEX after two rounds of cell counting showed a
consistent increase in cell number. Thus, after 4 to 12 weeks,
DEX-resistant sublines were obtained that grew stably in DEX (1
µM)-containing medium and these resistant cell lines were named
697DR and MB-YUDR.
Cell proliferation and viability
assays
Cell proliferation was measured using a conventional
hemocytometer counting chamber. A trypan blue exclusion test was
conducted to assess cell viability, where the relative percentages
of live and dead cells were quantified using a hemocytometer.
Apoptosis assay
Detection and quantification of hypodiploid DNA
content of apoptotic cells was performed by flow cytometric
analysis after staining cells with propidium iodide (PI) as
previously described (22). The
apoptotic cell nuclei (sub-G1 peak in the DNA fluorescence
histogram) were counted using CellQuest software version 3.0 (BD
Biosciences, San Jose, CA, USA). Data are expressed as the
percentage of the sub-G1 fraction.
RNA isolation, reverse transcription
and quantitative real-time PCR (RT-qPCR)
Total RNA was isolated using RNeasy Mini kit
(Qiagen, Inc., Valencia, CA, USA). RNA samples were resuspended in
RNase-free water and quantified by measuring absorbance at 260 and
280 nm. cDNA was synthesized using the SuperScript®
VILO™ cDNA Synthesis kit (Invitrogen; Thermo Fisher Scientific,
Inc.). The mRNA expression levels of BIM and GRα were
analyzed using GAPDH as a reference gene. RT-qPCR was
performed using QuantiFast SYBR® Green PCR (Qiagen,
Inc.). The primers used are listed in Table I. The reaction mixtures were incubated
at 95°C for 5 min, followed by 45 cycles of 95°C for 15 sec and
57°C for 30 sec in a Stratagene RT-qPCR instrument (Agilent
Technologies, Inc., Santa Clara, CA, USA). The data were analyzed
using the ΔCq (cycle threshold) method.
 | Table I.Primers sequences of GRα, Bim and
GAPDH. |
Table I.
Primers sequences of GRα, Bim and
GAPDH.
| Gene | Primer | Base sequence |
|---|
| GRα | Forward primer |
5′-CGGTCTGAAGAGCCAAGAG-3′ |
|
| Reverse primer |
5′-CAGCTAACATCTCGGGGAAT-3′ |
| Bim | Forward primer |
5′-CGATCCTCCAGTGGGTATTTCTCT-3′ |
|
| Reverse primer |
5′-ATACCCTCCTTGCATAGTAAGCG-3′ |
| GAPDH | Forward primer |
5′-GAAGGTGAAGGTCGGAGTC-3′ |
|
| Reverse primer |
5′-GAAGATGGTGATGGGATTTC-3′ |
Analysis of miRNAs by RT-qPCR
Total miRNAs were isolated from ALL cell lines using
the miRNeasy Μini Κit (Qiagen, Inc.). The miScript SYBR®
Green PCR kit (Qiagen, Inc.) and miScript Primer Assays (Qiagen,
Inc.) were used on a real-time PCR system to quantify the plasma
miRNAs. Qiagen miScript primers (hsa-miR-142-3p, hsa-miR-17-5p,
hsa-miR-18a-5p, hsa-miR-19a-3p, hsa-miR-19b-3p, hsa-miR-20a-5p and
hsa-miR-92-3p) used are listed in Table
II. The data were analyzed using the ΔCq (cycle threshold)
method.
| Table II.miScript primer sequences. |
Table II.
miScript primer sequences.
| miRNA primer | Base sequence |
|---|
| hsa-miR-142-3p |
5′-UGUAGUGUUUCCUACUUUAUGGA-3′ |
| hsa-miR-17-5p |
5′-CAAAGUGCUUACAGUGCAGGUAG-3′ |
| hsa-miR-18a-5p |
5′-UAAGGUGCAUCUAGUGCAGAUAG-3′ |
| hsa-miR-19a-3p |
5′-UGUGCAAAUCUAUGCAAAACUGA-3′ |
| hsa-miR-19b-3p |
5′-UGUGCAAAUCCAUGCAAAACUGA-3′ |
| hsa-miR-20a-5p |
5′-UAAAGUGCUUAUAGUGCAGGUAG-3′ |
| hsa-miR-92a-3p |
5′-UAUUGCACUUGUCCCGGCCUGU-3′ |
Western blot analysis
Western blotting was performed with whole-cell
extracts prepared by lysing cells (1×107) in lysis
buffer (95% Laemmli sample buffer and 5% 2-mercaptoethanol).
Protein concentration was determined using BCA protein assay
reagents (Pierce; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. The proteins (20 µg/lane) were separated
on a 10% sodium dodecyl sulfate-polyacrylamide gel by
electrophoresis followed by semi-dry transfer onto PVDF membranes
(Invitrogen; Thermo Fisher Scientific, Inc.). Transferred PVDF
membranes were pretreated with 5% non-fat dry milk in TBST (10 mM
Tris-HCl, pH 7.6, 137 mM NaCl, 0.1% Tween-20) and incubated with
the primary antibody (dilution 1:1,000-3,000) at 4°C overnight. The
membrane was then washed 3 times with TBST and incubated with
horseradish peroxidase-conjugated secondary antibody (dilution
1:1,000-3,000) for 1 h at room temperature. After washing three
times again, antibodies bound to protein blots were detected by
using Western Lightening Chemiluminescence Reagent Plus
(PerkinElmer, Inc., Waltham, MA, USA) and visualized on LAS-3000
Mini (Fujifilm, Snizuoka, Japan).
Statistical analysis
Results are expressed as mean ± standard deviation
of at least three independent experiments performed in triplicate.
To test differences between two populations the unpaired Student's
t-test was applied. To test the differences among more than two
populations, one-way ANOVA was performed followed by Dunnett's post
hoc test. Differences were considered to be statistically
significant at P<0.05.
Results
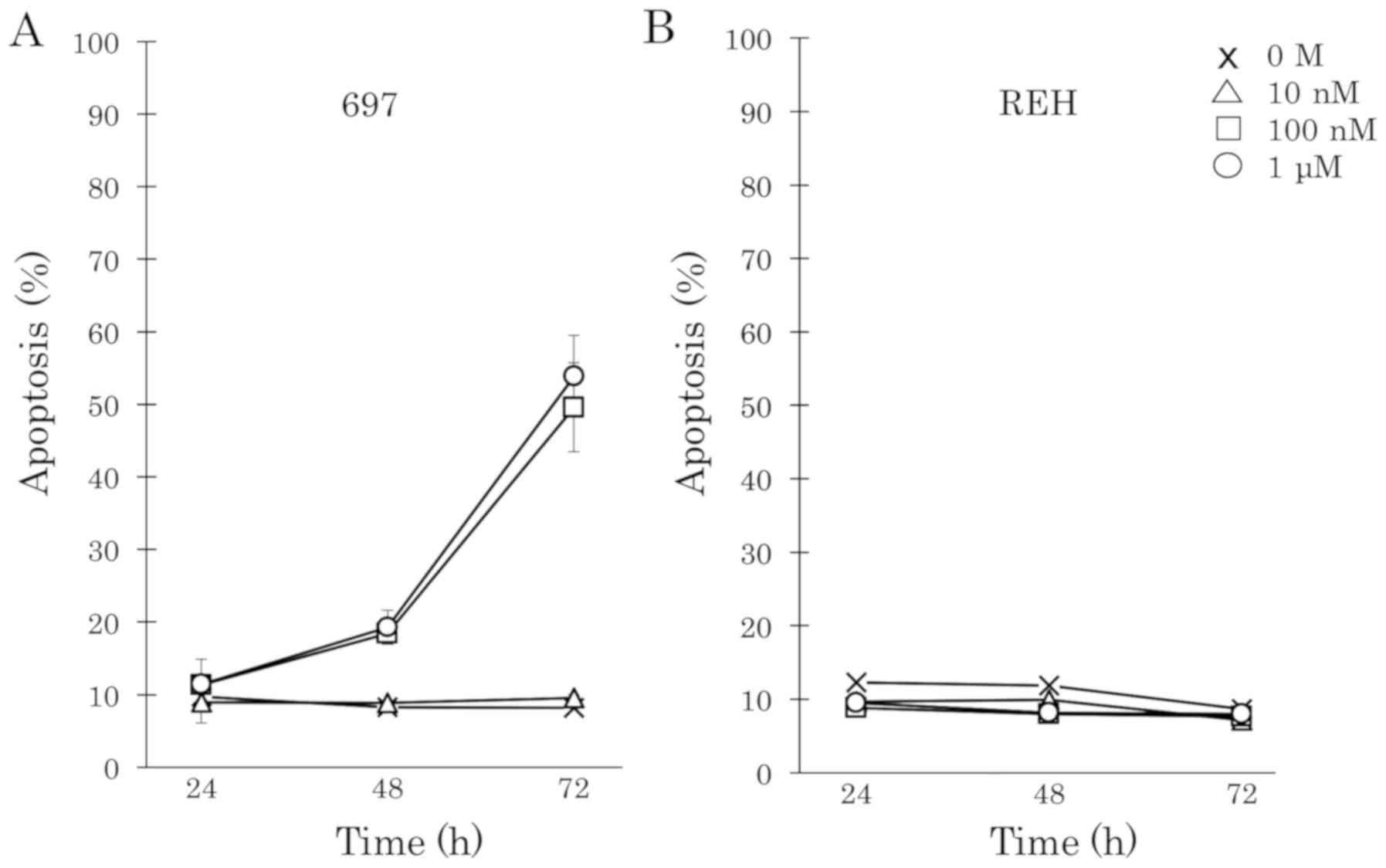
DEX induces apoptosis in cell line 697
but not in cell line REH
To examine whether DEX induces cell death in BCP-ALL
cells, two human BCP-ALL cell lines, 697 and REH were treated with
DEX at increasing concentrations from 0.1 nM to 1 µM. DEX strongly
induced cell apoptosis in a time- and dose-dependent manner in the
697 cell line (Fig. 1A). However, the
REH cell line was completely resistant to DEX (Fig. 1B). The cell proliferation of 697 and
REH cells upon DEX treatment was examined. Fifty percent of
DEX-sensitive 697 cells showed cell death and the proliferation of
cells which survived was completely inhibited by 100 nM DEX
treatment. However, higher concentrations of DEX (>1 µM) did not
suppress the proliferation of DEX-resistant REH cells.
DEX upregulates glucocorticoid
receptor and BIM expression and induces apoptosis in the 697 cell
line
RT-qPCR and western blot analyses were performed to
detect the effect of DEX on the expression levels of GR and BIM in
DEX-sensitive and -resistant cell lines. GRα and BIM exhibited
differential expression of mRNA and protein between the
DEX-sensitive and -resistant cell line. DEX treatment upregulated
both mRNA and protein expression levels of GRα in the DEX-sensitive
697 cell line, but not in the DEX-resistant REH cell line (Figs. 2A, C and 3). After 6 h of DEX treatment, mRNA and
protein expression levels of BIM were upregulated in the
DEX-sensitive 697 cell line, but not in the DEX-resistant REH cell
line (Figs. 2B, D and 3). Western blot analysis was performed using
caspase-3 and cleaved PARP antibodies to further validate that
cells had undergone apoptosis. As shown in Fig. 3, activation of caspase-3 and cleavage
of PARP were observed in the DEX-sensitive 697 cell line in a
time-dependent manner, but not in the DEX-resistant cell line
REH.
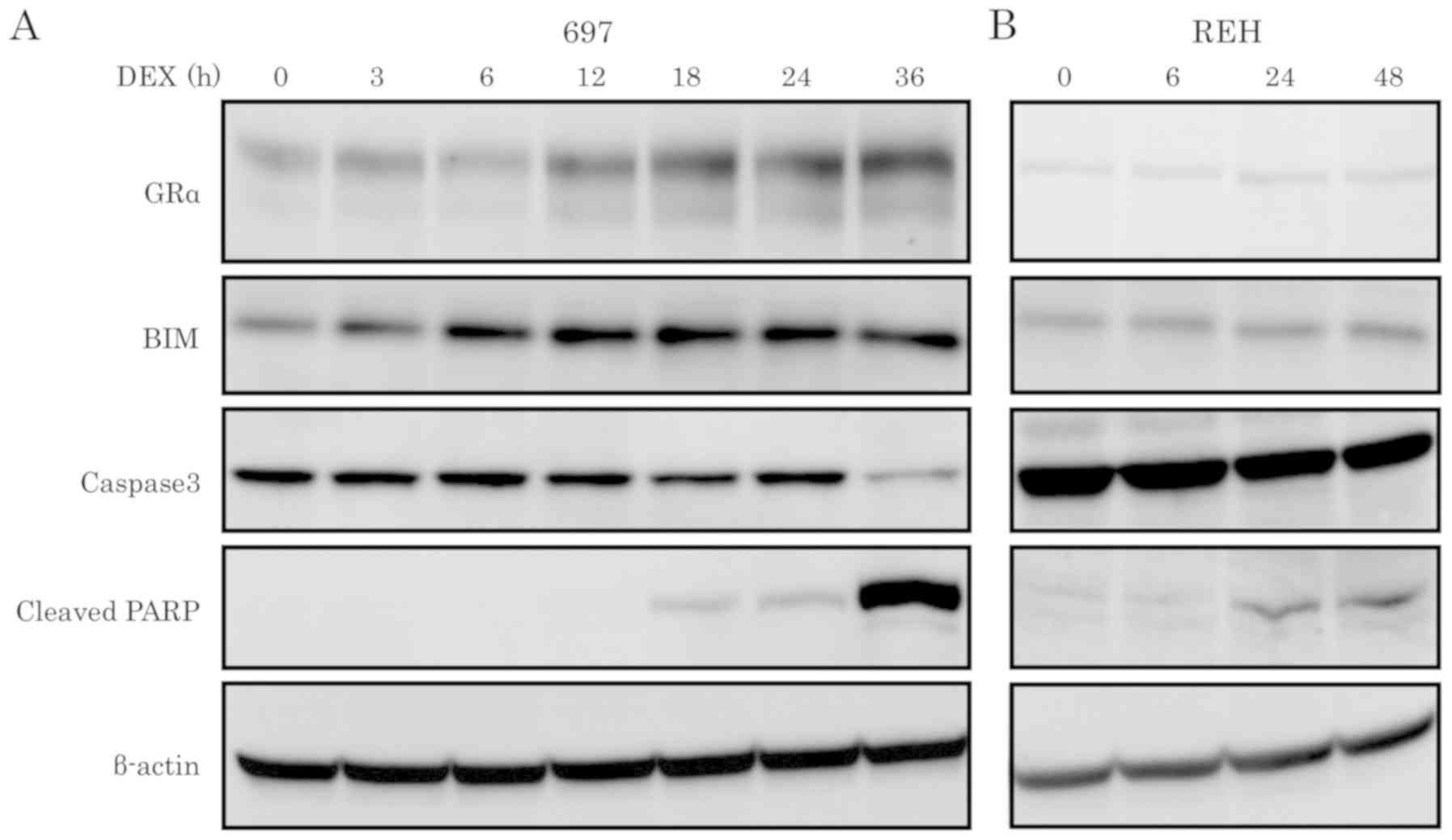 | Figure 3.Protein expression levels of GRα,
BIM, caspase-3 and cleaved PARP in ALL cell lines upon DEX
treatment. ALL cell lines, (A) 697 and (B) REH, were treated with
100 nM of DEX at the indicated time points and the protein
expression levels of GRα, BIM, caspase-3 and cleaved PARP were
determined by western blotting. DEX, dexamethasone; ALL, acute
lymphoblastic leukemia; BIM, Bcl-2-interacting mediator of cell
death; GR, glucocorticoid receptor. |
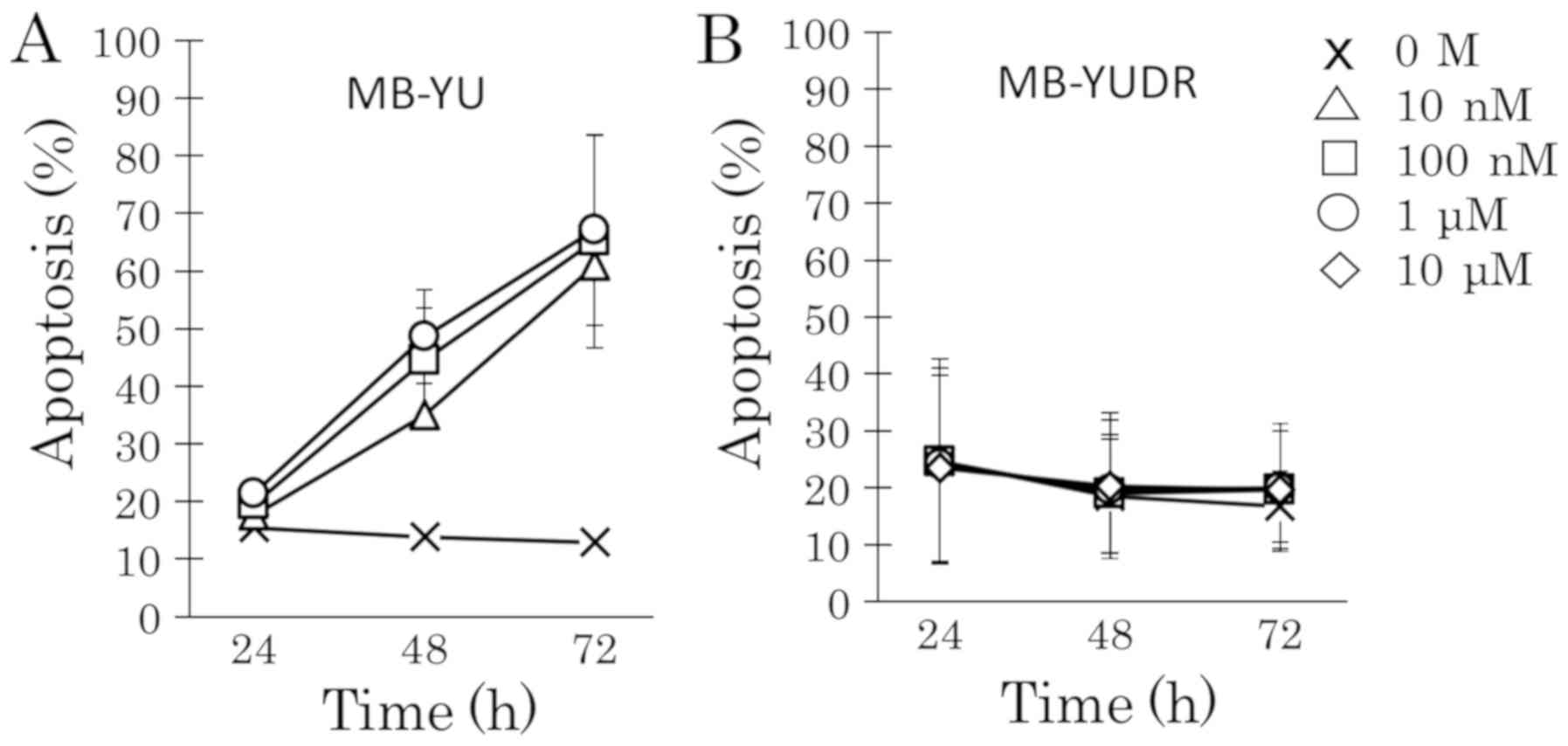
Long-term exposure induces DEX
resistance in BCP-ALL cells
To explore acquired DEX resistance in the 697 cell
line, cells were treated with increasing concentrations of DEX
(from 0.1 nM to 1 µM). DEX-sensitive 697 cells that grew stably in
a low DEX concentration underwent apoptosis at 100 nM DEX (Fig. 4B and C). However, 697 cells that grew
stably in a high DEX concentration did not undergo apoptosis even
at 1 µM DEX (Fig. 4D and E). After
incubation with DEX for 4 to 12 weeks, acquired DEX-resistant
sublines (697DR and MB-YUDR) could proliferate stably in RPMI-1640
medium with 10% FBS in the presence of DEX (10 µM). The
DEX-resistant sublines (697DR and MB-YUDR) (Figs. 4F and 5B) exhibited marked resistance to DEX
compared to the parental cells (697 and MB-YU) (Figs. 4A, F and 5). RT-qPCR and western blot analyses showed
that GRα mRNA and GRα protein were downregulated in both 697DR
(Fig. 6) and MB-YUDR cell lines
(Fig. 7).
 | Figure 4.Development of DEX-resistant BCP-ALL
cell lines. Aliquots of parental cells were seeded into 25 cm2
culture bottles, and cultured in RPMI-1640 medium supplemented with
10% FBS and treated with increasing concentrations of DEX (from 0.1
nM to 1 µM). Fresh medium with DEX was changed every 48 h. Cells
were transferred into new culture bottles every 7 days. We
continued this process while observing cell death every day, and
performing cell counting regularly by using Invitrogen Cell
Counter. Cells were treated with increasing concentrations of DEX
after two rounds of cell counting and showed a consistent increase
in cell number. (A) Parental 697 cells were treated with 0 M, 10,
100 nM, 1 and 10 µM of DEX for 24, 48 and 72 h. Apoptotic
progression was monitored via flow cytometric analysis of propidium
iodide-stained nuclei. (B) 697 cells that grew stably in 0.1 nM DEX
were treated with 0 M, 10, 100 nM, 1 and 10 µM of DEX for 24, 48
and 72 h. Apoptotic progression was monitored. (C) 697 cells that
grew stably in 1 nM DEX were treated with 0 M, 10, 100 nM, 1 and 10
µM of DEX for 24, 48 and 72 h. Apoptotic progression was monitored.
(D) 697 cells that grew stably in 10 nM DEX were treated with 0 M,
10, 100 nM, 1 and 10 µM of DEX for 24, 48 and 72 h. Apoptotic
progression was monitored. (E) 697 cells that grew stably in 1 µM
DEX were treated with 0 M, 10, 100 nM, 1 and 10 µM of DEX for 24,
48 and 72 h. Apoptotic progression was monitored. (F) 697DR cells
were treated with 0 M, 10, 100 nM, 1 and 10 µM of DEX for 24, 48
and 72 h. Apoptotic progression was monitored. BCP-ALL, B-cell
precursor acute lymphoblastic leukemia; DEX, dexamethasone. |
Expression of GR and BIM in
DEX-resistant BCP-ALL cell lines by DEX treatment
RT-qPCR was performed to detect mRNA expression
levels of GRα and BIM in DEX-resistant BCP-ALL cell
lines in response to 100 nM DEX treatment. Although DEX treatment
upregulated mRNA expression levels of GRα and BIM in
the DEX-sensitive 697 cells in a time-dependent manner, mRNA
expression levels of GRα and BIM were not altered by
DEX treatment in 697DR cells (Fig. 8A and
B).
Expression of miR-142-3p and miR-17~92
in DEX-resistant BCP-ALL cell lines
miR-142-3p regulates the expression of GRα (14) and the expression level of miR-17~92 is
an important regulator of GC-induced apoptosis (18). Expression levels of miR-142-3p and
miR-17~92 were examined by RT-qPCR. miR-142-3p and miR-17~92 were
upregulated in acquired DEX-resistant 697DR cells (Fig. 9A) and MB-YUDR cells (Fig. 9B). Although miR-17-5p, miR-18a-5p and
miR-19a-3p were upregulated in DEX-resistant cell line REH,
miR-142-3p, miR-19b-3p, miR-20a-5p and miR-92a-3p were not
upregulated (Fig. 9C).
Discussion
Although the mechanisms regulating glucocorticoid
(GC) resistance are being unraveled, resistance to GCs still
remains a major hurdle in the treatment of B-cell precursor acute
lymphoblastic leukemia (BCP-ALL). In the present study, it was
demonstrated that GC-sensitive BCP-ALL cells exhibit upregulation
of the GRα gene and protein expression in response to GC treatment.
However, GRα mRNA and protein expression was downregulated in the
GC-resistant cell line (REH) and dexamethasone (DEX)-resistant cell
lines (697DR and MB-YUDR). It is controversial whether GC
resistance occurs at the level of the GR (23). Previous research has shown that there
was no significant difference in GR mRNA and protein expression
when comparing GC-resistant and GC-sensitive leukemia cells
(24,25). However, our observations are
consistent with the notion that low cellular levels of functional
GR in leukemia cells decreases sensitivity to GCs (26). Although GCs induce auto upregulation
of GR expression in GC-sensitive ALL cells (27), GCs give rise to therapeutic resistance
to themselves with high GR-β levels that cause an imbalance in the
GR-α/β ratio (28). In addition to
downregulated receptors, miRNA-mediated gene dysfunction may relate
to GC resistance. miR-142-3p decreased GRα protein expression by
directly targeting the 3′-untranslated region of GRα mRNA,
leading to GC resistance (14).
Furthermore, Lv et al found that the miR-142-3p inhibitor
effectively reversed GC resistance to GC sensitivity, due to the
resultant increase in GRα expression (14). Although miR-142-3p was found to affect
the expression of GRα protein but not GRα mRNA in Jurkat
cells (14), both GRα mRNA as well as
protein were decreased in our study. This disparity could be due to
differences between T-ALL and BCP-ALL cells. miR-142-3p may
regulate the expression of GRα at the transcriptional level in
BCP-ALL. Although both GRα mRNA and protein were downregulated in
DEX-resistant REH cells (Fig. 6),
miR142-3p was not upregulated (Fig.
9B). It has been reported that DNA methylation status could
affect GC-sensitivity (29) and
mutation in the GR gene decreased sensitivity to GCs
(30). Therefore, low cellular levels
of GRα due to DNA methylation or GR gene mutations may
result in GC resistance in REH cells.
Upregulation of BIM, a pro-apoptotic member of the
B-cell lymphoma-2 family, is an important mediator of GC-induced
apoptosis. It was recently identified that GR binding at the
intronic region of the BIM gene enhances BIM transcription
(11). We observed that BIM mRNA and
protein were upregulated by DEX stimulation in the GC-sensitive ALL
cell line. However, both GR and BIM were downregulated in
GC-resistant ALL cell line. These findings support that the absence
of GR binding at the BIM intronic region was associated with
BIM gene silencing and DEX resistance (11). The miR-17~92 cluster, containing six
individual miRNAs, has been strongly implicated in hematopoietic
malignancies (31). Overexpression of
miR-17~92 causes BIM reduction, resulting in the inhibition of
induction of apoptosis (32). We
observed upregulation of miR-17~92 in acquired DEX-resistant
BCP-ALL cell lines (697DR and MB-YUDR). miR17-5p, miR18a-5p and
miR19a-3p were upregulated in DEX-resistant REH cells. Our data are
consistent, therefore, with a model in which DEX-resistance is
mediated through the sequential upregulation of miR-17 and
downregulation of BIM. GCs are known to downregulate miR-17~92
expression resulting in elevation of BIM mRNA and protein levels
(18). Since the promotor of
miR-17~92 is a prime target of GCs (19), its binding to the miR-17~92 promotor
region might be altered in our DEX-resistant cells. The BET
(bromodomain and extraterminal domain) proteins, such as BRD4,
directly regulate miR-17~92 expression by binding to the miR-17~92
promoter (33). Therefore, the
expression level of BET protein might be upregulated in
DEX-resistant cells. This might be another mechanism of developing
GC resistance, which needs further investigation.
Although intracellular levels of miR-142-3p and
miR-17~92 cluster were increased in GC-resistant BCP-ALL cells, it
is not known whether miR-142-3p and miR-17~92 cluster directly
regulate GR expression and GC sensitivity. Thus, further study in
overexpression and/or downregulation of these microRNAs should be
carried out to confirm the role of miR-142-3p and miR-17~92 cluster
in GC resistance. Our data may provide an additional predictive
marker in BCP-ALL. A large cohort of patient samples should be
analyzed together with the in vivo response to therapy.
There are several other microRNAs that have been
shown to modulate GC sensitivity in lymphoid malignancies.
Downregulation of miR-128b and miR-221 has been implicated in GC
resistance (34). miR-182 functions
in promoting resistance to GCs in lymphoblastic malignancies via
negative regulation of FOXO3A (35).
With increasing miRNA expression patterns emerging into the vision
of researchers, they are worth more investigation as promising
predictive biomarkers for chemotherapy effectiveness and disease
prognosis.
In conclusion, GC resistance is associated with GR
reduction and increased intracellular levels of miR-142-3p and
miR-17~92 cluster. This is the first report to show that elevation
of miR-142-3p and miR-17~92 plays an important role in acquired GC
resistance of BCP-ALL. The potential of miR-142-3p and miR-17~92 as
critical therapeutic targets for BCP-ALL requires further
investigation.
Acknowledgements
We thank Dr Takao Deguchi (National Center for Child
Health and Development) for valuable discussions and
suggestions.
Funding
The present study was funded by the Japanese
Ministry of Health (15K096500K).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
NS, YK and MH conceived and designed the study. NS
performed the experiments and organized all data. NS, YK, RH, MM,
TI, DN and MH analyzed and interpreted the data. NS, YK and MH
wrote the paper. NS, YK, RH, MM, TI, DN and MH reviewed and edited
the manuscript. All authors read and approved the manuscript and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
GC
|
glucocorticoid
|
|
GR
|
glucocorticoid receptor
|
|
BCP-ALL
|
B-cell precursor acute lymphoblastic
leukemia
|
|
DEX
|
dexamethasone
|
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pui CH, Campana D, Pei D, Bowman WP,
Sandlund JT, Kaste SC, Ribeiro RC, Rubnitz JE, Raimondi SC, Onciu
M, et al: Treating childhood acute lymphoblastic leukemia without
cranial irradiation. N Engl J Med. 360:2730–2741. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Den Boer ML, Harms DO, Pieters R, Kazemier
KM, Gobel U, Körholz D, Graubner U, Haas RJ, Jorch N, Spaar HJ, et
al: Patient stratification based on
prednisolone-vincristine-asparaginase resistance profiles in
children with acute lymphoblastic leukemia. J Clin Oncol.
21:3262–3268. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dördelmann M, Reiter A, Borkhardt A,
Ludwig WD, Götz N, Viehmann S, Gadner H, Riehm H and Schrappe M:
Prednisone response is the strongest predictor of treatment outcome
in infant acute lymphoblastic leukemia. Blood. 94:1209–1217.
1999.PubMed/NCBI
|
|
5
|
Kaspers GJ, Veerman AJ, Pieters R, Van
Zantwijk CH, Smets LA, Van Wering ER and Van Der Does-Van Den Berg
A: In vitro cellular drug resistance and prognosis in newly
diagnosed childhood acute lymphoblastic leukemia. Blood.
90:2723–2729. 1997.PubMed/NCBI
|
|
6
|
Pieters R, Huismans DR, Loonen AH, Hählen
K, van der Does-van den Berg A, van Wering ER and Veerman AJ:
Relation of cellular drug resistance to long-term clinical outcome
in childhood acute lymphoblastic leukaemia. Lancet. 338:399–403.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zou YF, Xu JH, Wang F, Liu S, Tao JH, Cai
J, Lian L, Xiao H, Chen PL, Tian G, et al: Association study of
glucocorticoid receptor genetic polymorphisms with efficacy of
glucocorticoids in systemic lupus erythematosus: A prospective
cohort study. Autoimmunity. 46:531–536. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sousa AR, Lane SJ, Cidlowski JA, Staynov
DZ and Lee TH: Glucocorticoid resistance in asthma is associated
with elevated in vivo expression of the glucocorticoid receptor
beta-isoform. J Allergy Clin Immunol. 105:943–950. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang H, Gou X, Jiang T and Ouyang J: The
effects of microRNAs on glucocorticoid responsiveness. J Cancer Res
Clin Oncol. 143:1005–1011. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bouillet P, Huang DC, O'Reilly LA,
Puthalakath H, O'Connor L, Cory S, Adams JM and Strasser A: The
role of the pro-apoptotic Bcl-2 family member bim in physiological
cell death. Ann N Y Acad Sci. 926:83–89. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jing D, Bhadri VA, Beck D, Thoms JA, Yakob
NA, Wong JW, Knezevic K, Pimanda JE and Lock RB: Opposing
regulation of BIM and BCL2 controls glucocorticoid-induced
apoptosis of pediatric acute lymphoblastic leukemia cells. Blood.
125:273–283. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Carthew RW and Sontheimer EJ: Origins and
mechanisms of miRNAs and siRNAs. Cell. 136:642–655. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Filipowicz W, Bhattacharyya SN and
Sonenberg N: Mechanisms of post-transcriptional regulation by
microRNAs: Are the answers in sight? Nat Rev Genet. 9:102–114.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lv M, Zhang X, Jia H, Li D, Zhang B, Zhang
H, Hong M, Jiang T, Jiang Q, Lu J, et al: An oncogenic role of
miR-142-3p in human T-cell acute lymphoblastic leukemia (T-ALL) by
targeting glucocorticoid receptor-α and cAMP/PKA pathways.
Leukemia. 26:769–777. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liang YN, Tang YL, Ke ZY, Chen YQ, Luo XQ,
Zhang H and Huang LB: miR-124 contributes to glucocorticoid
resistance in acute lymphoblastic leukemia by promoting
proliferation, inhibiting apoptosis and targeting the
glucocorticoid receptor. J Steroid Biochem Mol Biol. 172:62–68.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tessel MA, Benham AL, Krett NL, Rosen ST
and Gunaratne PH: Role for microRNAs in regulating glucocorticoid
response and resistance in multiple myeloma. Horm Cancer.
2:182–189. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang X, Li C, Ju S, Wang Y, Wang H and
Zhong R: Myeloma cell adhesion to bone marrow stromal cells confers
drug resistance by microRNA-21 up-regulation. Leuk Lymphoma.
52:1991–1998. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Molitoris JK, McColl KS and Distelhorst
CW: Glucocorticoid-mediated repression of the oncogenic microRNA
cluster miR-17~92 contributes to the induction of Bim and
initiation of apoptosis. Mol Endocrinol. 25:409–420. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Harada M, Pokrovskaja-Tamm K, Söderhäll S,
Heyman M, Grander D and Corcoran M: Involvement of miR17 pathway in
glucocorticoid-induced cell death in pediatric acute lymphoblastic
leukemia. Leuk Lymphoma. 53:2041–2050. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen H, Zhang D, Zhang G, Li X, Liang Y,
Kasukurthi MV, Li S, Borchert GM and Huang J: A semantics-oriented
computational approach to investigate microRNA regulation on
glucocorticoid resistance in pediatric acute lymphoblastic
leukemia. BMC Med Inform Decis Mak. 18 (Suppl 2):S572018.
View Article : Google Scholar
|
|
21
|
Kang J, Kisenge RR, Toyoda H, Tanaka S, Bu
J, Azuma E and Komada Y: Chemical sensitization and regulation of
TRAIL-induced apoptosis in a panel of B-lymphocytic leukaemia cell
lines. Br J Haematol. 123:921–932. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Toyoda H, Ido M, Hayashi T, Gabazza EC,
Suzuki K, Kisenge RR, Kang J, Hori H and Komada Y: Experimental
treatment of human neuroblastoma using live-attenuated poliovirus.
Int J Oncol. 24:49–58. 2004.PubMed/NCBI
|
|
23
|
Bhadri VA, Trahair TN and Lock RB:
Glucocorticoid resistance in paediatric acute lymphoblastic
leukaemia. J Paediatr Child Health. 48:634–640. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lauten M, Cario G, Asgedom G, Welte K and
Schrappe M: Protein expression of the glucocorticoid receptor in
childhood acute lymphoblastic leukemia. Haematologica.
88:1253–1258. 2003.PubMed/NCBI
|
|
25
|
Tissing WJ, Meijerink JP, Brinkhof B,
Broekhuis MJ, Menezes RX, den Boer ML and Pieters R:
Glucocorticoid-induced glucocorticoid-receptor expression and
promoter usage is not linked to glucocorticoid resistance in
childhood ALL. Blood. 108:1045–1049. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Paugh SW, Bonten EJ, Savic D, Ramsey LB,
Thierfelder WE, Gurung P, Malireddi RK, Actis M, Mayasundari A, Min
J, et al: NALP3 inflammasome upregulation and CASP1 cleavage of the
glucocorticoid receptor cause glucocorticoid resistance in leukemia
cells. Nat Genet. 47:607–614. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Geng CD and Vedeckis WV: A new, lineage
specific, autoup-regulation mechanism for human glucocorticoid
receptor gene expression in 697 pre-B-acute lymphoblastic leukemia
cells. Mol Endocrinol. 25:44–57. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ledderose C, Möhnle P, Limbeck E, Schütz
S, Weis F, Rink J, Briegel J and Kreth S: Corticosteroid resistance
in sepsis is influenced by microRNA-124-induced downregulation of
glucocorticoid receptor-alpha. Crit Care Med. 40:2745–2753. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gasson JC, Ryden T and Bourgeois S: Role
of de novo DNA methylation in the glucocorticoid resistance of a
T-lymphoid cell line. Nature. 302:621–623. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Charmandari E, Kino T, Ichijo T, Jubiz W,
Mejia L, Zachman K and Chrousos GP: A novel point mutation in helix
11 of the ligand-binding domain of the human glucocorticoid
receptor gene causing generalized glucocorticoid resistance. J Clin
Endocrinol Metab. 92:3986–3990. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mavrakis KJ, Wolfe AL, Oricchio E,
Palomero T, de Keersmaecker K, McJunkin K, Zuber J, James T, Khan
AA, Leslie CS, et al: Genome-wide RNA-mediated interference screen
identifies miR-19 targets in Notch-induced T-cell acute
lymphoblastic leukaemia. Nat Cell Biol. 12:372–379. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Scherr M, Elder A, Battmer K, Barzan D,
Bomken S, Ricke-Hoch M, Schröder A, Venturini L, Blair HJ, Vormoor
J, et al: Differential expression of miR-17~92 identifies BCL2 as a
therapeutic target in BCR-ABL-positive B-lineage acute
lymphoblastic leukemia. Leukemia. 28:554–565. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xu Z, Sharp PP, Yao Y, Segal D, Ang CH,
Khaw SL, Aubrey BJ, Gong J, Kelly GL, Herold MJ, et al: BET
inhibition represses miR17-92 to drive BIM-initiated apoptosis of
normal and transformed hematopoietic cells. Leukemia. 30:1531–1541.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kotani A, Ha D, Hsieh J, Rao PK, Schotte
D, den Boer ML, Armstrong SA and Lodish HF: miR-128b is a potent
glucocorticoid sensitizer in MLL-AF4 acute lymphocytic leukemia
cells and exerts cooperative effects with miR-221. Blood.
114:4169–4178. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang A, Ma J, Wu M, Qin W, Zhao B, Shi Y,
Jin Y and Xie Y: Aberrant microRNA-182 expression is associated
with glucocorticoid resistance in lymphoblastic malignancies. Leuk
Lymphoma. 53:2465–2473. 2012. View Article : Google Scholar : PubMed/NCBI
|