Introduction
Cancer, a multi-step disease, is characterized by
increased cell division, dysregulation of cell growth and
resistance to cell death. Increases in oxygen consumption rates in
cancer cells leads to hypoxia and consequently stimulates
angiogenesis. In particular, breast cancer is both a genetic
disease as well as a multiple factor-associated disease (1–4). It is the
foremost cause of cancer-related death corresponding to 23% of all
cancer incidents in women, and worldwide, one million women are
diagnosed with breast cancer and about half of them succumb to the
disease every year (5,6). More than 8% of women present with
invasive breast cancer that can be associated with a high death
rate, and unfortunately, this percentage is expected to rise to 26%
in 2020 (7,8). For this reason, it is necessary to
develop new strategies for the treatment of breast cancer. However,
it is difficult to discover new strategies for breast cancer due to
its high potential of metastasis (9).
Apoptosis, programmed cell death, plays a crucial
role in the development and maintenance of cellular homeostasis.
However, dysregulation of this process has been implicated in
various diseases including cancer (10,11).
Cancer cells employ many strategies to resist apoptotic cell death
and to combat the host immune system, and many anticancer therapies
have been targeted to the activation of cell survival signals
(12). Protein kinase A (PKA),
composed of two regulatory and two catalytic subunits, is activated
upon stimulation by various extracellular or intracellular signals.
Cyclic AMP (cAMP), a key intracellular mediator, binds to the PKA
regulatory subunits, and the consequent dissociation of catalytic
subunits results in PKA activation. PKA activity is specifically
blocked by the H-89 inhibitor, derived from H-8
N-[2-(methylamino)ethyl]- 5-isoquinoline-sulfonamide (13,14).
Besides its general role in antiapoptotic cell death, PKA is also
involved in the enhancement of apoptosis (15–17). In
particular, in certain cell death mechanisms, PKA directly induces
apoptosis even in the absence of cAMP (18). The role of cAMP-dependent signals has
also been investigated in regards to both cell death and survival
(19,20). Moreover, various protein regulators in
the cAMP signaling pathway have been proposed as possible
therapeutic targets to stimulate apoptosis for the treatment of
certain cancers (15–17). PKA-dependent apoptosis is mediated via
either phosphorylation of targeted proteins involved in cell death
or by activation of an intrinsic mitochondrial cell death pathway,
in many cancers (17,21–26).
Furthermore, cAMP can also sensitize cells to the proapoptotic
action of agents, such as DNA damaging agents, via a non-cAMP
pathway (27).
β-Lapachone (β-Lap), a quinone compound derived from
the Lapacho tree (Tabebula impetiginosa), has been shown to
be highly effective in treating various types of cancer in
experimental models, including liver cancer and melanoma (28–33). β-Lap
reacts with a cellular enzyme, NAD(P)H:quinone oxidoreductase 1
(NQO1), which is overexpressed in many cancers (34–38). In
particular, 84.7% of breast cancer tissues showed positive
expression of NQO1 while only 30.8% of adjacent non-tumor tissues
showed NQO1 expression (39). The
reaction of β-Lap with NQO1 activates a futile cycle by consuming
NADPH and generating ROS (33–36). ROS
from the redox cycle of β-Lap also contribute to cellular toxicity
in cancer cells (34,35). This demonstrates the importance of
NQO1 in the anticancer action of β-Lap, especially in cancer cells
highly expressing NQO1 (38–41). The differential role of NQO1-mediating
redox activation by β-Lap, from the mitochondria to produce ROS,
suggests this approach could be useful as a potential anticancer
treatment (41,42). However, the mechanism underlying how
β-Lap induces apoptosis is still unknown.
In the present study, we showed that production of
ROS in NQO1-overexpressing breast cancer cells with highly
activated PKA led to apoptotic cell death. In contrast, inhibition
of PKA activity caused decreased apoptosis. We suggest that β-Lap
may be a potential treatment strategy for NQO1-positive cancer
cells as β-Lap-dependent PKA activation is specific to cancer
cells, and is different from other cAMP-PKA activation pathway
treatments that have harmful effects on normal cells.
Materials and methods
Reagents
Roswell Park Memorial Institute (RPMI)-1640 medium
and fetal bovine serum (FBS) were purchased from Gibco Life
Technologies/Thermo Fisher Scientific, Inc. (Waltham, MA, USA).
β-Lapachone, NAC (N-acetylcysteine), DCFDA
(2′,7′-dichloroflourescin diacetate), forskolin, dibutyryl-cAMP
(cAMP-analog) and H89 were obtained from Sigma-Aldrich/Merck KGaA.
The CCK-8 (Cell Counting Kit-8) was purchased from Dojindo (Tokyo,
Japan). Primary antibodies against caspase-3 (cat. no. 9662;
dilution 1:1,000), cleaved caspase-3 (cat. no. 9661; dilution
1:1,000), PARP (cat. no. 9542; dilution 1:1,000), and cleaved PARP
(cat. no. 95411; dilution 1:1,000) were from Cell Signaling
Technology (Beverly, MA, USA), and Bcl-2 (cat. no. 7382; dilution
1:1,000), Bcl-xL (cat. no. 56021; dilution 1:1,000), cytochrome
c (cat. no. 13560; dilution 1:1,000), Bak (cat. no. 832;
dilution 1:1,000), p-PKAα/β/γ T198 (cat. no. 32968; dilution
1:1,000) and PKAα (cat. no. 903; dilution 1:1,000) were purchased
from Santa Cruz Biotechnology (Santa Cruz, CA, USA). β-actin was
obtained from Sigma-Aldrich/Merck KGaA. Secondary antibodies
against rabbit, mouse and goat were purchased from Bio-Rad
Laboratories, Inc. (Hercules, CA, USA).
Cell culture
Two breast cancer cell lines, MDA-MB-231
overexpressing NQO1 (231-NQO1+/+) and MDA-MB-231 lacking
NQO1 (231-NQO1−/−), were described previously (43) and were provided by Dr David Boothman
(UT Southwestern Medical Center, Dallas, TX, USA). These cells were
cultured in RPMI medium supplemented with 5% (v/v) FBS, 100 U/ml
penicillin, and 100 µg/ml streptomycin at 37°C in a 5%
CO2 humidified atmosphere.
Cell viability
Cell viability was determined by the CCK-8 kit. In
brief, both 231-NQO1+/+ and 231-NQO1−/− cells
(5×103 cells/well) were treated with β-Lap in the
presence or absence of inhibitors or activators for 2 h in 96-well
plates, and after removal of medium, were further incubated in
fresh RPMI medium with 5% FBS for 4 h. CCK-8 reagent (10 µl) was
added into each well and the cells were further incubated for 4 h
at 37°C in a 5% CO2 humidified atmosphere. Absorbance
was measured at 485 nm using a microplate reader (Hidex 1
FN/Chameleon; Turku, Finland).
Determination of intracellular
ROS
Intracellular ROS were determined by a DCFDA
cellular ROS detection assay kit (cat. no. ab113851; Abcam,
Burlingame, CA, USA). Briefly, both 231-NQO1+/+ and
231-NQO1−/− cells (5×103 cells/well) were
seeded into 96-well plates containing RPMI medium (200 µl)
supplemented with 5% FBS. After incubation for 24 h at 37°C, cells
were treated with different concentrations (0, 2, 3 and 4 µM) of
β-Lap for 2 h, under the same conditions. After subsequent
incubation for 4 h, 30 µM DCFDA dissolved in DMSO/PBS was added to
each well, followed by incubation for 30 min under light-free
conditions. The plates were read using a GloMax®
detection system (Model #E 8032; Promega, Sunnyvale, CA, USA) at
485/535 nm.
Western blot analysis
Cells were collected and washed twice with ice-cold
1X PBS. Total proteins were extracted with cell lysis buffer (cat.
no. 87788; Pierce; Thermo Fisher Scientific, Inc.) containing
protease and phosphatase inhibitor cocktails (Halt™ Proteases &
Phosphatase Single-Use Inhibitor Cocktail (100X; Thermo Scientific,
Inc.), and the protein concentration was determined using the
Pierce protein assay kit (Thermo Scientific, Inc.). Total protein
lysates (30 µg) were separated on a 10% SDS-PAGE gel, and the
target proteins were specifically detected by western blotting
using the indicated antibodies by incubating with primary
antibodies at 4°C overnight and subsequently with secondary
antibodies at room temperature for 1 h. Proteins were visualized
with ECL Detection Reagent (Thermo Scientific, Inc.), and
quantified using ImageJ software (National Institutes of Health,
Bethesda, MD, USA). Protein level was corrected by β-actin
normalization as the control value.
Statistical analysis
Each experiment was conducted independently at least
three times, and values are expressed as the mean value ± standard
deviation (SD). The difference between two groups was assessed by a
two-tailed Student's t-test. One-way analysis of variance (ANOVA)
was used to compare the means of three groups or more, and each
comparison of these groups was followed by multiple comparison
Tukey's tests. Probability values of *P<0.05 and **P<0.01
were considered significant (as indicated by the relevant symbols
in the figure).
Results
Cell death by β-Lap in MDA-MB-231
breast cancer cells is dependent on NQO1 and increases in
intracellular ROS
β-Lap, an anticancer drug that reacts against many
cancers, selectively targets only those cells highly expressing
NQO1 proteins, and β-Lap-induced cytotoxicity in these cancer cells
is dependent on the accumulation of ROS produced from the futile
cycle reactions of β-Lap and NQO1 (31–40). To
further investigate the mechanistic details of β-Lap-induced cell
death in breast cancer cells, two syngeneic breast cancer cell
lines, MDA-MB-231 overexpressing NQO1 (231-NQO1+/+) and
MDA-MB-231 lacking NQO1 (231-NQO1−/−), were treated with
different concentrations of β-Lap (0, 2, 3 and 4 µM) for 2 h. The
cell viability of 231-NQO1+/+ cells was significantly
decreased in a β-Lap dose-dependent manner, while
231-NQO1−/− cells maintained their viability regardless
of β-Lap concentration (Fig. 1A and
B) and microscopic phenotype (Fig.
1C). According to the western blot analysis,
231-NQO1+/+ cells showed a high level expression of
NQO1, while there was no expression of NQO1 in the
231-NQO1−/− cells (Fig. 1D and
E).
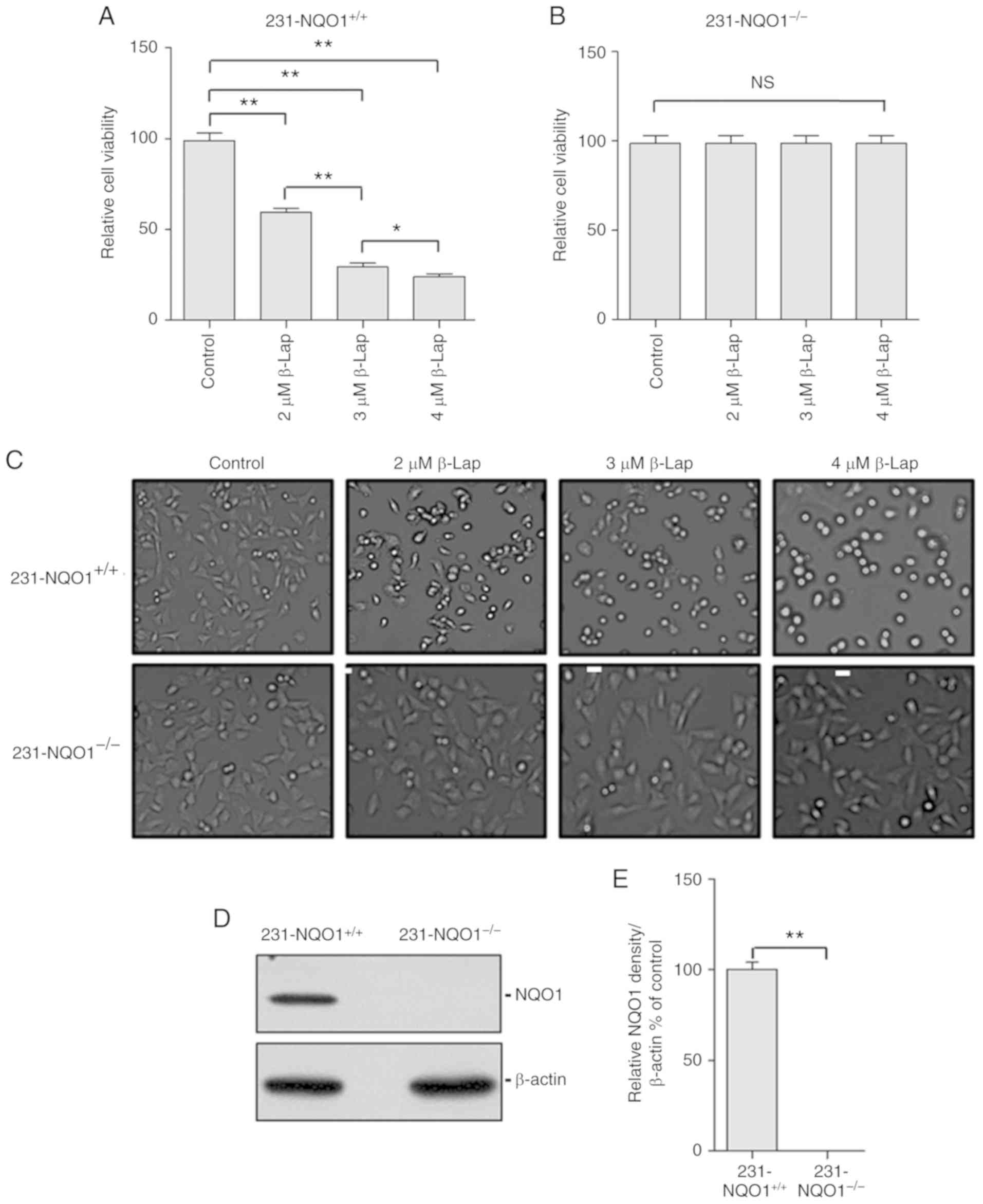 | Figure 1.β-Lapachone (β-Lap) decreases the
cell viability of NQO1-overexpressing human breast cancer cells in
a dose-dependent manner. (A and B) Determination of cell viability.
The 231-NQO1+/+ (A) and 231-NQO1−/− (B) cells
were treated with β-Lap (0, 2, 3 and 4 µM) for 2 h. After further
incubation in fresh RPMI medium with 5% FBS for 4 h, cell viability
was determined by CCK-8 assay. (C) Cell morphological change. The
231-NQO1+/+ (upper panels) and 231-NQO1−/−
(lower panels) cells were treated with β-Lap at different
concentrations as indicated. After incubation for 4 h, images were
taken for cell morphology (×100 magnification) using bright-field
microscopy. Scale bar, 10 µm. (D and E) Determination of NQO1
expression. The 231-NQO1+/+ and 231-NQO1−/−
cells were subjected to western blot analysis using anti-NQO1
antibody (D) and results were quantified (E) NQO1, NAD(P)H:quinone
oxidoreductase; 1231-NQO1+/+, NQO1-overexpressing
MDA-MB-231 cells; 231-NQO1−/−, MDA-MB-231 cells lacking
NQO1. Data represent the mean (±SD) of three independent
experiments (*P<0.05, **P<0.01; NS, not significant). |
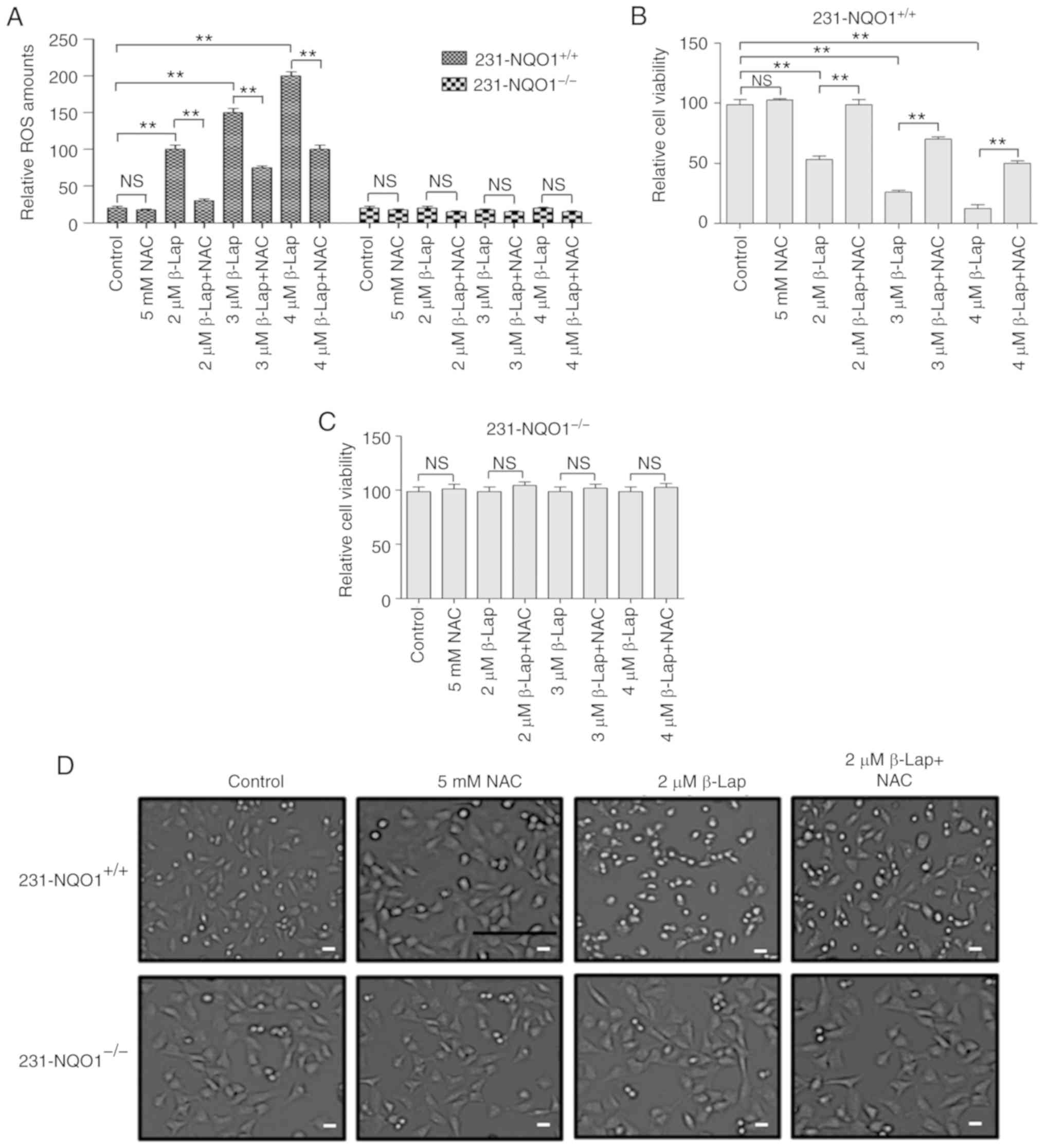
We further investigated whether a decrease in cell
viability of 231-NQO1+/+ cells treated with β-Lap is due
to the accumulation of cellular ROS. When 231-NQO1+/+
cells were treated with β-Lap (0, 2, 3 and 4 µM), intracellular ROS
levels were gradually increased in a dose-dependent manner, and
were specifically inhibited by treatment with the ROS scavenger [5
mM NAC (N-acetyl cysteine)] (Fig.
2A). However, 231-NQO1−/− cells showed no
accumulation of ROS with β-Lap treatment (Fig. 2A). Cell viability and morphological
changes under light microscopy were similarly observed in both
231-NQO1+/+ and 231-NQO1−/− cells treated
with β-Lap in the presence of NAC, as shown by cell viability
results (Fig. 2B-D). These data
suggested that β-Lap specifically induced cell death in
231-NQO1+/+ breast cancer cells through stimulation of
cellular ROS production.
β-Lap treatment in
231-NQO1+/+ cells induces apoptotic cell death via
activation of PKA
According to many previous studies, β-Lap can induce
apoptosis in a variety of cancer cells (31–38). In
order to examine the mechanisms underlying β-Lap-mediated cell
death, we compared the expression levels of antiapoptotic or
apoptotic proteins in both 231-NQO1+/+ and
231-NQO1−/− cells upon treatment with different
concentrations of β-Lap (0, 2, 3 and 4 µM). As expected, while the
expression levels of antiapoptotic Bcl-2, and Bcl-xL proteins were
gradually decreased with increased concentrations of β-Lap in
231-NQO1+/+ cells, the expression levels in
NQO1-deficient cells (231-NQO1−/−) were unchanged
regardless of the level of β-Lap used (Fig. 3A and C). In contrast, proapoptotic Bak
and cytochrome c were significantly increased in the
231-NQO1+/+ cells in a β-Lap dose-dependent manner, but
this was not observed in 231-NQO1−/− cells (Fig. 3A and D). In addition, caspase-3
activation (cleaved caspase-3) and PARP cleavage was significantly
stimulated when 231-NQO1+/+ cells were incubated with
different concentrations of β-Lap (Fig.
3A and E). Importantly, we observed a significant increase in
PKA activation (phosphorylated-PKAα/β/γ at T198) in β-Lap-treated
231-NQO1+/+ cells, but not in 231-NQO1−/−
cells (Fig. 3A and B). In addition,
the pattern of PKA activation was comparable with induction of
proapoptotic events, such as caspase-3 activation and PARP
cleavage, as shown in Fig. 3.
Moreover, this PKA activation was ROS-dependent. Indeed, the
phosphorylation of PKAα/β/γ at T198 was significantly inhibited by
5 mM NAC in β-Lap-treated 231-NQO1+/+ cells, but not in
231-NQO1−/− cells (Fig.
4), indicating that PKA activation is closely associated with
apoptotic cell death induced by β-Lap-dependent ROS in
231-NQO1+/+ cells.
 | Figure 3.PKA is highly activated during
β-Lapachone (β-Lap)-induced apoptosis. (A) Western blots. The
231-NQO1+/+ (left panel) and 231-NQO1−/−
(right panel) cells were treated with β-Lap (0, 2, 3 and 4 µM) for
2 h and further incubated in fresh medium with 5% FBS for 4 h.
After cell lysis, total cell extracts (30 µg) were separated on 8
or 10% SDS-PAGE and analyzed by western blotting using primary
antibodies against proteins (Bcl-2, Bcl-xL, cytochrome c,
Bak, p-PKAα/β/γ cat, PKA cat, caspase-3, cleaved caspase-3, PARP
and cleaved PARP). β-actin was used as a loading control. (B-E)
Quantification of protein expression and activation. The relative
amount of all proteins are shown in western blot analyses and were
quantified by NIH ImageJ software and represented as a graph,
respectively. The relative amount of p-PKAα/β/γ to PKA cat was
determined first and subsequently normalized to β-actin. Data
represent the mean (±SD) of three independent experiments
(**P<0.01; NS, not significant). NQO1, NAD(P)H:quinone
oxidoreductase; PKA, protein kinase A; Bcl-2, B-cell lymphoma-2;
Bcl-xL, B-cell lymphoma-extra large; PARP, Poly(ADP-ribose)
polymerase; 1231-NQO1+/+, NQO1-overexpressing MDA-MB-231
cells; 231-NQO1−/−, MDA-MB-231 cells lacking NQO1. |
PKA activation deteriorates the
β-Lap-induced cell death
As shown above, PKA significantly activated
β-Lap-induced cell death in MDA-MB-231 breast cancer cells
overexpressing NQO1 (231-NQO1+/+). Additionally, in some
studies, PKA has been suggested to be a potential apoptotic cell
death activator (15–26). In order to further investigate the
possible role of PKA activation during β-Lap-induced cell death, we
treated 231-NQO1+/+ and 231-NQO1−/− cells
with PKA activators (dibutyryl-cAMP, an analog of cAMP, and
forskolin) or PKA inhibitor (H89) and examined the effects on cell
viability. The viability of 231-NQO1+/+ cells was
dramatically decreased when cells were treated with both β-Lap and
dibutyryl-cAMP (di-cAMP) or forskolin when compared to the control.
In addition, treatment with PKA activators, either di-cAMP or
forskolin, caused a significant decrease in cell viability in both
231-NQO1+/+ and 231-NQO1−/− cells in the
absence of β-Lap when compared to the control (Fig. 5A and B). Furthermore, a decrease in
cell viability by β-Lap treatment in 231-NQO1+/+ cells
was significantly recovered by PKA inhibitor H89 (Fig. 5A), but not completely, suggesting that
ROS-mediated cell death might be additionally regulated by some
other mechanisms. H89 had a positive influence on the recovery of
cell viability, and this was slightly decreased by di-cAMP even in
231-NQO1−/− cells (Fig. 5A and
B).
We next examined the molecular levels of cell
death-related proteins and PKA activation in both
231-NQO1+/+ and 231-NQO1−/− cells treated
with β-Lap in the presence of PKA inhibitors or activators. When
231-NQO1+/+ cells were treated with either di-cAMP or
forskolin in combination with β-Lap, we observed a significant
decrease in antiapoptotic proteins, such as Bcl-2 and Bcl-xL
(Fig. 6A and C), and also a large
increase in proapoptotic cytochrome c, Bak expression,
caspase-3 activation, and PARP cleavage (Fig. 6A, E and G), with a substantial
activation of PKA (p-PKAα/β/γ at T198) (Fig. 6A and B), compared to treatment with
PKA activators alone, either di-cAMP or forskolin without β-Lap.
Furthermore, we also observed a slight increase in proapoptotic
proteins and a decrease in antiapoptotic proteins in
231-NQO1−/− cells treated with either cAMP or forskolin,
regardless of β-Lap treatment (Fig. 6A,
D, F and H). On the contrary, antiapoptotic proteins and
proapoptotic events were significantly increased and decreased,
respectively, in 231-NQO1+/+ cells treated with β-Lap in
the presence of H89, a PKA inhibitor (Fig. 6). Furthermore, combination of β-Lap
and H89 in NQO1-negative cells (231-NQO1−/−) caused a
slight increase in antiapoptotic proteins and a substantial
decrease in cytochrome c and Bak expression (Fig. 6).
 | Figure 6.Treatment with PKA activators further
increases β-Lapachone (β-Lap)-induced apoptotic cell death through
activation of the PKA signaling pathway. (A) Western blots. The
231-NQO1+/+ (left panel) and 231-NQO1−/−
(right panel) cells were treated with 2 µM β-Lap in combination
with PKA activators (10 µM dibutyryl-cAMP (di-cAMP), 10 µM
Forskolin) or PKA inhibitor (5 µM H89) for 2 h, and additionally
incubated in fresh medium + 5% FBS containing PKA activators or
inhibitor for 4 h. Cells were lysed, and total cell extracts (30
µg) were separated by 8 or 10% SDS-PAGE and analyzed by western
blotting using primary antibodies to Bcl-2, Bcl-xL, cytochrome
c, Bak, p-PKAα/β/γ cat, PKA cat, caspase-3, cleaved
caspase-3, PARP, and cleaved PARP. β-actin was used as a loading
control. (B-H) The relative amounts of all proteins shown in
western blot analyses were quantified by NIH ImageJ software and
represented as a graph: p-PKA (B) in 231-NQO1+/+ (left)
and 231-NQO1−/− (right) cells, Bcl2/Bcl-xL in
231-NQO1+/+ (C) and 231-NQO1−/− (D) cells,
cytochrome c/Bak in 231-NQO1+/+ (E) and
231-NQO1−/− (F) cells, and cleaved caspase 3/cleaved
PARP in 231-NQO1+/+ (G) and 231-NQO1−/− (H)
cells, respectively. The relative amount of p-PKAα/β/γ to PKA cat
was determined first and subsequently normalized to β-actin. Data
represent the mean (± SD) of three independent experiments
(*P<0.05, **P<0.01; NS, not significant). NQO1, NAD(P)
H:quinone oxidoreductase; PKA, protein kinase A; Bcl-2, B-cell
lymphoma-2; Bcl-xL, B-cell lymphoma-extra large; PARP,
Poly(ADP-ribose) polymerase; 1231-NQO1+/+,
NQO1-overexpressing MDA-MB-231 cells; 231-NQO1−/−,
MDA-MB-231 cells lacking NQO1. |
Discussion
In the present study, we showed that β-Lap induced
apoptotic cell death via the PKA pathway in human breast cancer
MDA-MB-231 cells overexpressing NQO1 protein. According to previous
studies, β-Lap-induced cell death is mostly dependent on ROS
production by intracellular metabolism combined with a futile cycle
in a NQO1-dependent manner (34–38). We
also observed an increase in intracellular ROS when
231-NQO1+/+ cells were treated with β-Lap, in a
dose-dependent manner. Additionally, we observed activation of PKA
in the β-Lap-induced cell death of 231-NQO1+/+ cells,
and we also showed that PKA activators further exacerbated
β-Lap-induced cell death; PKA inhibitor substantially recovered
cell survival even in cases of β-Lap-treated 231-NQO1+/+
cells. These results suggested that the PKA activity activated by
β-Lap was associated with apoptotic cell death in these breast
cancer cell lines.
Although cAMP-induced PKA activation is an essential
pathway used to maintain cell viability in normal and cancer cells,
PKA activation has also been suggested to induce apoptotic cell
death in many cancer cells (15–17).
Indeed, activation of cAMP and PKA signaling pathways stimulates
apoptosis through phosphorylation of apoptosis-associated proteins
such as Bim under stress conditions, such as during DNA damage
(25). Most of these cell death
mechanisms are associated with an intrinsic mitochondrial pathway
(22). Furthermore, PKA-induced cell
death in cancer cells is observed in not only a cAMP-dependent but
also a cAMP-independent manner (18).
In particular, despite the potent anticancer effects of cAMP/PKA
activation by cancer drugs, these substances causing PKA activation
via increase of intracellular cAMP are not recommended to be used
as anticancer drugs because of their high cytotoxicity via the
cAMP/PKA/CREB signaling pathway in normal cells. Therefore, the
discovery of novel drugs that induce cAMP-independent PKA
activation in cancer cells could be a beneficial strategy for
future cancer treatment.
According to our data, β-Lap, a quinone compound,
highly activated PKA and consequently induced apoptotic cell death
in NQO1-positive breast cancer cells but not in NQO1-negative
cells, suggesting that β-Lap-mediated ROS production in
NQO1-positive cells might be essential for cell death via both PKA
activation and other mechanisms. In particular, although direct
treatment with PKA activators such as dibutyryl-cAMP or forskolin
of 231-NQO1−/− cells substantially activated PKA, it led
to less cell death even in the presence or absence of β-Lap, when
compared with 231-NQO1+/+ cells treated with β-Lap,
indicating that PKA activation in β-Lap-treated
231-NQO1+/+ cells might not be solely cAMP-dependent;
rather, some other cAMP-independent mechanisms may activate PKA, as
shown in another study (18).
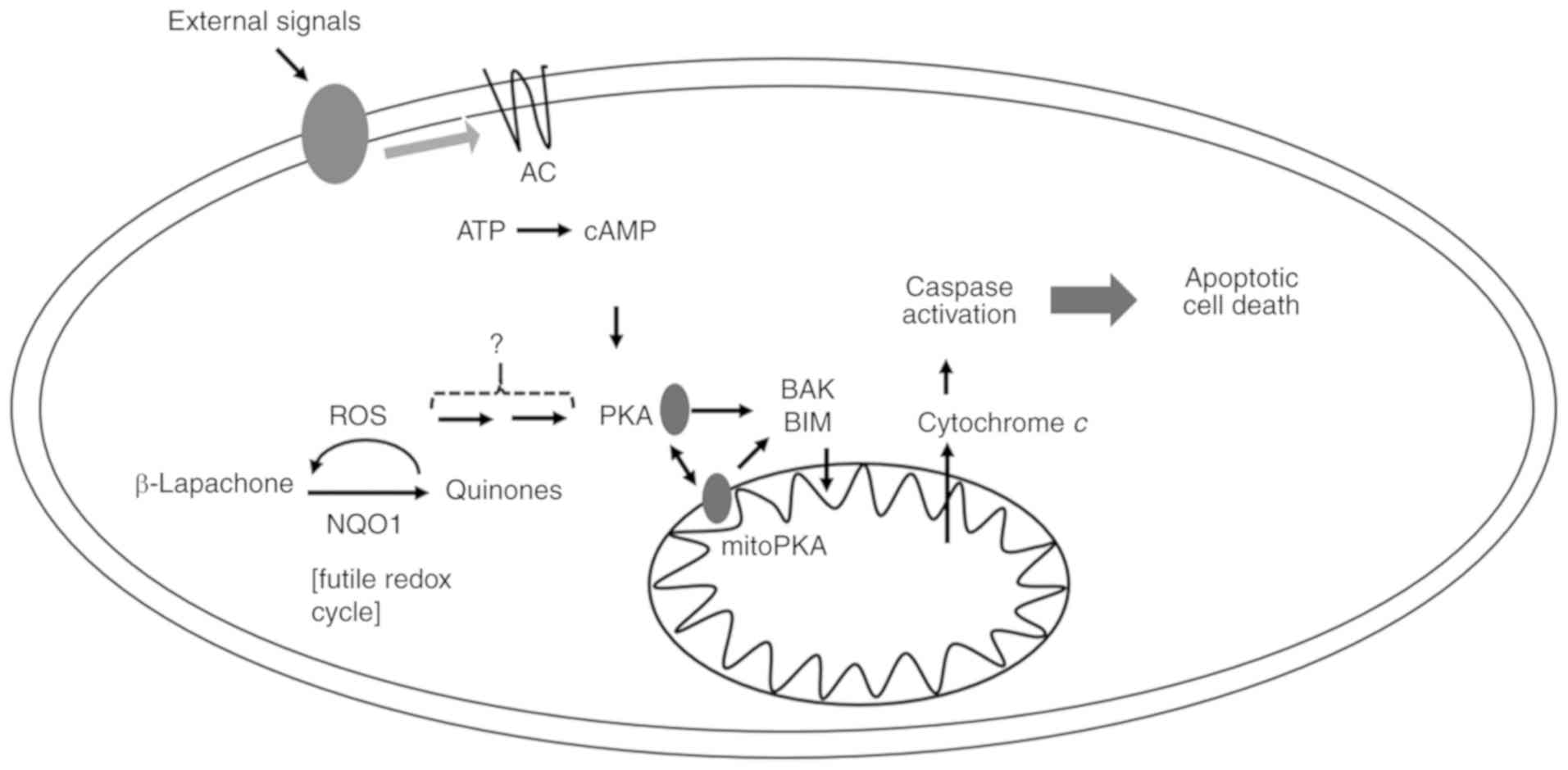
In addition to cAMP-independent PKA activation,
β-Lap might also mediate the coordinative regulation of cytosolic
and mitochondrial PKA activation through high production of ROS in
mitochondria. β-Lap-mediated ROS were increased and inhibited by
antioxidant NAC in 231-NQO1+/+ cells, and coordinatively
PKA activation was observed. PKA was more highly activated upon
treatment with β-Lap, than by direct treatment with PKA activators.
We observed a cumulative upregulation of PKA with treatment of the
PKA activator and β-Lap, suggesting that β-Lap-mediated ROS were
additionally related to PKA activation involved in cell death
(Fig. 7). Indeed, some studies have
suggested that increases in intracellular ROS activate the PKA
signaling pathway (44–46).
In agreement with previous studies (31–38), this
study also showed that β-Lap induced apoptotic cell death by
NQO1-mediated ROS in a dose-dependent manner. β-Lap treatment
caused a significant decrease in antiapoptotic proteins, such as
Bcl-2 and Bcl-xL. In contrast, β-Lap caused a large increase in
proapoptotic events including caspase activation, PARP cleavage,
and cytochrome c. Nevertheless, our study still has some
limitations to make a solid conclusion. In this study we only used
a single type of breast cancer cell, and we did not examined the
effect of β-Lap on normal breast cells with or without NQO1
overexpression, which will be investigated in the future.
As mentioned previously, a combination of different
processes may be effective for the treatment of cancer. However, a
new strategy targeting apoptosis in cancer cells still remains to
be developed. In particular, in this study we suggest a novel
mechanism by which β-Lap can induce apoptosis via activation of
PKA, and use of β-Lap as a selective treatment in cancer
patients.
Acknowledgements
We thank Dr David Boothman (UT Southwestern Medical
Center, Dallas, TX, USA) for providing MDA-MB-231 cells
overexpressing NQO1 (231-NQO1+/+) and MDA-MB-231 cells
lacking NQO1 (231-NQO1−/−).
Funding
This study was supported by the Basic Research
Program through the National Research Foundation of Korea (NRF)
funded by the Ministry of Education Science and Technology
[2018R1D1A1B07043715 (to DRK), 2018R1D1A1B07049185 (to D-HK)] and
by the Ministry of Science, ICT, and Future Planning
(NRF-2015R1A5A2008833).
Availability of data and materials
The datasets used during the present study are
available from corresponding author upon request.
Authors' contributions
SZ and DRK conceived and designed the study. SZ
carried out experiments, analyzed the data, and wrote the primary
manuscript. JSH, THL, TMP, DHK and MA analyzed the data and edited
the manuscript. DHK and DRK edited the manuscript and received the
research funding. All authors read and approved the manuscript and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
β-Lap
|
β-Lapachone
|
|
NQO1
|
NAD(P)H:quinone oxidoreductase 1
|
|
PKA
|
protein kinase A
|
|
ROS
|
reactive oxygen species
|
|
NAC
|
N-acetylcysteine
|
|
cAMP
|
cyclic AMP
|
|
RPMI
|
Roswell Park Memorial Institute 1640
medium
|
|
FBS
|
fetal bovine serum
|
|
DCFDA
|
2′,7′-dichloroflourescin diacetate
|
|
CCK-8
|
Cell Counting Kit-8
|
|
PBS
|
phosphate-buffered saline
|
|
DMSO
|
dimethyl sulfoxide
|
|
SDS-PAGE
|
sodium dodecyl sulfate-polyacrylamide
gel electrophoresis
|
References
|
1
|
Semenza GL: Molecular mechanisms mediating
metastasis of hypoxic breast cancer cells. Trends Mol Med.
18:534–543. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Valastyan S and Weinberg RA: Tumor
metastasis: Molecular insights and evolving paradigms. Cell.
147:275–292. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lambert AW, Pattabiraman DR and Weinberg
RA: Emerging biological principles of metastasis. Cell.
168:670–691. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lerebours F and Lidereau R: Molecular
alterations in sporadic breast cancer. Crit Rev Oncol Hematol.
44:121–141. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li CI, Beaber EF, Tang MC, Porter PL,
Daling JR and Malone KE: Reproductive factors and risk of estrogen
receptor positive, triple-negative, and HER2-neu overexpressing
breast cancer among women 20–44 years of age. Breast Cancer Res
Treat. 137:579–587. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Coughlin SS and Ekwueme DU: Breast cancer
as a global health concern. Cancer Epidemiol. 33:315–318. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tangjitgamol S, Anderson BO, See HT,
Lertbutsayanukul C, Sirisabya N, Manchana T, Ilancheran A, Lee KM,
Lim SE, Chia YN, et al: Management of endometrial cancer in Asia:
Consensus statement from the Asian Oncology Summit 2009. Lancet
Oncol. 10:1119–1127. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mark HF, Aswad B, Bassily N, Taylor W,
Brown S, Sun CL, Samy M, Zolnierz K, Wong E, Bland KI and Hsu PH:
HER-2/neu gene amplification in stages I–IV breast cancer detected
by fluorescent in situ hybridization. Genet Med. 1:98–103. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thompson CB: Apoptosis in the pathogenesis
and treatment of disease. Science. 267:1456–1462. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fulda S: Apoptosis pathways and their
therapeutic exploitation in pancreatic cancer. J Cell Mol Med.
13:1221–1227. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Varfolomeev E and Vucic D: Inhibitor of
apoptosis proteins: Fascinating biology leads to attractive tumor
therapeutic targets. Future Oncol. 7:633–648. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Taskén K and Aandahl EM: Localized effects
of cAMP mediated by distinct routes of protein kinase A. Physiol
Rev. 84:137–167. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fimia GM and Sassone-Corsi P: Cyclic AMP
signalling. J Cell Sci. 114:1971–1972. 2001.PubMed/NCBI
|
|
15
|
Cross TG, Scheel-Toellner D, Henriquez NV,
Deacon E, Salmon M and Lord JM: Serine/threonine protein kinases
and apoptosis. Exp Cell Res. 256:34–41. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lerner A, Kim DH and Lee R: The cAMP
signaling pathway as a therapeutic target in lymphoid malignancies.
Leuk Lymphoma. 37:39–51. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Insel PA, Bourne HR, Coffino P and Tomkins
GM: Cyclic AMP-dependent protein kinase: Pivotal role in regulation
of enzyme induction and growth. Science. 190:896–898. 1975.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hedrick ED, Agarwal E, Leiphrakpam PD,
Haferbier KL, Brattain MG and Chowdhury S: Differential PKA
activation and AKAP association determines cell fate in cancer
cells. J Mol Signal. 8:102013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Almeida MQ and Stratakis CA: How does
cAMP/protein kinase A signaling lead to tumors in the adrenal
cortex and other tissues? Mol Cell Endocrinol. 336:162–168. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chowdhury S, Howell GM, Rajput A, Teggart
CA, Brattain LE, Weber HR, Chowdhury A and Brattain MG:
Identification of a novel TGFβ/PKA signaling transduceome in
mediating control of cell survival and metastasis in colon cancer.
PLoS One. 6:e193352011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zambon AC, Zhang L, Minovitsky S, Kanter
JR, Prabhakar S, Salomonis N, Vranizan K, Dubchak I, Conklin BR and
Insel PA: Gene expression patterns define key transcriptional
events in cell-cycle regulation by cAMP and protein kinase A. Proc
Natl Acad Sci USA. 102:8561–8566. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang L, Zambon AC, Vranizan K, Pothula K,
Conklin BR and Insel PA: Gene expression signatures of cAMP/Protein
Kinase A (PKA)-promoted, Mitochondrial-dependent Apoptosis:
Comparative analysis of wild-type and camp-deathless s49 lymphoma
cells. J Biol Chem. 283:4304–4313. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Coffino P, Bourne HR and Tomkins GM:
Mechanism of lymphoma cell death induced by cyclic AMP. Am J
Pathol. 81:199–204. 1975.PubMed/NCBI
|
|
24
|
Yan L, Herrmann V, Hofer JK and Insel PA:
beta-Adrenergic receptor/cAMP-mediated signaling and apoptosis of
S49 lymphoma cells. Am J Physiol Cell Physio. 279:C1665–C1674.
2000. View Article : Google Scholar
|
|
25
|
Zhang L and Insel PA: The pro-apoptotic
protein Bim is a convergence point for cAMP/protein kinase A-and
glucocorticoid-promoted apoptosis of lymphoid cells. J Biol Chem.
279:20858–20865. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Naviglio S, Di Gesto D, Romano M,
Sorrentino A, Illiano F, Sorvillo L, Abbruzzese A, Marra M,
Caraglia M, Chiosi E, et al: Leptin enhances growth inhibition by
cAMP elevating agents through apoptosis of MDA-MB-231 breast cancer
cells. Cancer Biol Ther. 8:1183–1190. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ugland H, Boquest AC, Naderi S, Collas P
and Blomhoff HK: cAMP-mediated induction of cyclin E sensitizes
growth-arrested adipose stem cells to DNA damage-induced apoptosis.
Mol Biol Cell. 19:5082–5092. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Blanco E, Bey EA, Khemtong C, Yang SG,
Setti-Guthi J, Chen H, Kessinger CW, Carnevale KA, Bornmann WG,
Boothman DA and Gao J: Beta-lapachone micellar nanotherapeutics for
non-small cell lung cancer therapy. Cancer Res. 70:3896–3904. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li CJ, Li YZ, Pinto AV and Pardee AB:
Potent inhibition of tumor survival in vivo by beta-lapachone plus
taxol: Combining drugs imposes different artificial checkpoints.
Proc Natl Acad Sci USA. 96:13369–13374. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dong Y, Chin SF, Blanco E, Bey EA, Kabbani
W, Xie XJ, Bornmann WG, Boothman DA and Gao J: Intratumoral
delivery of beta-lapachone via polymer implants for prostate cancer
therapy. Clin cancer Res. 15:131–139. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pardee AB, Li YZ and Li CJ: Cancer therapy
with beta-lapachone. Curr Cancer Drug Targets. 2:2002.227–242.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Reinicke KE, Bey EA, Bentle MS, Pink JJ,
Ingalls ST, Hoppel CL, Misico RI, Arzac GM, Burton G, Bornmann WG,
et al: Development of beta-lapachone prodrugs for therapy against
human cancer cells with elevated NAD(P)H:Quinone oxidoreductase 1
levels. Clin Cancer Res. 11:3055–3064. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Trachootham D, Alexandre J and Huang P:
Targeting cancer cells by ROS-mediated mechanisms: A radical
therapeutic approach? Nat Rev Drug Discov. 8:579–591. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pink JJ, Planchon SM, Tagliarino C, Varnes
ME, Siegel D and Boothman DA: NAD(P)H:Quinone oxidoreductase
activity is the principal determinant of beta-lapachone
cytotoxicity. J Biol Chem. 275:5416–5424. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Siegel D, Yan C and Ross D:
NAD(P)H:Quinone oxidoreductase 1 (NQO1) in the sensitivity and
resistance to antitumor quinones. Biochem Pharmacol. 83:1033–1040.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Park EJ, Choi KS and Kwon TK:
β-Lapachone-induced reactive oxygen species (ROS) generation
mediates autophagic cell death in glioma U87 MG cells. Chem Biol
Interact. 189:37–44. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Choi EK, Terai K, Ji IM, Kook YH, Park KH,
Oh ET, Griffin RJ, Lim BU, Kim JS, Lee DS, et al: Upregulation of
NAD(P)H:Quinone oxidoreductase by radiation potentiates the effect
of bioreductive beta-lapachone on cancer cells. Neoplasia.
9:634–642. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Garate M, Wani AA and Li G: The
NAD(P)H:Quinone oxidoreductase 1 induces cell cycle progression and
proliferation of melanoma cells. Free Radic Biol Med. 48:1601–1609.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang Y, Zhang Y, Wu Q, Cui X, Lin Z, Liu S
and Chen L: Clinical implications of high NQO1 expression in breast
cancers. J Exp Clin Cancer Res. 33:142014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lewis AM, Ough M, Hinkhouse MM, Tsao MS,
Oberley LW and Cullen JJ: Targeting NAD(P)H:quinone oxidoreductase
(NQO1) in pancreatic cancer. Mol Carcinog. 43:215–224. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lyn-Cook BD, Yan-Sanders Y, Moore S,
Taylor S, Word B and Hammons GJ: Increased levels of
NAD(P)H:Quinone oxidoreductase 1 (NQO1) in pancreatic tissues from
smokers and pancreatic adenocarcinomas: A potential biomarker of
early damage in the pancreas. Cell Biol Toxicol. 22:73–80. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li JZ, Ke Y, Misra HP, Trush MA, Li YR,
Zhu H and Jia Z: Mechanistic studies of cancer cell mitochondria-
and NQO1-mediated redox activation of beta-lapachone, a potentially
novel anticancer agent. Toxicol Appl Pharmacol. 281:285–293. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cao L, Li LS, Spruell C, Xiao L,
Chakraberti G, Bey EA, Reinicke KE, Srougi MC, Moore Z, Dong Y, et
al: Tumor-selective, futile redox cycle-induced bystander effects
elicited by NQO1 bioactivatable radiosensitizing drugs in
triple-negative breast cancers. Antioxid Redox Signal. 21:237–250.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu Y, Yang F, Li S, Dai J and Deng H:
Glutaredoxin deletion shortens chronological life span in
saccharomyces cerevisiae via ROS-Mediated Ras/PKA activation. J
Proteome Res. 17:2318–2327. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li Z, Ji G and Neugebauer V: Mitochondrial
reactive oxygen species are activated by mGluR5 through IP3 and
activate ERK and PKA to increase excitability of amygdala neurons
and pain behavior. J Neurosci. 31:1114–1127. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bovo E, Lipsius SL and Zima AV: Reactive
oxygen species contribute to the development of arrhythmogenic
Ca2+ waves during β-adrenergic receptor stimulation in
rabbit cardiomyocytes. J Physiol. 590:3291–3304. 2012. View Article : Google Scholar : PubMed/NCBI
|