Introduction
It is well established that a variety of protein
kinases induce cell transformation by phosphorylating downstream
protein substrates. For example, Aurora-A is a Serine/Threonine
(Ser/Thr) kinase and a widely known oncoprotein associated with the
development of numerous human cancers. Aurora-A transforms cells
(1–3)
by phosphorylating a wide range of downstream substrates (4,5). For
example, Aurora-A phosphorylates hepatoma upregulated protein
(HURP) at four serine residues (6,7). This
process relays the cell transforming activity from Aurora-A to HURP
(6,7).
In contrast to these findings, protein phosphatases,
which remove phosphate groups from protein substrates result in the
prevention or termination of kinase-induced cell proliferative
signals and therefore are usually considered as tumor suppressors
(8). However, the role of the Ser/Thr
phosphatase protein phosphatase 1α (PP1α) in tumor progression
remains undetermined. PP1α has been revealed to prevent oncogenic
transformation in NIH3T3 fibroblasts (9). PP1α activity is detrimental to cell
growth by causing activation of Rb-dependent G1 arrest in bone
osteosarcoma cells (10,11) and induction of apoptosis in T and
fibroblast cells (12–15). In contrast to these observations, PP1α
is overexpressed in ascites hepatoma (16–18),
prostate cancer (19), diffuse large
B-cell lymphoma (20), and oral
squamous cell carcinoma cell lines (21). These studies seemingly indicate a
differential influence of PP1α on different cancer types. Moreover,
the contribution of PP1α to lung cancer remains ambiguous. For
example, PP1α was revealed to be involved in the RIF1-induced tumor
growth of the lung cancer cell line H1299 (22), while overexpression of PP1α in H1299
cells restricted tumor formation in vivo (23). Furthermore, downregulation or
inactivation of PP1α was revealed to be associated with poor
prognosis (24) and radioresistance
(25) of lung cancer cells.
Numerous signaling cascades are regulated from
protein kinases or protein phosphatases during cancer formation.
The MAPK pathway and the AKT cascade are frequently altered in
human cancer types (26,27). The mitogen activated protein kinase
(MAPK) superfamily consists of the ERK, p38 and Jun N-terminal
kinase (JNK) proteins, and functions by transmitting upstream
signals to its downstream effectors, thereby regulating several
aspects of cancer development (26,28). ERK
and p38 are engaged in oncogenic processes, whereas JNK
demonstrates an oncosuppressive role. Furthermore, AKT is activated
by PI3K/PDK1 and inactivated by PTEN. The activation of AKT leads
to the activation and/or the inactivation of a variety of
downstream effectors, such as cyclin D1, and GSK3β or c-Raf,
respectively (27,29–31).
To explore the role of PP1α in lung cancer and
determine whether PP1α contributes to cell proliferation or
transformation according to each tissue type, it was examined by
literature review whether the DNA region covering the PP1α
locus was subjected to frequent alterations, such as gain or loss
in various types of cancer. Certain studies demonstrated that PP1α
was upregulated in lung cancer tissues, and that it was required
for the cell transforming activity of the lung cancer cell line
A549 by activating the AKT signaling pathway. However, PP1α
inhibited 293T cell proliferation by regulating the JNK and AKT
cascades. Collectively, the data indicated that PP1α contributes to
lung cancer formation and that it exhibits differential
growth-stimulating effects in different cell types via distinct
mechanisms of action.
Materials and methods
Chemicals, antibodies, plasmids and
shRNAs
Fetal bovine serum (FBS), Dulbecco's modified
Eagle's medium (DMEM), penicillin, streptomycin, and Lipofectamine™
were purchased from GIBCO-BRL; Thermo Fisher Scientific, Inc. The
catalog numbers and suppliers for the antibodies used in the
present study are as follows: PP1α (cat. no. sc-271762), GFP (cat.
no. sc-9996), tubulin (cat. no. sc-5286), actin (cat. no. sc-8432)
and NF-κB (cat. no. sc-8008; all from Santa Cruz Biotechnology,
Inc.); cyclin E1 (HPA018169; Sigma-Aldrich; Merck KGaA); GAPDH
(cat. no. sc-32233; Santa Cruz Biotechnology, Inc.); phospho-AKT
pathway sampler kit, (AKT, AKT p308, AKT p473, phospho-c-Raf,
phospho-GSK3β, phospho-PTEN, phospho-PDK1; cat. no. 9916), MAPK
family sampler kit, (ERK, p38, JNK; cat. no. 9926), and
phospho-MAPK family antibody sampler kit, (phospho-ERK,
phospho-p38, phospho-JNK; cat. no. 9910; all from Cell Signaling
Technology, Inc.); GSK3β (cat. no. sc-53931), c-Raf (cat. no.
sc-52827) and PTEN (cat. no. sc-7974; all from Santa Cruz
Biotechnology, Inc.). The antibodies against PP1α, tubulin, and
actin, were purchased from Santa Cruz Biotechnology, Inc., and the
antibodies against AKT or MAPK pathways were purchased from Cell
Signaling Technology, Inc. The p-HURP antibodies were generated by
immunizing rabbits with synthesized peptides containing phospho-Ser
as follows: p627, VKLFSGLSVSSEGP; p725, CLSSERMSLPLLA; p757,
EGMELNSSITSQDV; p830, EHARH ISFGGNLI. All the antisera were further
purified by antigenic peptide-bound column. EGFP-PP1α was obtained
from Dr Monique Beullens (51). The
shRNAs used in the study were obtained from the National RNAi Core
Facility at Academia Sinica in Taiwan and the targeting sequence
for LacZ and PP1α was CGCGATCGTAATCACCCGAGT and
TGAGTGCAAGAGACGCTACAA respectively.
Tissue procurement
The study protocol in terms of the collection of the
biopsies of patients was approved by the Ethics Committee at
Taichung Veterans General Hospital. No patient had previously
received any neoadjuvant treatment such as chemotherapy before
surgery. Patients (31) were
recruited from 2004/4/7 to 2005/12/28 in the study with males
accounting for 71%, and a mean age of 65 ranging from 41 to 87. All
patients provided informed consent and signed the consent form
individually. The study samples were obtained after surgery from a
non-necrotic area of the tumor and from adjacent non-tumorous
tissue from neighboring sites outside the tumor. Both tumor and
adjacent non-tumor tissues were confirmed by pathologists. The
tissue samples were placed immediately in cryovials, frozen in
liquid nitrogen, and stored at −80°C until analysis by western
blotting.
Cell culture
The cell lines used in this study were purchased
from the American Type Culture Collection. The culture medium for
293T cells was Dulbecco/Vogt Modified Eagle's Minimal Essential
Medium with 5% fetal bovine serum, and for A549 cells it was
Roswell Park Memorial Institute (RPMI)-1640 medium with 10% fetal
bovine serum, 1% nonessential amino acids, and 1% sodium pyruvate.
Moreover, 2 mM glutamine, 100 U/ml penicillin, and 100 g/ml
streptomycin were added in all media. All cell culture-related
reagents were purchased from Invitrogen; Gibco; Life Technologies;
Thermo Fisher Scientific, Inc. Cells were maintained in a
humidified incubator at 37°C in the presence of 5%
CO2.
Single cell proliferation assay
The assay was based on our previous study (22). Briefly, cells were seeded in 24-well
plates and transfected with desired constructs tagged with EGFP.
The following day, the cells were replated with low density to
10-cm dishes, to prevent cell contact. The cells were allowed to
proliferate for 1–4 days, and the percentage of cells with
proliferation judged by formation of ‘mini-colonies’ with cell
numbers ≥2 was counted. If cells could not proliferate, they
remained single in the 10-cm dish after 36-h culture.
Colony formation assay
Cells (2×103) were seeded in 10-cm dishes
and cultured for 10 days. The cell foci were fixed with methanol
and stained with 5% Giemsa solution. The focus number was then
counted.
PolyHEMA-based anchorage-independent
growth
PolyHEMA (2.5 mg/ml) was added to 6-cm dishes and
heated on a hotplate until most of the solution evaporated. The
coated dishes were sterilized under UV irradiation before use.
Cells (5×105) were then seeded onto the dishes and
allowed attachment and proliferation for 2 days. The cells were
trypsinized and counted with a hemocytometer.
Migration wound healing assays
Cells with full confluence were incubated with
serum-free medium for overnight. The cells were then scraped with a
20-µl gel-loading tip to create an empty space which allows for
cell migration. Serum containing the medium was then added to cells
and the migrated cells were photographed at 0, 3, 6 and 9 h with an
CCD camera (Olympus, Model DP71) attached to an inverted microscope
(Olympus Optical Co., Ltd., Tokyo, Japan). The cells with migration
were counted.
Preparation of cell extracts and
western blot analysis
To prepare the cell-free extracts, cells were lysed
in extraction buffer (20 mM PIPES, pH 7.2, 100 mM NaCl, 1 mM EDTA,
0.1% CHAPS, 10% sucrose, 1 mM PMSF, 1 mM DTT, 1 mM Na3VO4, and 10
µg/ml each of leupeptin, aprotinin, chymostatin, and pepstatin).
After incubation at 4°C for 30 min, cellular debris was removed by
centrifugation at 15,856 × g for 90 min in an Eppendorf centrifuge.
Protein concentrations were determined using the Bradford assay
(Bio-Rad Laboratories, Inc.). The resulting samples were heated at
95°C for 10 min and loaded onto a 10% SDS-polyacrylamide
electrophoresis gel (SDS-PAGE) and then transferred onto a
polyvinylidene difluoride membrane (PVDF; EMD Millipore). The PVDF
membrane was then blocked with 5% bovine serum albumin (BSA)/PBST
(0.1% Tween-20 in PBS). Various antibodies were incubated with the
membranes at 4°C for overnight. The membranes were washed with PBST
at room temperature for 30 min and repeated for 3 times. Secondary
antibodies against mouse (cat. no. sc-2005) or rabbit (cat. no.
sc-2004) IgG conjugated with horseradish peroxidase (Santa Cruz
Biotechnology, Inc.) were added for 1 h at room temperature
followed by washing with PBST for 3×30 min. NBT and BCIP substrates
(Zymed Laboratories, Inc.) were added to develop the membrane. The
protein level was assessed using the software (Gel-Pro analyzer
4.5), and the signal intensity of the protein of interest was
normalized against that of actin, tubulin, or GAPDH. When comparing
the changes of protein between the control and experimental groups,
a relative level of each protein of interest was used, where
[protein of interest/actin]experimental was normalized
with [protein of interest/actin]control.
Immunoprecipitation
One milligram of cell extracts with protease
inhibitors were incubated with protein A/G beads in 500 µl
immunoprecipitation washing buffer (50 mM HEPES, pH 7.6, 2 mM
MgCl2, 50 mM NaCl, 5 mM EGTA, 0.1% Triton X-100, 40 mM
glycerolphosphate) at 4°C, for 1 h, to preabsorb unwanted proteins.
Antibodies in the amount of 1 µg were then added to the cell
extracts for 4 h at 4°C. The cell extracts were then incubated with
protein A/G-beads for 1 h, followed by 6 washes with PBS for 3 h at
4°C. The resulting samples were heated at 95°C for 10 min and
applied to SDS-PAGE-based electrophoresis and western blotting.
Statistical analysis
All data were collected from three independent
experiments and presented as the mean ± SD. Statistical
significance determined by independent samples Student's t-tests
were represented as P<0.05, P<0.01, and P<0.001.
Results
PP1α is upregulated in lung cancer
tissues
In order to examine the potential involvement of
PP1α in cancer formation, we initially discovered that the DNA
region covering the PP1α locus, i.e. 11q13.2, was
subjected to DNA alteration in various types of human cancer
(Table I). The majority of these
alterations were DNA amplifications. For example, in the digestive
system the following cancer types were present: Esophageal squamous
cell carcinoma, hepatocellular carcinoma and colorectal cancer,
whereas in the immune system, myeloid leukemia and plasmablastic
lymphoma were noted. Moreover, PP1α protein levels were increased
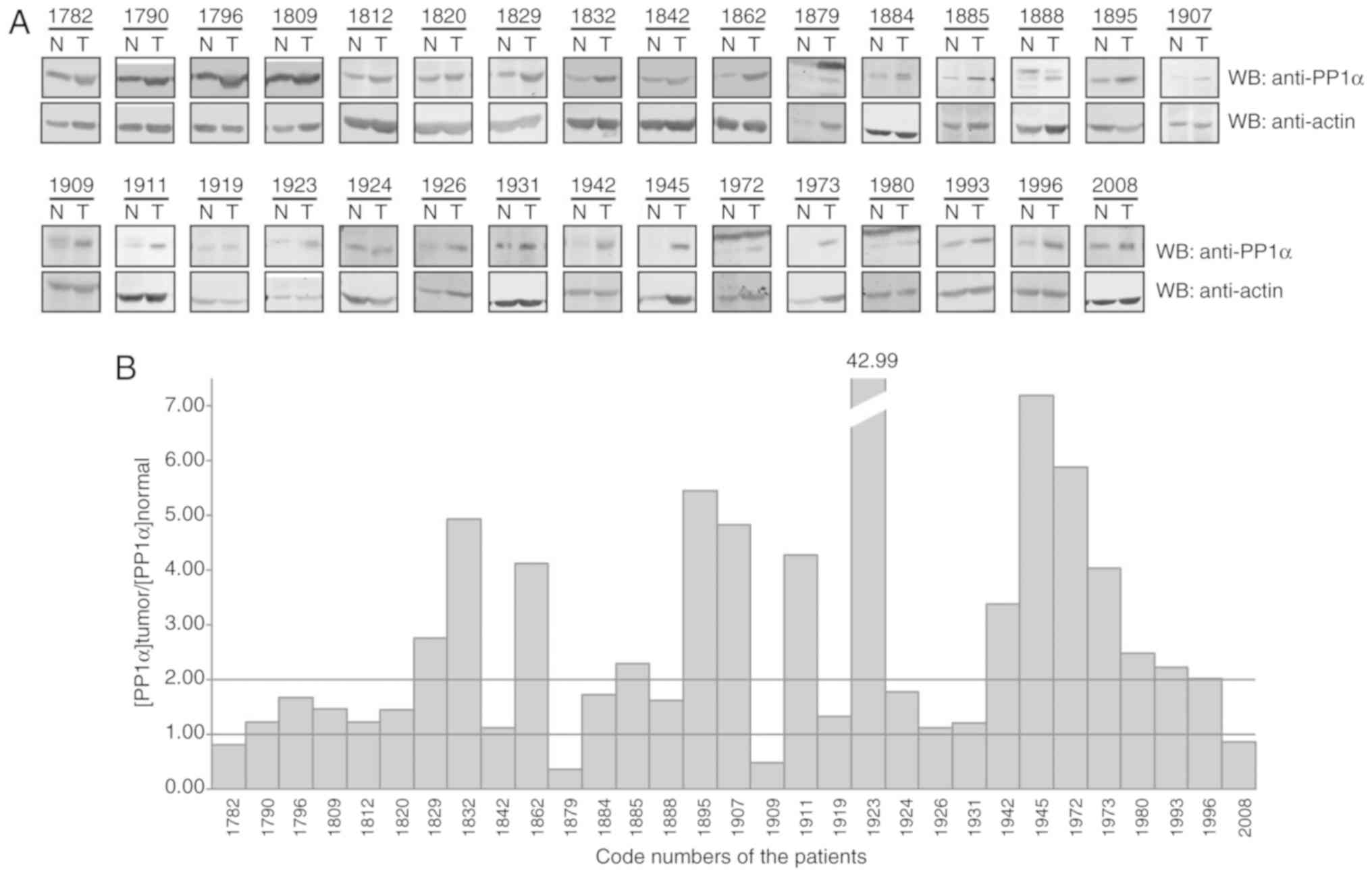
in lung cancer. At least, a 2-fold increase in PP1α protein levels
was observed in 15 out of 31 lung cancer pairs of cancerous and
adjacent normal tissues (Fig. 1).
These findings indicated that deregulation of PP1α occurred
concomitantly with cancer formation.
 | Table I.The DNA alteration covering PP1α
locus 11q13.2 in various cancer tissues. |
Table I.
The DNA alteration covering PP1α
locus 11q13.2 in various cancer tissues.
| Altered DNA
region | Cancer types | DNA alteration | (Refs.) |
|---|
| llql3.2 | esophageal squamous
cell carcinoma | amplification | (32) |
| llql3.2 | esophageal squamous
cell carcinoma | amplification | (33) |
| llql3.2 | esophageal squamous
cell carcinoma | amplification | (34) |
| llql3.2 | intrahepatic
cholangiocarcinoma | amplification | (43) |
| llql3.2 | hepatocellular
carcinoma | amplification | (44) |
| llql3.2 | colorectal
cancer | amplification | (49) |
| llql3.2 | myeloid
leukemia | amplification | (37) |
| llql3.2 | plasmablastic
lymphoma | amplification | (46) |
| llql3.2 | myeloid
leukemia | amplification | (47) |
| llql3.2 | ovanan cancer | amplification | (35) |
| llql3.2 | ovanan cancer | deletion | (36) |
| llql3.2 | prostate
cancer | amplification | (38) |
| llql3.2 | prostate
cancer | amplification | (39) |
| llql3.2 | breast cancer | amplification | (40) |
| llql3.2 | breast
carcinoma | deletion | (41) |
| llql3.2 | invasive lobular
carcinoma | amplification | (45) |
| llql3.2 | urothelial
carcinoma | amplification | (42) |
| llql3.2 | peritoneal
mesothelioma | deletion | (48) |
| llql3.2 | skin cancer | amplification | (50) |
PP1α is required for the
transformation of the A549 lung cancer cell line
To explore the role of PP1α in lung cancer
development, an ectopic overexpression model of PP1α was initially
performed in A549 cells, and it was further demonstrated that PP1α
could promote cell proliferation (Fig.
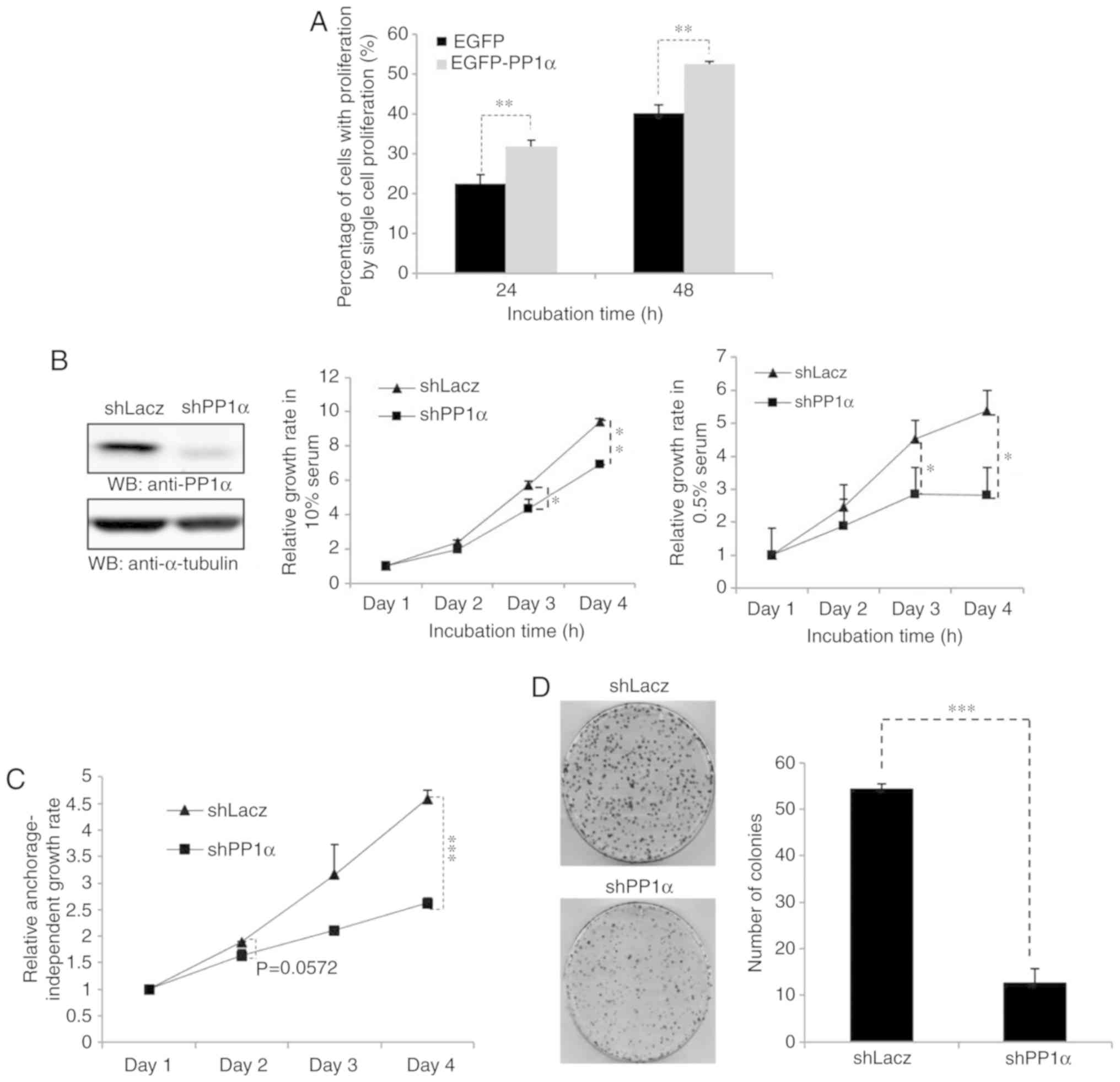
2A). Secondly, PP1α expression was knocked down in A549 cells
and the PP1α-depleted cells exhibited a considerably slower rate
under normal or low serum conditions (Fig. 2B). Low growth was also observed in the
presence of a cellular environment that would not allow strong
attachment of the cells (Fig. 2C) and
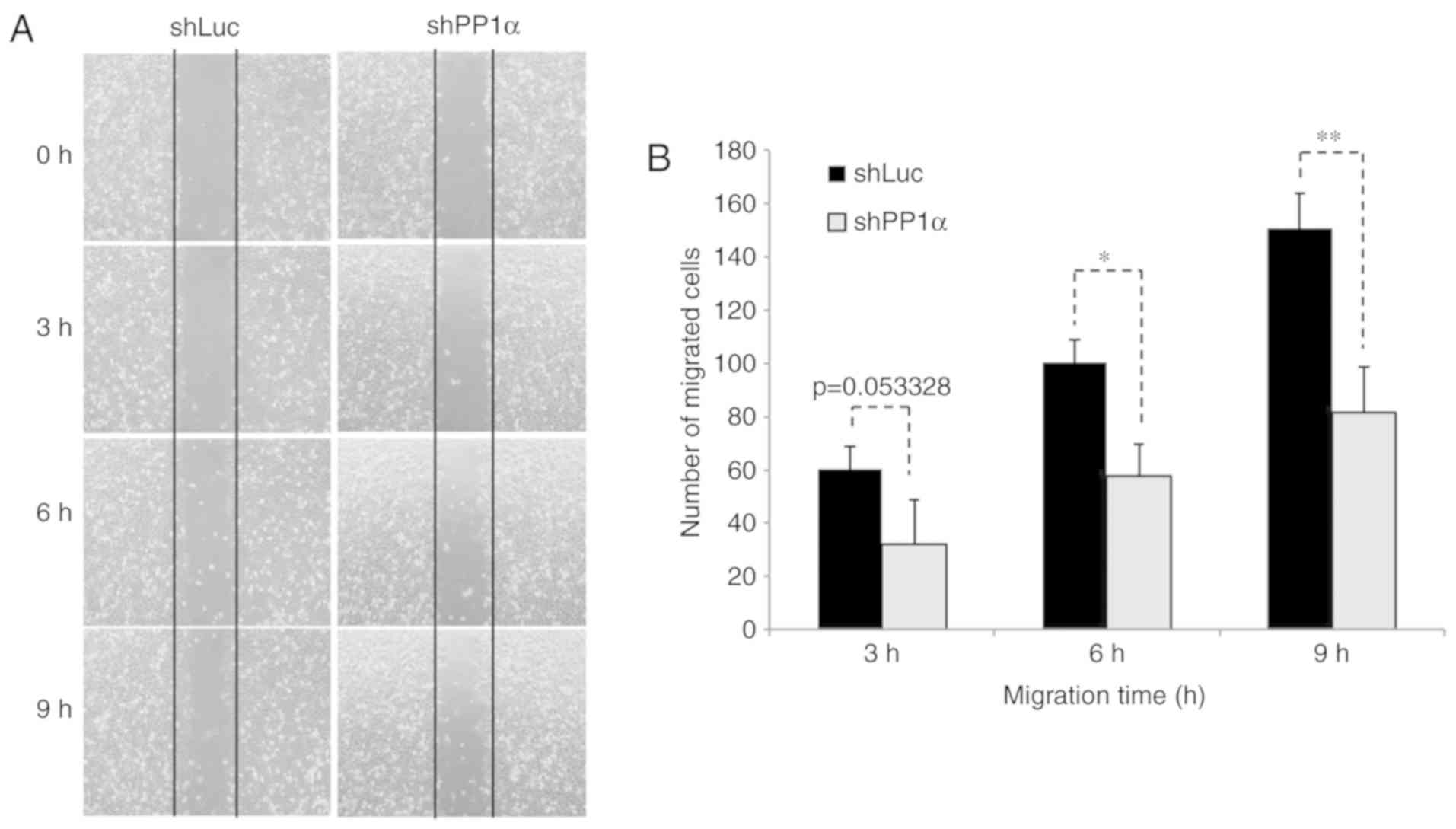
in the presence of a low number of cells (Fig. 2D). Moreover, A549 cells harboring a
PP1α shRNA sequence exhibited reduced migratory activity
(Fig. 3). All these observations
revealed that PP1α was a potential oncoprotein in lung cancer
cells.
PP1α activates the PDK1/AKT
pathway
To explore the underlying mechanisms by which PP1α
causes cell transformation, the expression levels of the proteins
involved in MAPK and/or the AKT signaling cascades were
investigated (Fig. 4). Although PP1α
did not affect the MAPK pathway, the protein phosphatase regulated
the AKT cascade. It was demonstrated that knockdown of PP1α
downregulated the levels of PDK1 and AKT p473, whereas it
upregulated the levels of p-Gsk3β and p-c-Raf.
PP1α inhibits cell proliferation of
293T cells
In addition to the amplification of the chromosomal
region nearby the PP1α locus 11q13.2, the data
further indicated that 11q13.2 was deleted in certain cancer
types of sex hormone-dependent tissues, such as ovarian and breast
cancer, and in other cancer types, such as peritoneal mesothelioma
(Table I). To examine whether PP1α
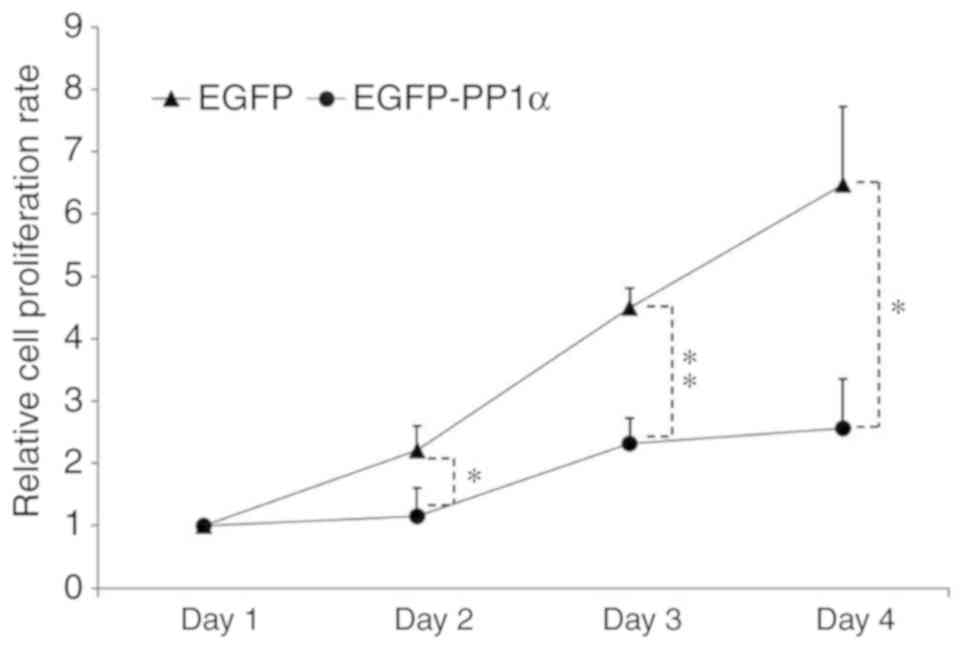
exerts a negative contribution to cell transformation, PP1α was
overexpressed in 293T cells and it was demonstrated that the cell
proliferative rate was largely impaired (Fig. 5).
PP1α regulates the dephosphorylation
of the Aurora-A downstream substrate HURP
To explore the mechanism by which PP1α retards cell
proliferation, we expanded our analysis on the finding that PP1α
acted against Aurora-A by dephosphorylating its substrates. The
previous studies conducted by our group established that Aurora-A
could phosphorylate HURP and promote cell transformation (6,7). Based on
these findings the potential involvement of HURP was investigated
in the PP1α-dependent inhibition of cell proliferation. It was
initially revealed that PP1α could interact with HURP (Fig. 6A). HURP is phosphorylated by Aurora-A
during mitosis (7). Therefore, it was
assessed whether the interaction of Aurora-A with PP1α could reduce
the phosphorylation levels of HURP. The data demonstrated that
overexpression of PP1α decreased the levels of the four p-HURP
isoforms catalyzed by Aurora-A in untreated cells or
nocodazole-induced mitosis (Fig. 6B),
whereas knockdown of PP1α exhibited the opposite effects (Fig. 6C). These isoforms were derived based
on the type of the amino acid residue that was phosphorylated each
time. It has been reported that the Aurora-A-induced
phosphorylation of HURP stimulates cell proliferation by
downregulating p-JNK (6). Therefore,
the expression levels of JNK were investigated and it was
demonstrated that PP1α significantly increased the levels of p-JNK
in 293T cells (Fig. 7A). Notably,
PP1α did not affect p-JNK levels in A549 cells (Fig. 4), while it reduced the levels of AKT
p473 in 293T cells (Fig. 7B),
revealing a differential action of PP1α in the two cell lines.
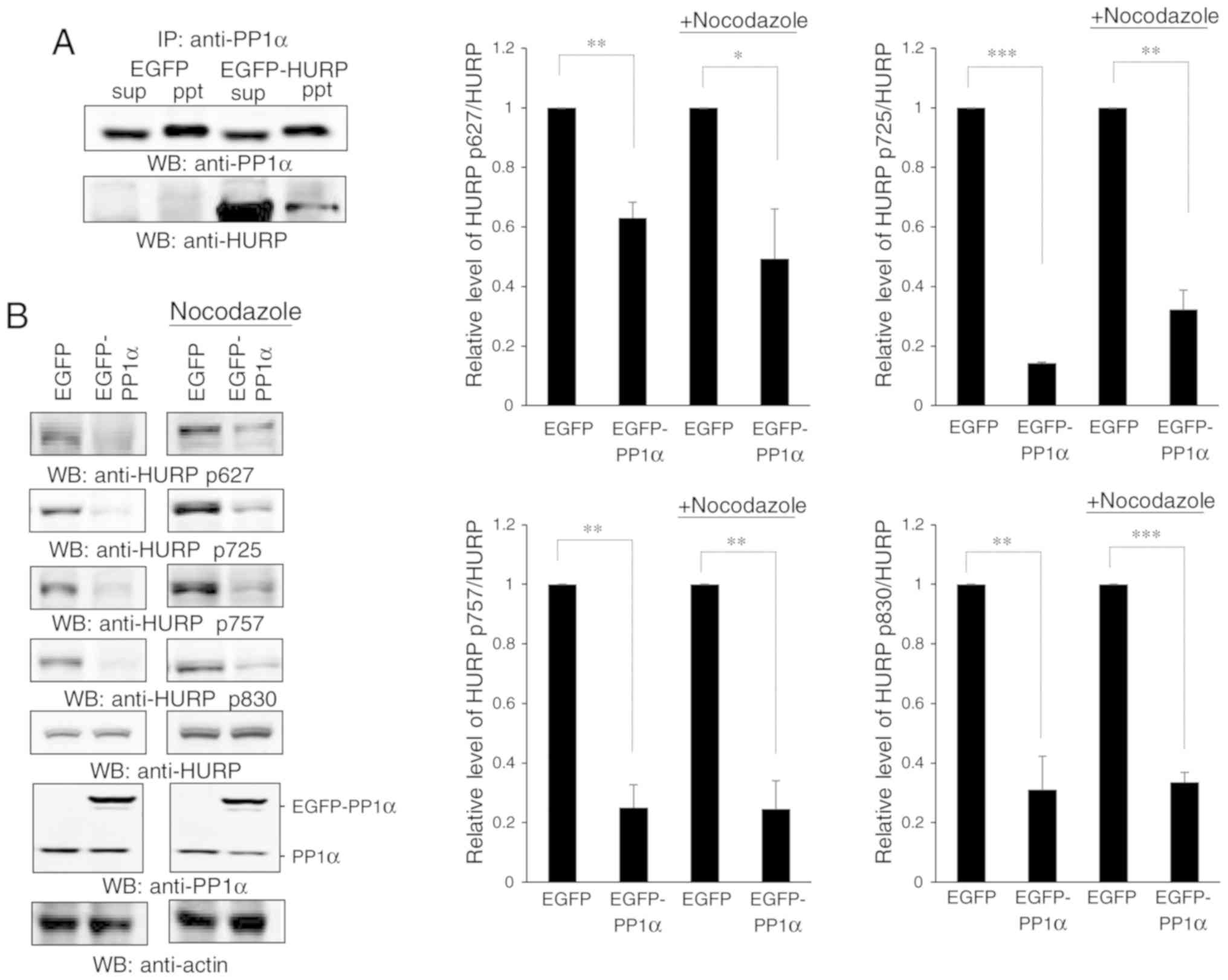 | Figure 6.PP1α interacts with HURP and controls
the dephosphorylation of HURP. (A) PP1α interacts with HURP.
Immunoprecipitation using an antibody against PP1α was conducted in
293T cells transfected with EGFP or EGFP-HURP. sup, supernatant;
ppt, pellet. (B) Overexpression of PP1α reduced the levels of
p-HURP. 293T cells transfected with EGFP or EGFP-PP1α were treated
with or without nocodazole and they were subsequently analyzed for
the expression levels of the four p-HURP isoforms at S627, S725,
S757 and S830 residues. The levels of each p-HURP isoform and HURP
in each treatment were measured, the ratio of p-HURP/HURP was
calculated, and
[p-HURP/HURP]EGFP-PP1α/[p-HURP/HURP]EGFP was
finally obtained and plotted. Since HURP may act at the mitotic
phase, cells were treated with nocodazole in order to examine
p-HURP levels during mitotic cell division. (C) Knockdown of PP1α
increased the levels of p-HURP. 293T cells harboring shLuc or
shPP1α were treated with or without nocodazole and subsequently
analyzed for the levels of various p-HURPs. The levels of each
p-HURP isoform and HURP in each treatment were measured, the ratio
of p-HURP/HURP was calculated, and
[p-HURP/HURP]EGFP-PP1α/[p-HURP/HURP]EGFP was
finally obtained and plotted. All data were collected from three
independent experiments with error bars representing the SD.
*P<0.05, **P<0.01 and ***P<0.001 represented significant
differences as determined using Student's t-test, respectively.
PP1α, protein phosphatase 1α; HURP, hepatoma upregulated
protein. |
Discussion
The chromosomal region covering PP1α is
subjected to DNA alterations in human cancers including
amplification and deletion, which reflects the distinct effect of
PP1α on the development of different cancer types. The present
study revealed that knockdown of PP1α attenuated the cell
transforming activity and inactivated the AKT signaling cascade.
This effect included the decrease in the p-AKT levels at the T473
residue, whereas the MAPK/JNK pathway was not affected in A549
cells. In contrast to these findings, overexpression of PP1α
inhibited cell proliferation, while enhancing the dephosphorylation
of HURP. Concomitantly, it inhibited p-JNK levels induced by p-HURP
and it increased the levels of p-AKT at the T473 residue in 293T
cells. Therefore, PP1α upregulated p-AKT levels and positively
regulated cell transformation in A549 cells. PP1α downregulation of
p-AKT levels increased the expression of p-JNK and inhibited 293T
cell proliferation. The data indicated that PP1α exerted
differential growth regulatory effects on A549 and 293T cells via
distinct or even opposite mechanisms. PP1α has been revealed to
activate the MAPK pathway in androgen-responsive cell lines, such
as PC3 cells (52). In the present
study, it was reported that PP1α inactivated the MAPK cascade by
increasing the levels of p-JNK in 293T cells, where the androgen
receptor was not expressed in these cell line models (53–55).
Therefore, the distinct effect of PP1α on the MAPK pathway may
depend on the androgen levels.
Furthermore, the present study demonstrated the
essential role of PP1α in the maintenance of the cell transforming
activity of lung cancer A549 cells. Initially, the overexpression
of ectopic PP1α promoted cell proliferation. Moreover, the
silencing of PP1α impaired cell migration and blocked cell
proliferation under normal and low serum conditions, and in the
presence of a low number of cells and a cell culture environment
that did not allow strong attachment. In addition, PP1α was
revealed to be overexpressed in 15 out of 31 lung cancer tissues.
Collectively, the data supported the oncogenic role of PP1α in lung
cancer formation, which is in line with previous studies
highlighting that downregulation or inactivation of PP1α is
associated with poor prognosis (24)
and radioresistance (25) of lung
cancer cells. However, negative correlation of PP1α with lung
cancer has also been reported (23).
Acknowledgements
We would like to thank Ms. Mei-Chun Liu from the
Instrument Center and Ms. Jen Miao at Taichung Veterans General
Hospital and Ms. Li-Wen Lee from the Department of Surgery at
Taichung Veterans General Hospital for their technical support.
Funding
The present research was supported by the Taichung
Veterans General Hospital-National Chi Nan University Joint
Research Program (TCVGH-NCNU1077902 and TCVGH-NCNU1067902),
Taichung Veterans General Hospital (TCVGH-1083207C and
TCVGH-1083208D), the Ministry of Science and Technology (MOST
105-2320-B-260-001-MY3), and the China Medical University and
Hospital grant (DMR-107-116, 108-119) awarded to SCC.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Ethics approval and consent to
participate
The Institutional Review Board and Human Ethics
Committee of Taichung Veterans General Hospital (Taichung, Taiwan)
approved the study. All patients signed informed consent for
participation in the present study.
Authors' contributions
KCC, SCC, JYH and CTRY designed the study, collected
the literature, analyzed and interpreted the data. CTRY prepared
the manuscript and arranged the manpower. JMMC, RYC and YRJH
performed the experiments. All authors approved the final
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bischoff JR, Anderson L, Zhu Y, Mossie K,
Ng L, Souza B, Schryver B, Flanagan P, Clairvoyant F, Ginther C, et
al: A homologue of Drosophila aurora kinase is oncogenic and
amplified in human colorectal cancers. EMBO J. 17:3052–3065. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang X, Zhou YX, Qiao W, Tominaga Y, Ouchi
M, Ouchi T and Deng CX: Overexpression of aurora kinase A in mouse
mammary epithelium induces genetic instability preceding mammary
tumor formation. Oncogene. 25:7148–7158. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW,
Sahin A, Brinkley BR and Sen S: Tumour amplified kinase STK15/BTAK
induces centrosome amplification, aneuploidy and transformation.
Nat Genet. 20:189–193. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fu J, Bian M, Jiang Q and Zhang C: Roles
of Aurora kinases in mitosis and tumorigenesis. Mol Cancer Res.
5:1–10. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Katayama H, Brinkley WR and Sen S: The
Aurora kinases: Role in cell transformation and tumorigenesis.
Cancer Metastasis Rev. 22:451–464. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen JM, Chiu SC, Wei TY, Lin SY, Chong
CM, Wu CC, Huang JY, Yang ST, Ku CF, Hsia JY and Yu CT: The
involvement of nuclear factor-κappaB in the nuclear targeting and
cyclin E1 upregulating activities of hepatoma upregulated protein.
Cell Signal. 27:26–36. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yu CT, Hsu JM, Lee YC, Tsou AP, Chou CK
and Huang CY: Phosphorylation and stabilization of HURP by
Aurora-A: Implication of HURP as a transforming target of Aurora-A.
Mol Cell Biol. 25:5789–5800. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kopnin BP: Targets of oncogenes and tumor
suppressors: Key for understanding basic mechanisms of
carcinogenesis. Biochemistry (Mosc). 65:2–27. 2000.PubMed/NCBI
|
|
9
|
Liu CW, Wang RH and Berndt N: Protein
phosphatase 1alpha activity prevents oncogenic transformation. Mol
Carcinog. 45:648–656. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Berndt N, Dohadwala M and Liu CW:
Constitutively active protein phosphatase 1alpha causes
Rb-dependent G1 arrest in human cancer cells. Curr Biol. 7:375–386.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu CW, Wang RH, Dohadwala M, Schönthal
AH, Villa-Moruzzi E and Berndt N: Inhibitory phosphorylation of
PP1alpha catalytic subunit during the G(1)/S transition. J Biol
Chem. 274:29470–29475. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ayllón V, Martínez AC, García A, Cayla X
and Rebollo A: Protein phosphatase 1alpha is a Ras-activated Bad
phosphatase that regulates interleukin-2 deprivation-induced
apoptosis. EMBO J. 19:2237–2246. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dessauge F, Cayla X, Albar JP, Fleischer
A, Ghadiri A, Duhamel M and Rebollo A: Identification of PP1alpha
as a caspase-9 regulator in IL-2 deprivation-induced apoptosis. J
Immunol. 177:2441–2451. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Eke I, Koch U, Hehlgans S, Sandfort V,
Stanchi F, Zips D, Baumann M, Shevchenko A, Pilarsky C, Haase M, et
al: PINCH1 regulates Akt1 activation and enhances radioresistance
by inhibiting PP1alpha. J Clin Invest. 120:2516–2527. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang RH, Liu CW, Avramis VI and Berndt N:
Protein phosphatase 1alpha-mediated stimulation of apoptosis is
associated with dephosphorylation of the retinoblastoma protein.
Oncogene. 20:6111–6122. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Imai Y, Kakinoki Y, Takizawa N, Nakamura
K, Shima H and Kikuchi K: Up-regulation of nuclear PP1alpha and
PP1delta in hepatoma cells. Int J Oncol. 14:121–126.
1999.PubMed/NCBI
|
|
17
|
Nomoto K, Shibata N, Imai Y, Kitamura K,
Nakamura K, Mizuno Y and Kikuchi K: Activation of protein
phosphatase 1alpha promoter in ascites hepatoma cells. Int J Oncol.
13:331–334. 1998.PubMed/NCBI
|
|
18
|
Takizawa N, Mizuno Y, Saadat M and Kikuchi
K: Selective increases in isoform PP1 alpha of type-1 protein
phosphatase in ascites hepatoma cells. Jpn J Cancer Res.
85:274–278. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Prowatke I, Devens F, Benner A, Gröne EF,
Mertens D, Gröne HJ, Lichter P and Joos S: Expression analysis of
imbalanced genes in prostate carcinoma using tissue microarrays. Br
J Cancer. 96:82–88. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Troutaud D, Petit B, Bellanger C, Marin B,
Gourin-Chaury MP, Petit D, Olivrie A, Feuillard J, Jauberteau MO
and Bordessoule D: Prognostic significance of BAD and AIF apoptotic
pathways in diffuse large B-cell lymphoma. Clin Lymphoma Myeloma
Leuk. 10:118–124. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hsu LC, Huang X, Seasholtz S, Potter DM
and Gollin SM: Gene amplification and overexpression of protein
phosphatase 1alpha in oral squamous cell carcinoma cell lines.
Oncogene. 25:5517–5526. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mei Y, Liu YB, Cao S, Tian ZW and Zhou HH:
RIF1 promotes tumor growth and cancer stem cell-like traits in
NSCLC by protein phosphatase 1-mediated activation of Wnt/β-catenin
signaling. Cell Death Dis. 9:9422018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen PC, Li C, Wang D, Luo ZW, Fu SJ, Li
X, Li ZL, Chen XW, Li L, Huang ZX, et al: PP-1α and PP-1γ display
antagonism and differential roles in tumorigenicity of lung cancer
cells. Curr Mol Med. 13:220–227. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Verdugo-Sivianes EM, Navas L,
Molina-Pinelo S, Ferrer I, Quintanal-Villalonga A, Peinado J,
Garcia-Heredia JM, Felipe-Abrio B, Muñoz-Galvan S, Marin JJ, et al:
Coordinated downregulation of Spinophilin and the catalytic
subunits of PP1, PPP1CA/B/C, contributes to a worse prognosis in
lung cancer. Oncotarget. 8:105196–105210. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim W, Youn H, Kang C and Youn B:
Inflammation-induced radioresistance is mediated by ROS-dependent
inactivation of protein phosphatase 1 in non-small cell lung cancer
cells. Apoptosis. 20:1242–1252. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu F, Yang X, Geng M and Huang M:
Targeting ERK, an Achilles' Heel of the MAPK pathway, in cancer
therapy. Acta Pharm Sin B. 8:552–562. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mayer IA and Arteaga CL: The PI3K/AKT
pathway as a target for cancer treatment. Annu Rev Med. 67:11–28.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Selim KA, Abdelrasoul H, Aboelmagd M and
Tawila AM: The role of the MAPK signaling, topoisomerase and
dietary bioactives in controlling cancer incidence. Diseases.
5(pii): E132017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Carnero A and Paramio JM: The
PTEN/PI3K/AKT Pathway in vivo, cancer mouse models. Front Oncol.
4:2522014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Garcia-Echeverria C and Sellers WR: Drug
discovery approaches targeting the PI3K/Akt pathway in cancer.
Oncogene. 27:5511–5526. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Robertson BW, Bonsal L and Chellaiah MA:
Regulation of Erk1/2 activation by osteopontin in PC3 human
prostate cancer cells. Mol Cancer. 9:2602010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shi ZZ, Shang L, Jiang YY, Shi F, Xu X,
Wang MR and Hao JJ: Identification of genomic biomarkers associated
with the clinicopathological parameters and prognosis of esophageal
squamous cell carcinoma. Cancer Biomark. 15:755–761. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chunli W, Jiajie H, Lifei W, Beiqing P,
Xin X, Yan C, Mingrong W and Xuemei J: IGHMBP2 overexpression
promotes cell migration and invasion in esophageal squamous
carcinoma. Yi Chuan. 37:360–366. 2015.(In Chinese). PubMed/NCBI
|
|
34
|
Sawada G, Niida A, Hirata H, Komatsu H,
Uchi R, Shimamura T, Takahashi Y, Kurashige J, Matsumura T, Ueo H,
et al: An integrative analysis to identify driver genes in
esophageal squamous cell carcinoma. PLoS One. 10:e01398082015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Onkes W, Fredrik R, Micci F, Schönbeck BJ,
Martin-Subero JI, Ullmann R, Hilpert F, Bräutigam K, Janssen O,
Maass N, et al: Breakpoint characterization of the
der(19)t(11;19)(q13;p13) in the ovarian cancer cell line SKOV-3.
Genes Chromosomes Cancer. 52:512–522. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang L, Wenners A, Hilpert F, Fredrik R,
Micci F, Onkes W, Caliebe A, Maass N, Weimer J and Arnold N:
Frequent translocations of 11q13.2 and 19p13.2 in ovarian cancer.
Genes Chromosomes Cancer. 53:447–453. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sárová I, Březinová J, Zemanová Z,
Gančarčíková M, Vydra J, Cermák J and Michalová K: Rearrangement of
11q13.2 region in two patients with acute myeloid leukemia. Leuk
Res. 37:4792013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hooker S, Hernandez W, Chen H, Robbins C,
Torres JB, Ahaghotu C, Carpten J and Kittles RA: Replication of
prostate cancer risk loci on 8q24, 11q13, 17q12, 19q33, and Xp11 in
African Americans. Prostate. 70:270–275. 2010.PubMed/NCBI
|
|
39
|
Waters KM, Le Marchand L, Kolonel LN,
Monroe KR, Stram DO, Henderson BE and Haiman CA: Generalizability
of associations from prostate cancer genome-wide association
studies in multiple populations. Cancer Epidemiol Biomarkers Prev.
18:1285–1289. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bocanegra M, Bergamaschi A, Kim YH, Miller
MA, Rajput AB, Kao J, Langerød A, Han W, Noh DY, Jeffrey SS, et al:
Focal amplification and oncogene dependency of GAB2 in breast
cancer. Oncogene. 29:774–779. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chunder N, Mandal S, Roy A, Roychoudhury S
and Panda CK: Analysis of different deleted regions in chromosome
11 and their interrelations in early- and late-onset breast tumors:
Association with cyclin D1 amplification and survival. Diagn Mol
Pathol. 13:172–182. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Weilandt M, Koch A, Rieder H, Deenen R,
Schwender H, Niegisch G and Schulz WA: Target genes of recurrent
chromosomal amplification and deletion in urothelial carcinoma.
Cancer Genomics Proteomics. 11:141–153. 2014.PubMed/NCBI
|
|
43
|
Sia D, Hoshida Y, Villanueva A, Roayaie S,
Ferrer J, Tabak B, Peix J, Sole M, Tovar V, Alsinet C, et al:
Integrative molecular analysis of intrahepatic cholangiocarcinoma
reveals 2 classes that have different outcomes. Gastroenterology.
144:829–840. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chen CF, Hsu EC, Lin KT, Tu PH, Chang HW,
Lin CH, Chen YJ, Gu DL, Lin CH, Wu JY, et al: Overlapping
high-resolution copy number alterations in cancer genomes
identified putative cancer genes in hepatocellular carcinoma.
Hepatology. 52:1690–1701. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gruel N, Lucchesi C, Raynal V, Rodrigues
MJ, Pierron G, Goudefroye R, Cottu P, Reyal F, Sastre-Garau X,
Fourquet A, et al: Lobular invasive carcinoma of the breast is a
molecular entity distinct from luminal invasive ductal carcinoma.
Eur J Cancer. 46:2399–2407. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chang CC, Zhou X, Taylor JJ, Huang WT, Ren
X, Monzon F, Feng Y, Rao PH, Lu XY, Fabio F, et al: Genomic
profiling of plasmablastic lymphoma using array comparative genomic
hybridization (aCGH): Revealing significant overlapping genomic
lesions with diffuse large B-cell lymphoma. J Hematol Oncol.
2:472009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sarova I, Brezinova J, Zemanova Z,
Bystricka D, Krejcik Z, Soukup P, Vydra J, Cermak J, Jonasova A and
Michalova K: Characterization of chromosome 11 breakpoints and the
areas of deletion and amplification in patients with newly
diagnosed acute myeloid leukemia. Genes Chromosomes Cancer.
52:619–635. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Serio G, Gentile M, Pennella A, Marzullo
A, Buonadonna AL, Nazzaro P, Testini M, Musti M and Scattone A:
Characterization of a complex chromosome aberration in two cases of
peritoneal mesothelioma arising primarily in the hernial sac.
Pathol Int. 59:415–421. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Senda K, Goi T, Hirono Y, Katayama K and
Yamaguchi A: Analysis of RIN1 gene expression in colorectal cancer.
Oncol Rep. 17:1171–1175. 2007.PubMed/NCBI
|
|
50
|
Boukamp P, Popp S, Bleuel K, Tomakidi E,
Bürkle A and Fusenig NE: Tumorigenic conversion of immortal human
skin keratinocytes (HaCaT) by elevated temperature. Oncogene.
18:5638–5645. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Llorian M, Beullens M, Andrés I, Ortiz JM
and Bollen M: SIPP1, a novel pre-mRNA splicing factor and
interactor of protein phosphatase-1. Biochem J. 378:P229–P238.
2004. View Article : Google Scholar
|
|
52
|
Chen M, Wan L, Zhang J, Zhang J, Mendez L,
Clohessy JG, Berry K, Victor J, Yin Q, Zhu Y, et al: Deregulated
PP1α phosphatase activity towards MAPK activation is antagonized by
a tumor suppressive failsafe mechanism. Nat Commun. 9:1592018.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Chen PH, Tsao YP, Wang CC and Chen SL:
Nuclear receptor interaction protein, a coactivator of androgen
receptors (AR), is regulated by AR and Sp1 to feed forward and
activate its own gene expression through AR protein stability.
Nucleic Acids Res. 36:51–66. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yang Y, Tse AK, Li P, Ma Q, Xiang S,
Nicosia SV, Seto E, Zhang X and Bai W: Inhibition of androgen
receptor activity by Histone deacetylase 4 through receptor
SUMOylation. Oncogene. 30:2207–2218. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wei TY, Hsia JY, Chiu SC, Su LJ, Juan CC,
Lee YC, Chen JM, Chou HY, Huang JY, Huang HM and Yu CT: Methylosome
protein 50 promotes androgen- and estrogen-independent
tumorigenesis. Cell Signal. 26:2940–2950. 2014. View Article : Google Scholar : PubMed/NCBI
|