Introduction
Colorectal cancer (CRC), a commonly occurring
malignant tumor in the gastrointestinal tract, is the third leading
cause of cancer-related deaths worldwide, causing approximately
600,000 deaths each year (1,2). Each year, more than one million new CRC
patients are diagnosed worldwide (3),
and in China, especially in underdeveloped areas, the incidence of
CRC is increasing rapidly (4).
Currently, oxaliplatin-based chemotherapy is the primary strategy
for CRC treatment; however, patients ultimately relapse due to drug
resistance (5). Oxaliplatin is
reported to be the first platinum compound to be effective for the
treatment of CRC (6), through
inhibition of tumor cell growth and G2-phase cell cycle arrest
(7). However, resistance to
oxaliplatin poses a huge challenge to CRC treatment, and its
underlying mechanism remains unclear.
Phosphatase and tensin homolog deleted on chromosome
10 (PTEN) is a lipid phosphatase that frequently serves as a tumor
suppressor in multiple human cancers (8). It has been reported that PTEN is of
great significance in the progression of CRC (9,10). PTEN
can antagonize the action of the AKT pathway, and studies have
implicated the PTEN/AKT pathway to be associated with drug
resistance in several types of cancer (11,12). For
example, microRNA-21 was found to overcome sorafenib-resistance of
liver cancer cells through the PTEN/AKT pathway (11). In addition, overexpression of
microRNA-22 was able to reverse paclitaxel-resistance of CRC cells
by activating PTEN (12).
FK506-binding proteins (FKBPs), an immunophilin
family, bind immunosuppressive drugs and are involved in various
processes, including cancer progression and chemoresistance
(13). It has been shown that FKBP51
can inhibit the cell proliferation in CRC (14). Additionally, the tumorigenesis and
chemoresistance of cancers can be negatively regulated by USP49
through FKBP51/AKT signaling (15,16). FKBP3
(also known as FKBP25), a nuclear member of the FKBP family, has
been shown to transcriptionally regulate the expression of p53 and
p21 (17). Previously, it was also
reported to be associated with the activity of histone deacetylases
1/2 (HDAC1/2) (18). HDACs are an
ancient superfamily of enzymes containing multiple members
including HDAC1 and HDAC2, and are involved in the development and
progression of human cancers (19).
Overexpression of HDACs has been observed in a variety of human
cancers such as CRC (20), and
downregulation of HDAC2 was found to inhibit the cell growth of
liver cancer cells by the PTEN/PI3K/AKT pathway (21). In addition, a study revealed that
FKBP3 promotes lung cancer cell proliferation through the
regulation of Sp1/HDAC2/p27 signaling (22). However, the role of FKBP3 and HDAC2 in
oxaliplatin resistance in CRC and its potential molecular
mechanisms are still poorly understood.
Here we showed that FKBP3 and HDAC2 are highly
expressed in CRC tissues. Compared to CRC cells with low FKBP3
expression, CRC cells with high FKBP3 expression were more
insensitive to oxaliplatin treatment, concurrent with decreased
cleaved caspase-3 and increased B-cell lymphoma-2 (Bcl-2)
expression. Downregulation of FKBP3 significantly increased
apoptosis in oxaliplatin-resistant CRC cells, reduced the
expression levels of HDAC2, permeability glycoprotein (P-gp) and
phosphorylated AKT (p-AKT), and increased expression of PTEN and
cleaved caspase-3. Furthermore, downregulation of HDAC2 reversed
FKBP3-induced oxaliplatin resistance in CRC cells. Our data
demonstrated that FKBP3 and HDAC2 expression increased oxaliplatin
resistance in CRC cells, and that the downregulation of FKPB3
re-sensitized cells to oxaliplatin by reducing HDAC2 expression,
and possibly through the regulation of the PTEN/AKT pathway.
Materials and methods
CRC tissues and adjacent normal
tissues
Samples from a total of 58 CRC patients (age range,
40–70 years; 30 males and 28 females) were collected from June 2017
to June 2018). All patients enrolled in this study were treated at
Shanghai Municipal Hospital of Traditional Chinese Medicine
Affiliated to Shanghai TCM University (Shanghai, China. Tumor and
adjacent normal tissues were collected from these patients after
the receipt of written informed consent. Tissues were frozen and
stored in liquid nitrogen immediately before use. After total RNA
extraction, the expression of FKBP3 and HDAC2 in tissues was
detected. All experiments conducted in this study were approved by
the Ethics Committee of Shanghai Municipal Hospital of Traditional
Chinese Medicine Affiliated to Shanghai TCM University (Shanghai,
China).
Cell culture
After obtaining written informed consent, primary
cells were isolated from 12 CRC patients treated at Shanghai
Municipal Hospital of Traditional Chinese Medicine Affiliated to
Shanghai TCM University (Shanghai, China). The cells were cultured
in a 5% CO2 incubator (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) at 37°C with HyClone™ high glucose DMEM medium
(cat. no. SH30243.01; Thermo Fisher Scientific, Inc.) supplemented
with Gibco™ 10% fetal bovine serum (FBS; cat. no. 16000-044; Thermo
Fisher Scientific, Inc.) and 1% antibiotic (penicillin and
streptomycin; cat. no. P1400-100; Beijing Solarbio Science &
Technology Co., Ltd., Beijing, China). The medium was refreshed
every two days during culture.
Construction of lentiviral
plasmids
shRNA plasmids were constructed by synthesizing and
inserting sequences targeting FKBP3 (5′-GAGGTTCAATGTTGGATAT-3′) and
HDAC2 (5′-CCCAUAACUUGCUGUUAAA-3′) (22) into AgelI/EcoRI
restriction sites of a pLKO.1-puro vector. The coding DNA sequence
(CDS) region of FKBP3 (NM_002013.3) with the addition of
EcoRI and BamHI restriction sites was synthesized, by
Genewiz Co. (Shanghai, China), and inserted into the
EcoRI/BamHI sites of a pLVX-Puro vector (Clontech).
After confirmation by DNA sequencing (Shanghai Majorbio
Pharmaceutical Technology Co., Ltd., Shanghai, China),
pLKO.1-Puro-shFKBP3, pLKO.1-Puro-shHDAC2 or pLVX-Puro-FKBP3 was
co-transfected into 293T cells with viral packaging plasmids psPAX2
and pMD2G (Addgene, Inc., Cambridge, MA, USA) using Lipofectamine
2000 (Invitrogen; Thermo Fisher Scientific, Inc.). pLKO.1-Puro and
pLVX-Puro were used as negative control vectors. The virus
particles in the supernatant were collected by ultracentrifugation
48 h after transfection.
Experimental grouping
Primary CRC cells with high expression of FKBP3 were
divided into four groups and infected with FKBP3 overexpression
(oeFKBP3)/vector and FKBP3 interference (shFKBP3)/negative control
(shNC) lentiviruses. Forty-eight hours after infection, the
efficiency of oeFKBP3 and shFKBP3, as well as the expression of
HDAC2 were evaluated by real-time PCR and western blot
analysis.
To further investigate the role of FKBP3 and HDAC2
in oxaliplatin resistance in CRC, the primary CRC cells with high
expression of FKBP3 were treated with shNC+vector+40 µg/ml
oxaliplatin (O124003; Aladdin), shFKBP3+40 µg/ml oxaliplatin, or
oeFKBP3+40 µg/ml oxaliplatin. Primary cells were also treated with
shNC+vector+40 µg/ml oxaliplatin, oeFKBP3+40 µg/ml oxaliplatin,
shHDAC2+40 µg/ml oxaliplatin, or oeFKBP3+shHDAC2+40 µg/ml
oxaliplatin. Twenty-four hours after oxaliplatin treatment, cell
apoptosis and the expression of related-proteins were quantified,
and the 24-h treatment of oxaliplatin had a significant decrease in
apoptosis in CRC primary cells, and thus this time point was
selected for further experiments.
Real-time polymerase chain reaction
(qPCR) assay
Total RNA from primary CRC cells or treated cells
was extracted using TRIzol reagent (cat. no. 1596-026; Invitrogen;
Thermo Fisher Scientific, Inc.). After quantification, the
integrity of extracted RNA was confirmed by electrophoresis using a
1% gel. Subsequently, 1 µg of RNA was reverse transcribed into cDNA
using a Reverse Transcription Kit (#K1622; Fermentas; Thermo Fisher
Scientific, Inc.). Using the cDNA as a template, the qPCR reactions
were conducted on a Real-time PCR system (cat. no. ABI-7300;
Applied Biosystems; Thermo Fisher Scientific, Inc.) with a
SYBR-Green PCR kit (#K0223; Thermo Fisher Scientific, Inc.). The
mRNA expression levels of FKBP3 and HDAC2, normalized to GAPDH,
were calculated by the 2−ΔΔCq method (23). The primers used are listed as follows:
FKBP3 forward, 5′-ACCCAAAGAAACCAAGTC-3′ and reverse,
5′-ATACCAGCAGTGAACAAC-3′; HDAC2 forward, 5′-GCTGGGATTACAGGTGTGAG-3′
and reverse, 5′-AGGCTGAGGTGGGAGAATAC-3′; GAPDH forward,
5′-AATCCCATCACCATCTTC-3′ and reverse, 5′-AGGCTGTTGTCATACTTC-3′. The
qPCR program was as follows: 95°C for 10 min (95°C for 15 sec; 60°C
for 45 sec) ×40; 95°C for 15 sec; 60°C for 1 min; 95°C for 15 sec;
60°C for 15 sec (24).
Western blot analysis
Western blot analysis was performed as previously
described to determine the levels of related-proteins in CRC cells
(25). Total proteins from primary
CRC cells or treated cells were extracted using RIPA buffer
containing protease and phosphatase inhibitors (cat. no. R0010,
Solarbio, Beijing, China). Following quantification by a BCA Kit
(cat. no. PICPI23223; Thermo Fisher Scientific, Inc.), 25 µg of
protein were separated on 10 or 8% SDS-polyacrylamide gels.
Subsequently, proteins were transferred to polyvinylidene fluoride
(PVDF) membranes (cat. no. HATF00010; EMD Millipore, Billerica, MA,
USA) using a semi-dry transfer and blocked in 5% skim milk (cat.
no. BYL40422BD; Biosciences, Franklin Lakes, NJ, USA) at room
temperature for 1 h. The membranes were incubated with primary
antibodies against FKBP3 (dilution 1:2,000; cat. no. Ab16654;
Abcam, Cambridge, UK), HDAC2 (dilution 1:2,000; cat. no. Ab32117,
Abcam), PTEN (dilution 1:500; cat. no. Ab31392; Abcam), P-gp
(dilution 1:200; cat. no. 103477; Abcam), AKT [dilution 1:1,000;
cat. no. 4685; Cell Signaling Technology (CST), Inc., Danvers, MA,
USA], p-AKT (dilution 1:2,000; cat. no. 4060; CST), cleaved
caspase-3 (dilution 1:1,000; cat. no. Ab13847; Abcam), and GAPDH
(dilution 1:2,000; cat. no. 5174; CST) overnight at 4°C. The next
day, the membranes were washed 5–6 times and incubated with
HRP-labeled goat anti-rabbit (cat. no. A0208) and goat anti-mouse
(catalog no. A0216) secondary antibodies (dilution 1:1,000;
Beyotime Institute of Biotechnology, Haimen, China) at room
temperature for 2 h. After 5–6 washes, the blots were developed
with a chemiluminescent reagent (cat. no. WBKLS0100; EMD Millipore)
for 5 min prior to exposure on an ECL imaging system (Tanon-5200;
Tanon Science and Technology Co., Ltd., Shanghai, China). GAPDH
served as an internal control; the expression of proteins was
analyzed and calculated by ImageJ software (version 1.47v; National
Institutes of Health, Bethesda, MD, USA).
Cell apoptosis assay
Apoptosis in the treated primary CRC cells was
evaluated by flow cytometric (FCM) analysis. After treatment,
primary CRC cells were collected to conduct a Annexin V-fluorescein
isothiocyanate (FITC)/propidium iodide (PI) double stain (C1063;
Beyotime Institute of Biotechnology) according to the
manufacturer's protocol. Approximately
5×105−1×106 cells were counted and then
resuspended in 195 µl Annexin V-FITC binding buffer. Subsequently,
the cells were incubated with 5 µl Annexin V-FITC for 15 min at 4°C
in the dark prior to incubation in 5 µl PI for 5 min. Cells without
Annexin V-FITC and PI staining were used as a negative control.
Using BD Accuri™ C6 Software (version 1.0.264.21; BD Biosciences,
USA), the percentage of apoptosis in primary CRC cells was analyzed
and evaluated on a flow cytometer.
Statistical analysis
GraphPad Prism 7.0 software (GraphPad Software,
Inc., La Jolla, CA, USA) was applied in this study to analyze the
statistical significance. The significance between two groups was
analyzed by two-tailed Student's t-test, while one-way analysis of
variance (ANOVA) with a post-test of Tukey's multiple comparison
were used for three or more groups. Pearson's correlation analysis
was also applied for the correlation between the two groups. Based
on three independent experiments, data are expressed as mean ± SD
and P-values <0.05 were defined to be statistically
significant.
Results
Expression of FKBP3 and HDAC2 is
significantly increased in tumors of CRC patients
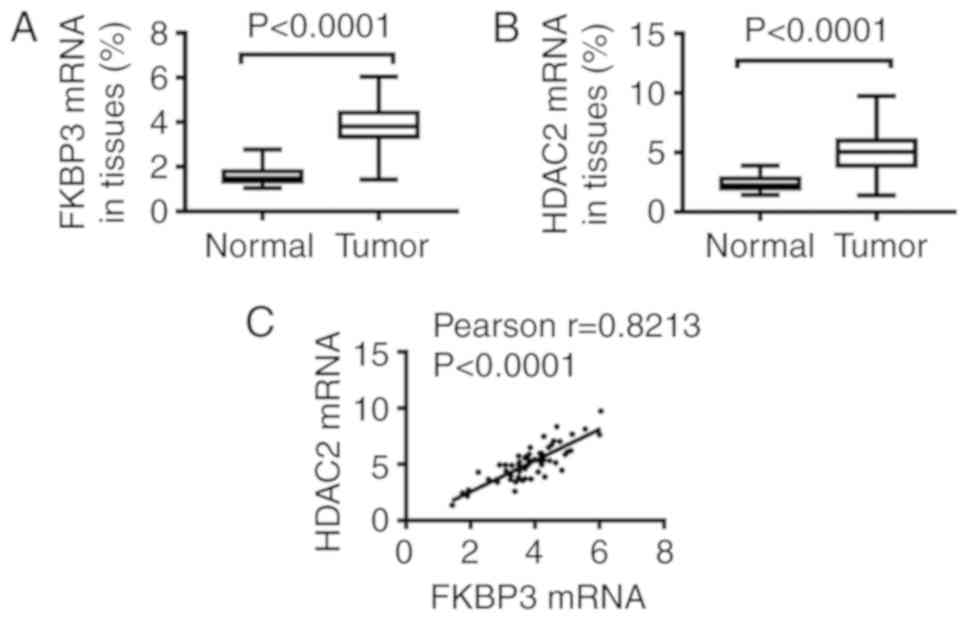
After RNA extraction of tissues, qPCR assay was
performed to detect FKBP3 and HDAC2 expression. As demonstrated in
Fig. 1, we found that, compared to
normal tissues, the expression of FKBP3 (Fig. 1A) and HDAC2 (Fig. 1B) at the mRNA level in tumors was
significantly increased. Importantly, Pearson analysis showed that
the expression of HDAC2 was positively correlated to FKBP3
expression (Fig. 1C). This suggests
that FKBP3 and HDAC2 are critically important in CRC
progression.
High expression of FKBP3 increases
resistance to oxaliplatin in primary CRC cells
Previous studies have shown that oxaliplatin
resistance in cancers displays different behaviors, including
inhibition of apoptosis (26). Here,
we evaluated apoptosis levels to assess Ooxaliplatin resistance in
CRC cells. We isolated primary cells from 12 CRC tissues and
examined the expression levels of FKBP3 and HDAC2. We found that
both FKBP3 (Fig. 2A and C) and HDAC2
(Fig. 2B and C) were highly expressed
in 6 primary CRC cell lines (G, H, I, J, K, L), and relatively
lowly expressed in another 6 cell lines (A, B, C, D, E, F).
Subsequently, the cells were treated with oxaliplatin at 40 µg/ml
for 24 h. The results in Fig. 2D
showed that, compared to CRC cells with low FKBP3 expression (A-F),
the percentage of apoptotic cells in CRC cells with high FKBP3
expression (G-L) was significantly decreased. Concurrently, the
expression of cleaved caspase-3, a cysteine protease that has been
reported to be an important regulator for programmed cell death or
apoptosis, was significantly decreased in CRC cells with high FKBP3
expression, and Bcl-2, an antiapoptotic protein, was increased,
while caspase-3 was unchanged (Fig.
2E) (27,28). This suggests that oxaliplatin
resistance in CRC cells may be positively associated with FKBP3 and
HDAC2 expression.
 | Figure 2.High expression of FKBP3 increases
oxaliplatin resistance in primary CRC cells. Expression analysis of
FKBP3 and HDAC2 was conducted in 58 primary CRC cell lines. Six
highly-expressing (G, H, I, J, K, L) and 6 lowly-expressing (A, B,
C, D, E, F) cell lines were selected for further study. (A and B)
The mRNA expression of FKBP3 and HDAC2 was detected by qPCR. (C)
The protein levels of FKBP3 and HDAC2 were detected by western blot
analysis. (D) After treatment with 40 µg/ml of oxaliplatin, the CRC
cells were incubated with Annexin V-FITC/PI, and the percentage of
apoptotic CRC cells was calculated by FCM analysis. The lower right
(LR) quadrant represents early apoptotic cells positively stained
with Annexin V, while the upper right (UR) quadrant represents late
apoptotic or necrotic cells doubly stained with Annexin V and PI.
The lower left (LL) quadrant shows living cells. (E) The protein
expression of caspase-3, cleaved caspase-3 and Bcl-2 in CRC cells
was detected. All data are presented as mean ± SD of three
experiments. **P<0.01 and ***P<0.001 compared to Low-FKBP3
(A-F: Low-FKBP3, G-L: High-FKBP3). CRC, colorectal cancer; FKBP3,
FK506-binding protein 3; HDAC2, histone deacetylase 2; Bcl-2,
B-cell lymphoma-2. |
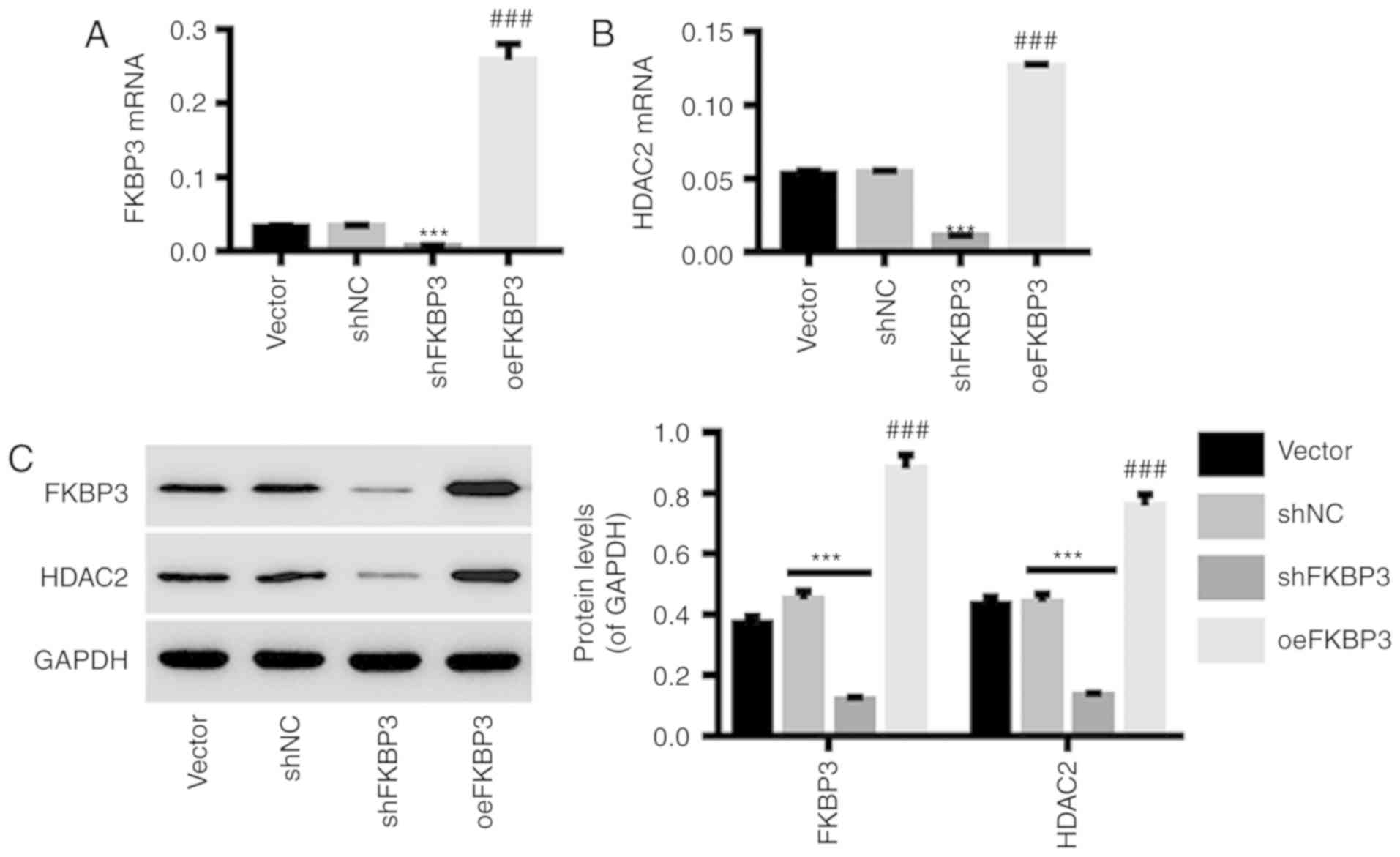
Downregulation and upregulation of
FKBP3 in primary CRC cells by lentiviral infection
Primary CRC cells were infected with shFKBP3/shNC or
oeFKBP3/vector lentiviruses. The results in Fig. 3 show that the expression of FKBP3
(Fig. 3A and C) at the mRNA and
protein levels in primary CRC cells was markedly downregulated by
shFKBP3 and upregulated by oeFKBP3. In addition, the expression of
HDAC2 in primary CRC cells was positively regulated by FKBP3
expression (Fig. 3B and C). This
indicates that HDAC2 is a possible downstream target of FKBP3,
which can be regulated by FKBP3. shFKBP3 and oeFKBP3 lentiviruses
were used for the following experiments.
Downregulation of FKBP3 decreases
oxaliplatin resistance in CRC cells and regulates the PTEN/AKT
pathway
The underlying mechanisms of FKBP3 in regulating
oxaliplatin resistance in CRC cells were investigated. We
downregulated or upregulated the expression of FKBP3 in a primary
CRC cell line and found that downregulation of FKBP3 significantly
promoted apoptosis in oxaliplatin-resistance CRC cells, whereas
FKBP3 upregulation had the opposite effect (Fig. 4A). Several related-proteins were also
assessed. PTEN, as a tumor suppressor, is a potent inhibitor of the
AKT pathway (29). It has been
reported that PTEN regulates cellular processes such as apoptosis
in cancers through the AKT pathway (30–32). P-gp,
encoded by multidrug resistance-associated protein (MRP), is a
major indicator of drug resistance, and caspase-3 is one of the
major apoptosis-executing enzymes. As shown in Fig. 4B, downregulation of FKBP3
significantly decreased HDAC2 and P-gp expression, and increased
cleaved caspase-3 expression, while full-length caspase-3 was
unchanged, and FKBP3 upregulation had the opposite effect.
Furthermore, downregulation of FKBP3 decreased AKT phosphorylation
and increased PTEN expression in oxaliplatin-resistant CRC cells,
while the expression of AKT was unchanged (Fig. 4C). This demonstrated that
downregulation of FKBP3 can increase the sensitivity of CRC cells
to oxaliplatin and FKBP3 possibly regulates the PTEN/AKT pathway in
oxaliplatin-resistant CRC cells.
 | Figure 4.Downregulation of FKBP3 decreases
oxaliplatin resistance in CRC cells and regulates the PTEN/AKT
pathway. CRC cells were treated with shNC+vector+40 µg/ml
oxaliplatin, shFKBP3+40 µg/ml oxaliplatin, or oeFKBP3+40 µg/ml
oxaliplatin for 48 h. (A) After incubation with Annexin V-FITC/PI,
the percentage of apoptotic cells was evaluated by FCM analysis. (B
and C) The levels of HDAC2, PTEN, P-gp, full-length caspase-3,
cleaved caspase-3, p-AKT and AKT were detected by western blot
analysis. All data are expressed as mean ± SD of three experiments.
**P<0.01, ***P<0.001 compared to shNC+vector. CRC, colorectal
cancer; FKBP3, FK506-binding protein 3; HDAC2, histone deacetylase
2; PTEN, phosphatase and tensin homolog deleted on chromosome 10;
P-gp, P-glycoprotein. |
FKBP3-induced resistance to
oxaliplatin in CRC cells is mediated by HDAC2
Next, we knocked down the expression HDAC2 in CRC
cells (Fig. 5A) and found that
downregulation of HDAC2 also significantly increased apoptosis in
oxaliplatin-resistant primary CRC cells (Fig. 5B), accompanied by decreased expression
of P-gp and p-AKT, and increased expression of PTEN and cleaved
caspase-3 (Fig. 5C and D), and
unchanged full-length caspase-3, which was similar to FKBP3
downregulation. In addition, FKBP3-induced inhibition of apoptosis
in oxaliplatin-treated CRC cells was significantly counteracted by
HDAC2 downregulation. These results further revealed that
downregulation of FKBP3 can attenuate the resistance of CRC cells
to oxaliplatin by reducing HDAC2 expression.
 | Figure 5.FKBP3-induced resistance to
oxaliplatin in CRC cells is mediated by HDAC2. CRC cells were
treated with shNC+vector+40 µg/ml oxaliplatin, oeFKBP3+40 µg/ml
oxaliplatin, shHDAC2+40 µg/ml oxaliplatin, or oeFKBP3+shHDAC2+40
µg/ml oxaliplatin for 48 h. (A) The expression of HDAC2 mRNA and
protein was detected. (B) After incubation with Annexin V-FITC/PI,
the percentage of apoptotic cells was evaluated by FCM analysis. (C
and D) The levels of HDAC2, PTEN, P-gp, full-length caspase-3,
cleaved caspase-3, p-AKT and AKT were detected by western blot
analysis. All data are shown as mean ± SD of three experiments.
**P<0.01, ***P<0.001 compared to shNC or shNC+vector;
###P<0.001 compared to oeFKBP3. CRC, colorectal
cancer; FKBP3, FK506-binding protein 3; HDAC2, histone deacetylase
2; PTEN, phosphatase and tensin homolog deleted on chromosome 10;
P-gp, P-glycoprotein. |
Discussion
Studies have demonstrated that several members of
the FK506-binding proteins (FKBPs) such as FKBP38, FKBP52 and
FKBP65 potentially play an important role in the tumor progression
and its acquisition of chemoresistance (33,34). For
example, FKBP38, a noncanonical member of the FKBP family,
contributes to tumorigenesis and chemoresistance by interacting
with B-cell lymphoma-2 (Bcl-2) (34).
In the present study, elevated expression of FKBP3 and histone
deacetylases 2 (HDAC2) was observed in colorectal cancer (CRC)
tissues. Primary CRC cells with high FKBP3 and HDAC2 expression
were insensitive to oxaliplatin, suggesting that oxaliplatin
resistance in CRC cells may be positively associated with FKBP3 and
HDAC2 expression. Furthermore, we showed that downregulation of
FKBP3 or HDAC2 significantly increased the sensitivity of CRC cells
to oxaliplatin by promoting apoptosis, which is in agreement with
previous reports (15,16,33,34),
suggesting that FKBP3 and HDAC2 may be involved in chemotherapy
resistance in primary CRC cells.
We also explored the underlying mechanisms by which
FKBP3 downregulation attenuates the resistance of CRC cells to
oxaliplatin. It has previously been reported that FKBP3 is
correlated with the activity of HDAC2 (18), which modulates histone acetylation,
thus regulating the expression of apoptosis-related genes (35,36).
Consistent with these reports, our study found that downregulation
of FKBP3 in CRC cells markedly decreased HDAC2 and P-glycoprotein
(P-gp) expression, and increased expression of pro-apoptotic
molecule, cleaved caspase-3, whereas FKBP3 upregulation had an
opposite effect. This suggests that HDAC2 is a possible downstream
effector of FKBP3 and may be involved in the carcinogenicity and
drug-resistant capabilities of FKBP3 in CRC. In addition,
downregulation of HDAC2 significantly increased the sensitivity of
primary CRC cells to oxaliplatin by promoting apoptosis,
accompanied by decreased expression of P-gp and p-AKT, and
increased expression of phosphatase and tensin homolog deleted on
chromosome 10 (PTEN) and cleaved caspase-3. Furthermore, HDAC2
downregulation significantly attenuated the effects of FKBP3
upregulation, providing further evidence that FKBP3 regulates the
resistance of CRC cells to oxaliplatin, possibly through modulation
of HDAC2 expression. It has been reported that FKBP3 functionally
associates with histone deacetylases, such as HDAC1 and HDAC2, and
through the recruitment of HDAC1 or HDAC2, FKBP3 could free up
acetylated lysine residues in mouse double minute 2 homolog (MDM2),
making them available for auto-ubiquitylation (18,37).
Finally, we found that downregulation of FKBP3 significantly
decreased AKT phosphorylation and increased PTEN expression in
oxaliplatin-resistant CRC cells. A recent study has shown that the
PI3K/AKT pathway is associated with chemotherapy resistance, mainly
through evasion of cellular apoptosis (38). PTEN can negatively regulate the
PI3K/AKT pathway by dephosphorylating PIP3 to PIP2, preventing all
downstream signaling events that are regulated by AKT (39,40). It is
well-known that the PI3K/AKT pathway is involved in the regulation
of cell proliferation and survival, and constitutively active AKT
can protect cells from apoptosis by suppressing caspase-3, as well
as decreasing the sensitivity of tumor cells to pro-apoptotic
agents (38,41). AKT signaling was also discovered to be
appreciably activated in tumors and functions in drug resistance
(42). Therefore, we inferred that
FKBP3 could regulate the PTEN/AKT pathway, and that the PTEN/AKT
pathway may be involved in FKBP3 regulation of chemoresistance in
CRC cells; however further experiments are needed to definitively
address this conclusion. For example, after inhibition or
activation of PTEN/AKT, FCM should be used to analyze the apoptosis
in FKBP3-overexpressing or -silenced CRC cells, as well as to
detect related proteins by western blotting. There are also
limitations in the present study; for example, the lack of time
points of oxaliplatin treatment, reference of shFKBP3 interference
sequence, as well as the lack of experiments for supporting our
hypothesis. If possible in the future, several types of shFKBP3
targeting different sequences will be tested to select shRNAs with
optimal interference effect, and more time points of oxaliplatin
treatment should be tested in the future to make our research more
credible. Moreover, an inhibitor of PETN/AKT can be applied to
further study the mechanisms of FKBP3, HDAC2 and the PTEN/AKT
pathway in CRC.
In conclusion, the present study demonstrated the
critically important role of FKBP3 and HDAC2 in the resistance of
CRC cells to oxaliplatin. Downregulation of FKBP3 can attenuate the
resistance of CRC cells to oxaliplatin by reducing HDAC2
expression. Moreover, FKBP3 can possibly regulate the PTEN/AKT
pathway, and the PTEN/AKT pathway may be involved in FKBP3
regulation of chemoresistance in CRC cells. Importantly, our study
suggests the possible clinical value and potential prognosis of
FKBP3 and HDAC2 in chemotherapeutic resistance of CRC, which may
contribute to CRC treatment.
Acknowledgements
Not applicable.
Funding
This study was funded by the Clinical Study on Chang
Ji Tai Combined with Auricular Acupuncture in Treating
Postoperative Cancer-Related Fatigue of Postoperative Colorectal
Cancer, Special Item of Shanghai Integrative Medicine [ZY
(2018–2020)-FWTX-3016].
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
ZZ and LH conceived and designed the study. JT, YS,
XC, RW, YH and XZ performed the experiments, and collected and
analyzed the data. ZZ and LH wrote the manuscript. All authors read
and approved the final manuscript and agree to be accountable for
all aspects of the research in ensuring that the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
All experiments conducted in this study were
approved by the Ethics Committee of Shanghai Municipal Hospital of
Traditional Chinese Medicine Affiliated to Shanghai TCM University
(Shanghai, China), and written informed consent was obtained.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ung L, Lam KY, Morris DL and Chua TC:
Tissue-based biomarkers predicting outcomes in metastatic
colorectal cancer: a review. Clin Transl Oncol. 16:425–435. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sostres C, Gargallo CJ and Lanas A:
Aspirin, cyclooxygenase inhibition and colorectal cancer. World J
Gastrointest Pharmacol Ther. 5:40–49. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Overman MJ, Morris V, Moinova H, Manyam G,
Ensor J, Lee MS, Eng C, Kee B, Fogelman D, Shroff RT, et al: Phase
I/II study of azacitidine and capecitabine/oxaliplatin (CAPOX) in
refractory CIMP-high metastatic colorectal cancer: Evaluation of
circulating methylated vimentin. Oncotarget. 7:67495–67506. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Meyerhardt JA and Mayer RJ: Systemic
therapy for colorectal cancer. N Engl J Med. 352:476–487. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kelland L: The resurgence of
platinum-based cancer chemotherapy. Nat Rev Cancer. 7:573–584.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Koul D, Shen R, Bergh S, Sheng X,
Shishodia S, Lafortune TA, Lu Y, de Groot JF, Mills GB and Yung WK:
Inhibition of Akt survival pathway by a small-molecule inhibitor in
human glioblastoma. Mol Cancer Ther. 5:637–644. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xiong F and Chen J: Effect of miR-3651 on
proliferation, apoptosis and expression of PTEN in colon cancer
cells. Chin Clin Oncol. 22:865–868. 2017.
|
|
10
|
Liang YF, Jian-Bo R, Wang LM, Chen C, Kang
DP, Lin BH, Chai XX, Zeng JC and Pathology DO: Expression and
clinical significance of PTEN in colon cancer. China Tropical Med.
15:966–969. 2015.
|
|
11
|
He C, Dong X, Zhai B, Jiang X, Dong D, Li
B, Jiang H, Xu S and Sun X: MiR-21 mediates sorafenib resistance of
hepatocellular carcinoma cells by inhibiting autophagy via the
PTEN/Akt pathway. Oncotarget. 6:28867–28881. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li J, Zhang Y, Zhao J, Kong F and Chen Y:
Overexpression of miR-22 reverses paclitaxel-induced
chemoresistance through activation of PTEN signaling in p53-mutated
colon cancer cells. Mol Cell Biochem. 357:31–38. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Solassol J, Mange A and Maudelonde T: FKBP
family proteins as promising new biomarkers for cancer. Curr Opin
Pharmacol. 11:320–325. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mukaide H, Adachi Y, Taketani S, Iwasaki
M, Koi-Kekiriyama N, Shigematsu A, Shi M, Yanai S, Yoshioka K,
Kamiyama Y and Ikehara S: FKBP51 expressed by both normal
epithelial cells and adenocarcinoma of colon suppresses
proliferation of colorectal adenocarcinoma. Cancer Invest.
26:385–390. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li L, Lou Z and Wang L: The role of FKBP5
in cancer aetiology and chemoresistance. Br J Cancer. 104:19–23.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Luo K, Li Y, Yin Y, Lei L, Wu C, Chen Y,
Nowsheen S, Qi H, Zhang L, Lou Z and Yuan J: USP49 negatively
regulates tumorigenesis and chemoresistance through FKBP51-AKT
signaling. EMBO J. 36:1434–1446. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ochocka AM, Kampanis P, Nicol S,
Allende-Vega N, Cox M, Marcar L, Milne D, Fuller-Pace F and Meek D:
FKBP25, a novel regulator of the p53 pathway, induces the
degradation of MDM2 and activation of p53. FEBS Lett. 583:621–626.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang WM, Yao YL and Seto E: The
FK506-binding protein 25 functionally associates with histone
deacetylases and with transcription factor YY1. EMBO J.
20:4814–4825. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Oiso H, Furukawa N, Suefuji M, Shimoda S,
Ito A, Furumai R, Nakagawa J, Yoshida M, Nishino N and Araki E: The
role of class I histone deacetylase (HDAC) on gluconeogenesis in
liver. Biochem Biophys Res Commun. 404:166–172. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jeong JB and Lee SH: Protocatechualdehyde
possesses anti- cancer activity through downregulating cyclin D1
and HDAC2 in human colorectal cancer cells. Biochem Biophys Res
Commun. 430:381–386. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang H, Zhao B, Huang C, Meng XM, Bian EB
and Li J: Melittin restores PTEN expression by down-regulating
HDAC2 in human hepatocelluar carcinoma HepG2 cells. PLoS One.
9:e955202014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhu W, Li Z, Xiong L, Yu X, Chen X and Lin
Q: FKBP3 promotes proliferation of non-small cell lung cancer cells
through regulating Sp1/HDAC2/p27. Theranostics. 7:3078–3089. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hong JY, Kang B, Kim A, Hwang S, Ahn J,
Lee S, Kim J, Park JH and Cheon DS: Development of a highly
sensitive real-time one step RT-PCR combined complementary locked
primer technology and conjugated minor groove binder probe. Virol
J. 8:3302011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xiong H, Hong J, Du W, Lin YW, Ren LL,
Wang YC, Su WY, Wang JL, Cui Y, Wang ZH and Fang JY: Roles of STAT3
and ZEB1 proteins in E-cadherin down-regulation and human
colorectal cancer epithelial-mesenchymal transition. J Biol Chem.
287:5819–5832. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Virag P, Brie I, Fischer-Fodor E,
Perde-Schrepler M, Tatomir C, Balacescu O, Irimie A and Postescu
ID: Assessment of cytotoxicity, apoptosis and DNA damages in
Colo320 colorectal cancer cells selected for oxaliplatin
resistance. Cell Biochem Funct. 29:351–355. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cohen GM: Caspases: The executioners of
apoptosis. Biochem J. 326:1–16. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gao G and Dou QP: G(1) phase-dependent
expression of bcl-2 mRNA and protein correlates with
chemoresistance of human cancer cells. Mol Pharmacol. 58:1001–1010.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee MS, Jeong MH, Lee HW, Han HJ, Ko A,
Hewitt SM, Kim JH, Chun KH, Chung JY, Lee C, et al: PI3K/AKT
activation induces PTEN ubiquitination and destabilization
accelerating tumourigenesis. Nat Commun. 6:77692015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Salmena L, Carracedo A and Pandolfi PP:
Tenets of PTEN tumor suppression. Cell. 133:403–414. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Davies MA, Lu Y, Sano T, Fang X, Tang P,
Lapushin R, Koul D, Bookstein R, Stokoe D, Yung WK, et al:
Adenoviral transgene expression of MMAC/PTEN in human glioma cells
inhibits akt activation and induces anoikis. Cancer Res.
58:5285–5290. 1998.PubMed/NCBI
|
|
32
|
Mehrian-Shai R, Chen CD, Shi T, Horvath S,
Nelson SF, Reichardt JK and Sawyers CL: Insulin growth
factor-binding protein 2 is a candidate biomarker for PTEN status
and PI3K/Akt pathway activation in glioblastoma and prostate
cancer. Proc Natl Acad Sci USA. 104:5563–5568. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Choi BH and Yoon HS: FKBP38-Bcl-2
interaction: A novel link to chemoresistance. Curr Opin Pharmacol.
11:354–359. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ma D, Bai X, Zou H, Lai Y and Jiang Y:
Rheb GTPase controls apoptosis by regulating interaction of FKBP38
with Bcl-2 and Bcl-XL. J Biol Chem. 285:8621–8627. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Krämer OH, Knauer SK, Zimmermann D,
Stauber RH and Heinzel T: Histone deacetylase inhibitors and
hydroxyurea modulate the cell cycle and cooperatively induce
apoptosis. Oncogene. 27:732–740. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jung KH, Noh JH, Kim JK, Eun JW, Bae HJ,
Xie HJ, Chang YG, Kim MG, Park H, Lee JY and Nam SW: HDAC2
overexpression confers oncogenic potential to human lung cancer
cells by deregulating expression of apoptosis and cell cycle
proteins. J Cell Biochem. 113:2167–2177. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang X, Taplick J, Geva N and Oren M:
Inhibition of p53 degradation by Mdm2 acetylation. FEBS Lett.
561:195–201. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fresno Vara JA, Casado E, de Castro J,
Cejas P, Belda-Iniesta C and González-Barón M: PI3K/Akt signalling
pathway and cancer. Cancer Treat Rev. 30:193–204. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gan YH and Zhang S: PTEN/AKT pathway
involved in histone deacetylases inhibitor induced cell growth
inhibition and apoptosis of oral squamous cell carcinoma cells.
Oral Oncol. 45:e150–e154. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Song MS, Salmena L and Pandolfi PP: The
functions and regulation of the PTEN tumour suppressor. Nat Rev Mol
Cell Biol. 13:283–296. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
van Echten-Deckert G, Zschoche A, Bär T,
Schmidt RR, Raths A, Heinemann T and Sandhoff K:
cis-4-Methylsphingosine decreases sphingolipid biosynthesis by
specifically interfering with serine palmitoyltransferase activity
in primary cultured neurons. J Biol Chem. 272:15825–15833. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Nemoto S, Nakamura M, Osawa Y, Kono S,
Itoh Y, Okano Y, Murate T, Hara A, Ueda H, Nozawa Y and Banno Y:
Sphingosine kinase isoforms regulate oxaliplatin sensitivity of
human colon cancer cells through ceramide accumulation and Akt
activation. J Biol Chem. 284:10422–10432. 2009. View Article : Google Scholar : PubMed/NCBI
|