Introduction
Prostate cancer (PCa) is one of the most prevalent
malignant tumors in males worldwide. In 2017, the estimated number
of new cases and deaths from PCa in the United States were 161,360
and 26,730, respectively (1).
Cancer cells often exhibit altered epigenetic signatures, which
results in dysregulation of expression of genes involved in
processes such as transcription, proliferation, apoptosis and DNA
repair (2). Radiation therapy (RT)
is an important treatment approach for PCa; it can be performed at
any stage of PCa as a curative monotherapy or as an adjuvant with
other therapeutic options (3).
However, 30–50% patients with high-risk PCa exhibit local relapse
following radiation therapy (4).
Therefore, determining the mechanisms underlying the development of
resistance to RT and a means of enhancing the sensitivity of PCa to
RT may improve patient outcomes.
Phospholipase Cε (PLCε) is a critical signaling hub
that regulates a variety of biological processes in cells (5). In contrast to the other members of the
PLC family, PLCε exhibits PLC enzyme activities and serves a role
as a guanine nucleotide exchange factor (6). Previous studies have reported that
high expression levels of PLCε are closely associated with the
initiation and progression of various types of cancer, particularly
in the upper gastrointestinal tract (7,8). This
finding has been further verified by two genome-wide studies
demonstrating that the PLCε gene is an essential tumor promoter in
esophageal squamous cell carcinoma (9). Our previous study demonstrated that
PLCε expression was significantly upregulated in PCa tissue
compared with normal prostate tissue and that PLCε was capable of
enhancing the proliferative ability and metastatic potential of PCa
cells via various pathways (10–12).
However, the association between PLCε and RT resistance in PCa has
not been determined.
Androgen receptor (AR) is crucial for the
development of PCa (13,14). Following ligand binding, the
AR-ligand complex translocates to the nucleus, binds to the DNA and
subsequently alters downstream gene transcription (15). Biologically, androgen deprivation
therapy (ADT) abrogates the ability of AR to promote cellular DNA
damage repair (DDR) (16,17). ADT-mediated blocking of the AR
pathway results in an inability of PCa cells to effectively
activate DDR; therefore, a combination of ADT and RT may promote
the lethality of RT and initiate DNA damage (16–18).
Poly (ADP-ribose) polymerase 1 (PARP1) is a member of the PARP
superfamily, and it exerts a large number of cellular activities,
such as gene transcription, chromatin remodeling and cell death
(19,20). In addition, PARP1 can be recruited
to AR functional sites and facilitate AR occupancy and function
(21).
The present study explored the underlying mechanisms
between PLCε and radiosensitivity in primary PCa (PPC) and
castration-resistant PCa (CRPC) using in vitro and in
vivo models. The aim of the present study was to establish
whether PLCε knockdown enhanced the radiosensitivity of CRPC via
the AR/PARP1/DNA-PKcs axis.
Materials and methods
Patients and specimens
A total of 30 samples of benign prostatic
hyperplasia (BPH), 35 samples of PPC and 27 samples of CRPC were
collected from patients who underwent prostate biopsy, TURP or
radical prostatectomy at the Department of Urology at the First
Affiliated Hospital of Chongqing Medical University (Chongqing,
China) between September 2015 and August 2017. All patients
provided written informed consent, and the protocol was approved by
the Ethical Committee of the First Affiliated Hospital of Chongqing
Medical University. PCa samples were histologically graded
according to the criteria of EAU guidelines (22). Adjacent normal prostate tissues (10
mm from the malignant locus) were also collected from patients with
unifocal lesions and were verified by pathologists. BPH samples
were verified by histological examination. Specimens were stored in
liquid nitrogen until further use.
Immunohistochemistry (IHC)
Prostate tumor and BPH tissues were fixed in 10%
neutral buffered formalin (Sigma-Aldrich; Merck KGaA) diluted with
0.01 M PBS buffer (pH 7.4; OriGene Technologies, Inc.) for 24 h at
4°C, embedded in paraffin and cut into 4-µm sections. Antigen
retrieval was performed in citrate buffer (pH 6.0) for 15 min at
98°C, and the sections were subsequently blocked with normal goat
serum (Beyotime Institute of Biotechnology) for 30 min at 37°C. The
sections were incubated overnight with the primary antibody
targeting PLCε (1:50; cat. no. sc-28402; Santa Cruz Biotechnology,
Inc.), AR (1:400; cat. no. 5153; CST Biological Reagents Co., Ltd.)
and DNA-dependent protein kinase catalytic subunit (DNA-PKcs;
1:100; cat. no. ab168854; Abcam) at 4°C, washed with PBS and
subsequently incubated with the biotin-streptavidin horseradish
peroxidase labeled Goat anti-rabbit IgG (1:300; cat. no. SP-9001;
Beijing Zhongshan Jinqiao Biotechnology Co., Ltd.) for 1 h at room
temperature. Signals were visualized using a peroxidase substrate
and hematoxylin counterstaining for 1 min at room temperature.
Sections were semi-quantitatively scored for staining intensity as
follows: 0, no staining; 1, weak staining; 2, light staining; 4,
moderate staining; and 6–8, strong staining. Staining scores ≥2
were regarded as positive expression, whereas scores <2 were
considered negative.
Cell lines, transfection and
agents
PPC cell line LNCaP was purchased from American Type
Culture Collection. Mycoplasma testing was performed using a
Mycoplasma Detection kit (Beijing Solarbio Science & Technology
Co., Ltd.). Bicalutamide®-resistant cells (Bica-R) and
Enzalutamide®-resistant cells (Enza-R) were developed by
treating LNCaP cells with Bicalutamide® and
Enzalutamide® in our laboratory between October 2016 and
February 2017 and were used as CRPC cell lines. To develop
resistance, LNCaP cells were cultured with 1, 5, 10 or 25 µM
bicalutamide or enzalutamide. After a month of screening, a dose of
10 µM was selected as the optimal concentration for subsequent
induction of CRPC cells. All cells were cultured in DMEM/F-12
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% FBS
(Gibco; Thermo Fisher Scientific, Inc.), 2 mM L-glutamine and 100
U/ml penicillin at 37°C with 5% CO2 in a humidified
incubator. The lentiviral vectors non-targeting LV-control
(LV-Ctrl) and LV-short hairpin (sh)RNA targeting PLCε (shPLCε) were
purchased from Shanghai GenePharma Co., Ltd. Other reagents used
were as follows: DMSO (Sigma-Aldrich; Merck KGaA); AR inhibitor
ARN-509 (38 nM; Medchem Express); PARP1 inhibitor AZD2281 (0.5 µM;
Selleck Chemicals); AR activator DHT (10 nM; Sigma-Aldrich; Merck
KGaA) and enhancer of zeste homolog 2 (EZH2) inhibitor
3-deazaneplanocin A (DZNeP; 1 µM; Selleck Chemicals).
Cell irradiation and MTT assays
LNCaP, Enza-R and Bica-R cells (3×103
cells/well) were plated in a 96-well plate in 100 µl medium.
Following adhesion, radiation was delivered at room temperature
using an x-6 MV photon linear accelerator (CD2300; Varian
Corporation). A total single dose of 2, 4, 6 or 8 Gy was delivered
with a dose rate of 300 cGy/sec using a source-to-surface distance
of 100 cm. Irradiated cells were incubated for 24 h at 37°C;
subsequently, 5 mg/ml MTT (Sigma-Aldrich; Merck KGaA) was added to
each well, and the cells were further incubated for 4 h at 37°C.
After removing the medium, 100 µl DMSO was added to each well, and
the plates were agitated on a rotator for 10 min at room
temperature. Absorbance was recorded using an ultraviolet
spectrophotometric reader at a wavelength of 490 nm. The MTT
experiments were repeated five times.
Flow cytometric analysis
LNCaP, Enza-R and Bica-R cells (1×106
cells/well) were plated in 6-well plates and cultured to 60%
confluence for cell cycle analysis. Serum-free medium was added,
and the cells were cultured for a further 24 h at 37°C. Following
the administration of 6 Gy radiation, cells were collected and
fixed with 75% ethanol overnight at 4°C. The distribution of cells
in different stages of the cell cycle was detected using a PN
B49007AD flow cytometer (Beckman Coulter Co., Ltd.) and analyzed
using FlowJo version 7.6.2 (FlowJo, LLC). Cell cycle distribution
analysis experiments were performed in triplicate.
Apoptosis analysis
LNCaP, Enza-R and Bica-R cells (1×106
cells/well) were plated in 6-well plates and cultured to 60%
confluence for apoptosis analysis. Cells were irradiated at room
temperature as described above. After 24 h, cells were washed with
PBS and detached with 2.5 mM EDTA in PBS. Annexin V and
7-amino-actinomycin D (7-AAD; both from Sungene Biotech Co., Ltd)
staining was performed according to the manufacturer's
instructions. Staining was quantified using flow cytometry
(CytoFLEX; Beckman Coulter, Inc.) by analyzing Annexin
V-fluorescein isothiocyanate (FITC) and 7-AAD fluorescence using
phycoerythrin emission signal detectors and FlowJo V10 software
(FlowJo LLC).
Colony formation assay
LNCaP, Enza-R and Bica-R cells (400 cells/well) were
seeded in 6-well plates, and the culture medium was changed every 2
days. Following 14 days of culture, cells were washed twice with
PBS and fixed with 4% paraformaldehyde for 30 min at room
temperature. Colonies were stained with Giemsa solution for 15 min
at room temperature, washed twice with PBS and air-dried. The
number of colonies were counted using a light microscope
(magnification, ×40). Colony formation efficiency (CFE) was
calculated as follows: CFE (%)=(number of colonies/400) ×100.
Colony formation assays were performed in triplicate.
Immunofluorescence
Enza-R and Bica-R cells (1×105
cells/well) were seeded on polylysine coated coverslips and
cultured for 24 h. Cells were washed with PBS, fixed in 4%
paraformaldehyde for 15 min at 4°C, permeabilized with PBS/0.1%
Triton X-100 and blocked with 5% BSA containing 1% Tween-20 for 30
min at 37°C. Immunostaining was performed with the primary antibody
targeting AR (1:200; cat. no. 5153; CST Biological Reagents Co.,
Ltd.) overnight at 4°C and a goat anti-rabbit IgG-AlexaFluor 488
secondary antibody diluted in goat serum (1:500; cat. no. SR134;
Beijing Solarbio Science & Technology Co., Ltd.) for 1 h at
37°C. Immunofluorescence images were acquired using fluorescence
microscopy using a Nikon Eclipse 80i microscope (Nikon Corporation;
magnification, ×400).
Western blotting
Patient tissues were ground repeatedly and lysed
using RIPA buffer (Beyotime Institute of Biotechnology) containing
0.1% PMSF. LNCaP, Enza-R and Bica-R cells were lysed using RIPA
lysis buffer containing protease inhibitors and 1 mM
Na3VO4. Nuclear and cytoplasmic proteins were
extracted with the Nuclear and Cytoplasmic Protein Extraction Kit
(Beyotime Institute of Biotechnology). Proteins were quantified
using the bicinchoninic acid assay (Beyotime Institute of
Biotechnology) and 3 µg protein/lane was resolved using SDS-PAGE
(NuPAGE 3–8% Tris-Acetate or NuPAGE 4–12% BisTris; Invitrogen;
Thermo Fisher Scientific, Inc.) and transferred to PVDF membranes
(EMD Millipore). Following blocking with 5% skimmed milk for 2 h at
room temperature, the membranes were incubated with primary
antibodies targeting PLCε (1:500; cat. no. sc-28402; Santa Cruz
Biotechnology, Inc.), AR (1:1,000; cat. no. 5153; CST Biological
Reagents Co., Ltd.), PARP1 (1:1,000; cat. no. ab32138; Abcam),
DNA-dependent protein kinase catalytic subunit (DNA-PKcs; 1:1,000;
cat. no. ab168854; Abcam), H2AX (1:1,000; cat. no. 7631; CST
Biological Reagents Co., Ltd.), γ-H2AX (1:1,000; cat. no. 7631; CST
Biological Reagents Co., Ltd.) and GAPDH (1:1,000; cat. no. 5174;
Cell Signaling Technology) overnight at 4°C, washed with TBS +
0.05% Tween-20 (Beijing Solarbio Science & Technology Co.,
Ltd.), and subsequently incubated with a horseradish
peroxidase-conjugated goat anti-rabbit immunoglobulin G secondary
antibody (1:2,000; cat. no. TA130015; OriGene Technologies, Inc.)
for 1 h at room temperature. The signals were visualized using the
Enhanced Chemiluminescence kit (EMD Millipore), and the intensity
of each band was quantified on X-ray films and densitometry
analysis was performed using imaging software Quantity One version
4.6.2 (Bio-Rad Laboratories, Inc.).
Reverse transcription-quantitative
(RT-q)PCR
Total RNA was extracted from LNCaP, Enza-R and
Bica-R cells and ground tissue using TRIzol® (Takara
Bio, Inc.) according to the manufacturer's protocol. RNA quality
was determined using a 2100 BioAnalyzer (Agilent Technologies
GmbH). cDNA synthesis was performed using a PrimeScript®
RT reagent kit (Takara Bio, Inc.) according to the manufacturer's
protocol, and the reverse transcription temperature protocol was 15
min at 37°C and 5 sec at 85°C. SYBR PremixExTaq™ II kit (Takara
Bio, Inc.) was used for RT-qPCR with the CFX96™Real-Time PCR
Detection System (Bio-Rad Laboratories, Inc.). The sequences of the
primers were as follows: PLCε forward,
5′-GCAACTACAACGCTGTCATGGAG-3′ and reverse,
5′-GCAACTACAACGCTGTCATGGAG-3′; AR forward, 5′-CCTACGGCTACACTCGG-3′
and reverse, 5′-CTGGCAGTCTCCAAACG-3′; H2AX forward,
5′-AGTGCTGGAGTACCTCACCG-3′ and reverse, 5′-CACGGCCTGGATGTTGG-3′;
PARP1 forward, 5′-TTTCCATCAAACATGGGCGAC-3′ and reverse,
5′-CGGAGTCTTCGGATAAGCTCT-3′; DNA-PKcs forward,
5′-CGGACCTACTACGACTG-3′ and reverse, 5′-AGACAAAGGGTGGAAA-3′; and
GAPDH forward, 5′-ACGGATTTGGTCGTATTGGGCG-3′ and reverse,
5′-CCTCCTGGAAGATGGTGATGG-3′. The thermocycling conditions were as
follows: 95°C for 30 sec, followed by 41 cycles of 95°C for 10 sec,
60°C for 30 sec and 72°C for 30 sec. The mRNA expression levels
were calculated using the comparative 2−ΔΔCq method
(10) and GAPDH served as the
reference. All gene expression experiments were repeated at least
three times.
Chromatin immunoprecipitation
(ChIP)
A SimpleChIP® Enzyme Chromatin IP kit
(Magnetic Beads; cat. no. 9003; CST Biological Reagents Co., Ltd.)
was used according to the manufacturer's protocol. Enza-R cells
(~4×106) were cross-linked with 1% formaldehyde and
uncross-linked using glycine. Chromatin was digested to ~500 bp
fragments and centrifuged at 9,400 × g for 10 min at 4°C. Chromatin
fragments were mixed with antibody targeting histone H3 lysine 27
(H3K27) trimethylation (H3K27me3; 1:50; cat. no. 9733) and control
antibody (rabbit IgG; 1:100; cat. no. both from 2729; CST
Biological Reagents Co., Ltd.), and incubated overnight at 4°C.
After DNA purification and elution, DNA enrichment was detected
using RT-qPCR.
Subcutaneous xenograft assay
Male 6-week-old NOD.Cg-Prkdcscid mice
were housed in a barrier facility, and the experiments were
approved by the Chongqing Medical University Institutional Animal
Care and Use Committee and the Animal Ethics Committee. A total of
5 mice/group were used, which was determined using power analysis.
For the xenograft assay, 1×106 Bica-R cells were
injected subcutaneously into the right flank of the mice. When the
tumors reached 1 mm3 in volume, mice were irradiated
with 2 Gy/day for 5 days. Body weight and tumor size were measured
using calipers twice a week for 20 days after irradiation. At the
end of the experiment, the mice were sacrificed by carbon dioxide
asphyxiation.
Statistical analysis
Statistical analysis was performed using SPSS
version 22.0 (IBM Corp.). Data are presented as the mean ± standard
deviation of at least three independent experiments. For analysis
of the differences between two groups, unpaired Student's t-test
were used to determine statistical significance. For multiple
comparisons, ANOVA and Tukey-Kramer (comparisons among multiple
groups) and Dunnett's (comparisons of multiple groups with the
control group) post-hoc analysis were used. Correlations were
calculated using Spearman's correlation coefficient. Mann-Whitney
test was used for the analysis of two independent variables.
P<0.05 was considered to indicate a statistically significant
difference.
Results
PLCε, AR and DNA-PKcs expression in
PCa
To identify potential associations among PLCε, AR
and DNA-PKcs during the development of PCa, the expression levels
of these proteins were determined in clinical samples of BPH, PPC
and CRPC by IHC. The PPC and CRPC tissues exhibited positive
staining for PLCε, AR and DNA-PKcs (Fig. 1A). The staining scores of PLCε, AR
and DNA-PKcs were significantly higher in CRPC compared with BPH
and PPC (Fig. 1B-D). A total of
four PPC and CRPC samples each, with their corresponding adjacent
normal tissues serving as controls, were used to determine the mRNA
and protein expression levels. The mRNA expression levels of PLCε
and AR were significantly increased in all PPC and CPRC samples
compared with the adjacent tissues (Fig. 1E and G). The western blotting
results demonstrated that the expression levels of PLCε and AR in
PPC and CRPC samples were upregulated compared with the adjacent
normal tissues (Fig. 1F and H). The
differences between the clinicopathological parameters and the
expression of activated PLCε or AR were determined; significant
differences in Gleason score between the positive and negative
staining groups of PLCε and AR were observed (10) in CRPC; no differences were observed
in the other clinicopathological characteristics (Table I).
 | Figure 1.Expression of PLCε, AR and DNA-PKcs
in PCa. (A) Hematoxylin and eosin and immunohistochemistry staining
of BPH, PPC and CRPC tissues. Magnification, ×200. (B-D) Staining
scores for (B) PLCε, (C) AR and (D) DNA-PKcs. (E and F) Expression
of PLCε and AR in four PPC specimens. (G and H) Expression of PLCε
and AR in four CRPC specimens. *P<0.05, **P<0.01 and
***P<0.001. PCa, prostate cancer; PLCε, phospholipase Cε; AR,
androgen receptor; BPH, benign prostatic hyperplasia; PPC, primary
PCa; CRPC, castration-resistant PCa; DNA-PKcs, DNA-dependent
protein kinase catalytic subunit. |
 | Table I.Clinicopathological characteristics
of patients with prostate cancer. |
Table I.
Clinicopathological characteristics
of patients with prostate cancer.
|
|
| PLCε | AR |
|---|
|
|
|
|
|
|---|
| Variable | Overall | Negative | Positive | P-value | Negative | Positive | P-value |
|---|
| Total, n (%) | 62 | 12 | 50 |
| 14 | 48 |
|
| Age, years |
|
|
| 0.375 |
|
| 0.647 |
|
Median | 75 | 76 | 73 |
| 75 | 74 |
|
|
IQR | 65–79 | 64–80 | 61–77 |
| 64–79 | 63–78 |
|
| PSA in PPC,
µg/l | 35 | 7/35 (20) | 28/35 (80) | 0.658 | 9/35 (26) | 26/35 (74) | 0.865 |
|
Median | 93.56 | 74.49 | 142.53 |
| 76.34 | 150.86 |
|
|
IQR | 32.46–243.79 | 16.53–175.26 | 75.38–250.16 |
| 18.61–197.95 | 86.32–264.78 |
|
| PSA in CRPC,
µg/l | 27 | 5/27 (19) | 22/27 (81) | 0.386 | 6/27 (22) | 21/27 (78) | 0.452 |
|
Median | 40.32 | 42.27 | 37.85 |
| 41.87 | 38.65 |
|
|
IQR | 20.18–156.97 | 22.45–187.35 | 18.94–100.32 |
| 20.98–169.76 | 19.67–97.48 |
|
| Gleason score in
PPC | 35 |
|
| 0.610 |
|
| >0.999 |
|
<7 | 25/35 (71) | 3/5
(60) | 22/30 (70) |
| 7/9
(77) | 18/26 (69) |
|
| ≥7 | 10/35 (29) | 2/5
(40) | 8/30
(30) |
| 2/9
(23) | 8/26
(31) |
|
| Gleason score in
CRPC | 27 |
|
| 0.011a |
|
| 0.017a |
|
<7 | 8/27
(30) | 5/7
(71) | 3/20
(15) |
| 4/5
(80) | 4/22
(18) |
|
| ≥7 | 19/27 (70) | 2/7
(29) | 17/20 (85) |
| 1/5
(20) | 18/22 (82) |
|
Upregulated expression levels of PLCε
and AR are associated with RT resistance in PCa
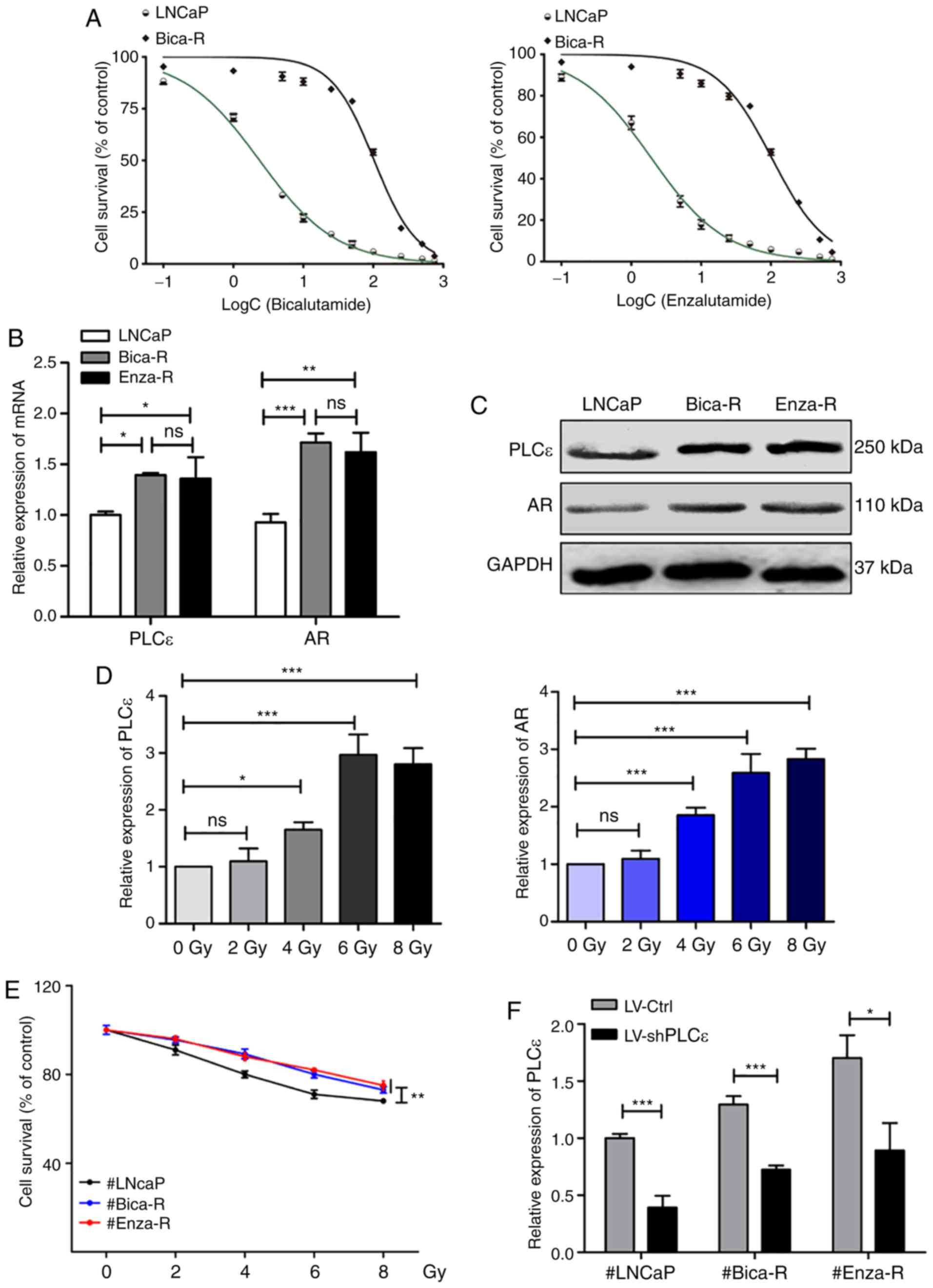
To verify the characteristics of the CRPC (Bica-R
and Enza-R) cells, MTT assays were performed to detect the half
maximal inhibitory concentration (IC50) of Bicalutamide
or Enzalutamide in Bica-R and Enza-R cells. The results
demonstrated that Bica-R cells exhibited a 43-fold increased
resistance to Bicalutamide, and Enza-R cells exhibited a 53-fold
increased resistance to Enzalutamide compared with the parental
LNCaP cells (Fig. 2A). The mRNA and
protein expression levels of PLCε and AR were determined by RT-qPCR
and western blot analysis in LNCaP, Bica-R and Enza-R cells; the
results revealed that the levels of PLCε and AR were increased in
the two CRPC cell lines compared with the LNCAP PPC cell line
(Fig. 2B and C). To investigate the
molecular mechanisms by which RT resistance occurred in CRPC,
LNCaP, Bica-R and Enza-R cells were treated with increasing doses
(0–8 Gy) of radiation. MTT assays were performed after 24 h of
irradiation. The results demonstrated that irradiation reduced cell
survival, and cell survival of Bica-R and Enza-R cells was
significantly increased compared with that of LNCaP cells at each
dose of radiation (Fig. 2E). To
probe the molecular mechanism underlying this phenomenon, RT-qPCR
was performed to measure the expression levels of PLCε and AR. The
results showed that expression of PLCε and AR increased as the dose
of radiation increased (Fig. 2D).
Based on the results of MTT and RT-qPCR assays, 6 Gy was selected
as the optimal dose of radiation for subsequent experiments. To
confirm the role of PLCε in RT resistance, PLCε was silenced in
LNCaP, Bica-R and Enza-R cells using shPLCε, and PLCε mRNA levels
were measured to evaluate the gene knockdown efficiency (Fig. 2F). Additionally, the mRNA levels of
AR were assessed, and the results demonstrated that the mRNA levels
of AR were decreased following shPLCε transfection in Enza-R, but
not LNCaP cells (Fig. 2G).
Inhibition of cell viability in the shPLCε-transfected cells
compared with the control cells following radiation was also
observed (Fig. 2H). In addition,
PLCε knockdown or radiation alone inhibited cell colony formation,
and PLCε knockdown enhanced the inhibitory effect of radiation on
cell colony formation (Fig. 2I and
J). Flow cytometry analysis revealed similar results; either
shPLCε knockdown or radiation alone resulted in an increase in the
percentage of cells in the G2/M phase (Fig. 3A) and increased apoptosis (Fig. 3B) compared with the control, and the
combination of the two treatments exhibited an enhanced effect.
Therefore, it was concluded that PLCε served a crucial role in RT
resistance of CRPC cells.
PLCε deficiency impairs DNA damage
repair by preventing unclear translocation of AR and inhibiting the
AR/DNA-PKcs pathway in CRPC
To determine whether PLCε downregulation enhanced
the sensitivity of CRPC to radiotherapy through DDR, the mRNA and
protein expression levels of AR, DNA-PKcs and H2AX were determined
following PLCε knockdown in CRPC cells. The results of RT-qPCR
demonstrated that PLCε knockdown significantly reduced AR and
DNA-PKcs expression, but exhibited no significant effects on H2AX
(Fig. 3C). Western blotting
revealed similar alterations in the expression of AR, DNA-PKcs and
H2AX at the protein level, but the level of γ-H2AX was
significantly downregulated in the shPLCε group compared with the
control group (Fig. 3D).
Immunofluorescence results demonstrated that shPLCε prevented AR
translocation from the cytoplasm to the nucleus in irradiated cells
(Fig. 4A). Western blotting results
also demonstrated a decrease in AR expression in the nucleus in the
shPLCε-transfected cells compared with the control cells (Fig. 4B).
An AR/PARP1 positive feedback loop
serves a vital role in DDR in CRPC
To verify whether expression of PARP1 and DNA-PKcs
were regulated by AR, CRPC cells were treated with the AR activator
DHT or the AR inhibitor ARN-509. Colony formation assay results
revealed that DHT increased the number of colonies formed by CRPC
cells, whereas ARN-509 decreased the number of colonies (Fig. 5A and B). A significant increase in
apoptosis was observed in CRPC cells treated with ARN-509 compared
with those treated with DHT (Fig.
5C). RT-qPCR results demonstrated that DHT treatment increased
the expression of AR, PARP1 and DNA-PKcs in CRPC cells compared
with the corresponding control groups; by contrast, treatment with
ARN-509 resulted in a decrease in AR, PARP1 and DNA-PKcs expression
levels (Fig. 5D). Western blotting
exhibited similar results (Fig.
5E). To determine the association between AR and PARP1, CRPC
cells were treated with the PARP1 inhibitor AZD2281, and the
expression levels of AR, PARP1 and DNA-PKcs were assessed. The
levels of AR were decreased in cells treated with AZD2281, as was
the expression of PARP1, DNA-PKcs (Fig.
5F) and γ-H2AX (Fig. 5G).
PLCε enhances the radiosensitivity of
CRPC by regulating H3K27 trimethylation levels in the PARP1
promoter region
EZH2, a histone-lysine N-methyltransferase enzyme
that participates in histone methylation, alters gene expression
patterns by specifically contributing to the H3K27me3 (23). It was hypothesized that the
regulatory effects of PLCε on PARP1 were associated with the levels
of H3K27me3 in the PARP1 promoter region. Bica-R cells were
transfected with shPLCε or treated with DZNeP (a specific inhibitor
of EZH2), and RT-qPCR results demonstrated that shPLCε reduced the
expression of PARP1, AR and DNA-PKcs, whereas DZNeP reversed the
effects of PLCε knockdown in Bica-R cells at the mRNA (Fig. 6A) and protein (Fig. 6B) levels. PLCε knockdown resulted in
an increase in the levels of EZH2 and H3K27me3 compared with the
control, and this effect was reversed by DZNep (Fig. 6B). ChIP-PCR was performed using a
H3K27me3 antibody, and the results demonstrated that shPLCε
increased the levels of H3K27me3 in the PARP1 promoter region
compared with the control group, and DZNep abrogated this effect
(Fig. 6C).
 | Figure 6.PLCε enhances the radiosensitivity of
CRPC by regulating H3K27me3 levels in the PARP1 promoter region.
(A) mRNA levels of PARP1, AR and DNA-PKcs in Bica-R cells were
transfected with shPLCε or treated with DZNeP. (B) Protein
expression levels of EZH2, H3K27me3, PARP1 and AR in Bica-R cells.
(C) ChIP-PCR was used to analyze the H3K27me3 levels in the PARP1
promoter region. *P<0.05, **P<0.01 and ***P<0.001. PLCε,
phospholipase Cε; CRPC, castration-resistant prostate cancer;
Bica-R, Bicalutamide-resistant; sh, short hairpin; EZH2, enhancer
of zeste homolog 2; PARP1, poly (ADP-ribose) polymerase 1; AR,
androgen receptor; ChIP, chromatin immunoprecipitation. |
PLCε knockdown inhibits CRPC growth in
vivo
To further evaluate the tumor-promoting role of PLCε
in CRPC and its association with RT, a mouse xenograft model was
used, where mice were injected with Bica-R cells or Bica-R cells
transfected with shPLCε. As demonstrated in Fig. 7A and B, PLCε-knockdown resulted in a
significant inhibition of tumor growth in animals following
radiation compared with the irradiated and non-irradiated control
groups. In addition, IHC results revealed that shPLCε transfection
significantly reduced the protein expression levels of PLCε, AR,
PARP1, DNA-PKcs and γ-H2AX in the tumors, evidenced by the positive
rate of IHC (Fig. 7C and D), which
was similar to the results obtained from analysis of clinical
samples.
 | Figure 7.PLCε knockdown inhibits the growth of
CRPC in vivo. (A and B) Immunocompromised mice were
subcutaneously injected with PLCε-knockdown or control Bica-R
cells, and the tumor sizes and weights of the two groups are
presented. (C) Comparison of positive rate of IHC in each group.
(D) Expression of PLCε, AR, PARP1, DNA-PKcs and γ-H2AX in each
group. Magnification, ×200. *P<0.05 and ***P<0.001. PLCε,
phospholipase Cε; CRPC, castration-resistant prostate cancer;
Bica-R, Bicalutamide-resistant; AR, Androgen receptor; PARP1, poly
(ADP-ribose) polymerase 1; H2AX, H2A histone family member X. |
Discussion
The prostate is highly dependent on the proper
functioning of AR (24). AR
signaling is essential for the development and physiological
functions of the prostate, which has been reported to be one of the
most important initiating and supporting factors underlying the
carcinogenesis and progression of PCa (13,15).
Therefore, ADT, which targets the AR signaling pathway, has become
a valuable and fundamental therapeutic method for treatment of PCa
(25). If PCa progresses to CRPC or
develops resistance to second-generation agents, such as
enzalutamide, abiraterone acetate or other therapeutics, clinicians
typically resort to alternative therapeutics that were initially
hypothesized to function through AR-independent mechanisms, among
which RT has exhibited great potential (14). RT alone has been demonstrated to be
effective in patients with PCa, and patients treated with a
combination of ADT and RT exhibit favorable survival outcomes
(26–28). The increased survival times were
previously hypothesized to be the result of the synergistic effects
of mechanistically different anti-cancer strategies (26). However, PCa of different
pathological grades or clinical stages displays heterogeneity in AR
expression levels, thus exhibiting varying levels of sensitivity to
ADT and different clinical benefits from combined ADT and RT
(28). In addition, resistance to
RT is frequently recurrent, particularly in CRPC (29). The results of the present study
suggested possible involvement of AR signaling in RT resistance. A
previous study has demonstrated that AR signaling is capable of
enhancing cellular DDR and consequently serves a pivotal role in
resistance to radiotherapy (30).
However, the underlying mechanism remains unclear.
In the present study, a novel mechanism for the
acquisition of RT resistance in CRPC was determined. RT induced the
upregulation of PLCε, which subsequently promoted AR translocation.
The increased presence of nuclear AR recruited PARP1 to specific
regions of the DNA, and PARP1 in turn facilitated AR
transcriptional activity and upregulated AR-mediated signaling,
constituting a positive feedback loop that enhanced AR signaling.
In addition, RT-induced increases in expression of PLCε reduced the
expression levels of EZH2, resulting in epigenetic upregulation of
PARP1 via the inhibition of H3K27me3 in its promoter region, which
further potentiated the AR/PARP1 loop. Finally, the elevated
activity of AR signaling induced alterations in the expression of
DDR-associated molecules towards a RT-resistant phenotype. The
clinical and in vivo data revealed that PLCε, AR and
DNA-PKcs were simultaneously increased in prostate cancer samples,
providing preliminary evidence for this mechanism. In the
cytological experiments, CRPC cell lines were used to confirm the
crucial role of the AR/PARP1/DNA-PKcs pathway in the development of
resistance to RT.
In the present study, PLCε was considered to be the
initiator of this novel signaling pathway in PCa following RT. PLCε
is the primary member of the PLC family of enzymes that catalyze
the hydrolysis of phosphatidylinositol 4,5-bisphosphate (5). PLCε is a unique member of this family
due to its ability to receive signal inputs from both the Ras and
Rho families of proteins and other heterotrimeric G proteins, thus
functioning as a hub in the regulation of signaling pathways
involved in tumor development and progression (5,31).
Numerous studies have demonstrated that PLCε is involved in the
initiation and progression of tumors, particularly in upper
gastrointestinal tract cancers (9,32–36).
Two genome-wide studies identified single nucleotide polymorphisms
in the PLCε gene in esophageal squamous cell carcinoma, providing
additional evidence that PLCε acts as an oncogene leading to cell
transformation (9,37). Our previous study demonstrated that
PLCε expression was significantly higher in PCa tissues compared
with normal prostate tissue, and PLCε knockdown resulted in reduced
proliferation of PCa through downregulation of AR expression
(10), and this result was
confirmed in the present study. Additionally, the results of the
present study revealed that PLCε regulation of AR may underlie the
development of RT resistance in PCa.
Since AR signaling has been implicated in RT
resistance, it was hypothesized in the present study that
preservation of AR functionality or AR-mediated signaling pathways
may promote the development of RT resistance. The results
demonstrated that RT upregulated the expression of PLCε and AR,
whereas RT combined with PLCε knockdown significantly decreased the
expression of AR and enhanced the radiosensitivity of LNCaP, Bica-R
and Enza-R cells. These results were further verified by the data
demonstrating that the expression of DDR-related molecules,
DNA-PKcs and γ-H2AX were reduced in cells or tissues following PLCε
knockdown.
Mechanistically, it was hypothesized that PARP1 was
involved in the AR signaling regulation of RT resistance based on
previous studies. PARP1 is recruited to AR functional sites on DNA
and consequently increases the transcriptional activities of AR and
promotes AR-dependent cellular biofunctions in PCa (20). Enhanced AR signaling activity has
also been demonstrated to be a prominent stimulator of PARP1 in
certain types of cancer (38).
Furthermore, increased expression of PARP1 is associated with
enhanced chromatin remodeling, DNA replication and cell survival,
all of which are related to phenotypic alterations observed in
RT-resistant cancer, including ovarian, breast and prostate cancer
(39). The results of the present
study demonstrated that activation of AR signaling using DHT
increased PARP1 expression, whereas inhibition of AR signaling,
both by knockdown of PLCε expression or treatment with an AR
antagonist, reduced PARP1 expression. In addition, a PARP1
inhibitor attenuated AR signaling. These results suggested an
intricate association between PLCε, AR and PARP1; PLCε knockdown
directly impaired AR expression and translocation or reduced PARP1
expression, disrupting the positive feedback loop that exists
between PARP1 and AR, which also resulted in the downregulation of
AR. The attenuation of AR was, in turn, capable of decreasing the
expression of PARP1. Therefore, upregulated expression of PLCε, as
a tumor promoter in PCa and CRPC, may act as an indispensable
protector of proper functioning of the AR/PARP1 loop.
However, the possibility that PLCε knockdown may
downregulate PARP1 expression via an AR-independent manner cannot
be ruled out. In the present study, EZH2 was demonstrated to be
upregulated following PLCε knockdown. Histone methylation serves an
important role in DNA repair and consequent cell survival
associated with RT resistance (23,40),
and EZH2 can directly initiate trimethylation of H3K27, a type of
histone methylation of the amino (N) terminal tail of the core
histone H3, which is associated with downregulation of nearby genes
through the formation of heterochromatic regions (41). Expression of H3K27me3 is associated
with radiation, and the loss of H3K27me3 is frequently observed in
radiation-associated angiosarcoma of the breast (42). In addition, the loss of H3K27me3
expression is a sensitive marker for the detection of malignant
tumors of peripheral nerve sheaths induced by radiation (43). Therefore, radiation or radiotherapy
may attenuate H3K27me3 expression and induce the expression of
genes associated with oncogenesis.
The results of the present study suggested that PLCε
knockdown mediated an increase in the presence of H3K27me3 in the
PARP1 promoter region by increasing EZH2 levels and reducing the
expression of PARP1, which may have disrupted the AR/PARP1 loop and
impaired downstream target genes, including the DDR associated
proteins, DNA-PKcs and γ-H2AX; this in turn resulted in a reduction
in DDR in CRPC, which serves a critical role in radiotherapy
(Fig. 8). However, the mechanism by
which PLCε regulated H3K27me3 levels in the PARP1 promoter region
remains unclear.
In conclusion, the results of the present study
suggested that PLCε knockdown enhanced the radiosensitivity of CRPC
by preventing nuclear translocation of AR and consequently
inhibiting AR signaling. In addition, PLCε knockdown impaired the
AR/PARP1 positive feedback loop by suppressing AR signaling or
epigenetically through regulating PARP1 expression, which
ultimately resulted in the downregulation of the DNA-PKcs axis and
reduction of DDR. This novel mechanism may represent a potential
therapeutic approach for treating patients with CRPC with RT
resistance.
Acknowledgements
The authors would like to thank Dr Shunhe Wang
(Department of Histology and Embryology, Chongqing Medical
University, Chongqing, China) for providing assistance in tissue
slicing and Dr Kui Liao (Department of Radiology, The First
Affiliated Hospital of Chongqing Medical University, Chongqing,
China) for providing technical assistance with radiation.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
JP and XW designed the experiments. JP and TL
collected the specimen and analyzed the clinical data. JP, TL, NL
and LL performed the experiments. JP, TL and ZQ wrote the
manuscript. XW and CL provided technical support during the
research project and supervised the progress of the experiments. TL
and ZQ analyzed the statistical data. JP and TL created the
figures. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the First Affiliated Hospital of Chongqing Medical
University (Chongqing, China). Informed consent was obtained from
the patients or their family members. The xenograft experiments
were approved by the Chongqing Medical University Institutional
Animal Care and Use Committee and the Animal Ethics Committee.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hayden AJ, Catton C and Pickles T:
Radiation therapy in prostate cancer: A risk-adapted strategy. Curr
Oncol. 2 (Suppl 17):S18–S24. 2010.
|
|
4
|
Paller CJ and Antonarakis ES: Management
of biochemically recurrent prostate cancer after local therapy:
Evolving standards of care and new directions. Clin Adv Hematol
Oncol. 11:14–23. 2013.PubMed/NCBI
|
|
5
|
Smrcka AV, Brown JH and Holz GG: Role of
phospholipase Cε in physiological phosphoinositide signaling
networks. Cell Signal. 24:1333–1343. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bunney TD and Katan M: PLC regulation:
Emerging pictures for molecular mechanisms. Trends Biochem Sci.
36:88–96. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhou RM, Li Y, Wang N, Liu BC, Chen ZF and
Zuo LF: PLC-ε1 Gene polymorphisms significantly enhance the risk of
esophageal squamous cell carcinoma in individuals with a family
history of upper gastrointestinal cancers. Arch Med Res.
43:578–584. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bunney TD, Baxendale RW and Katan M:
Regulatory links between PLC enzymes and ras superfamily GTPases:
Signalling via PLCepsilon. Adv Enzyme Regul. 49:54–58. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang LD, Zhou FY, Li XM, Sun LD, Song X,
Jin Y, Li JM, Kong GQ, Qi H, Cui J, et al: Genome-wide association
study of esophageal squamous cell carcinoma in Chinese subjects
identifies susceptibility loci at PLCE1 and C20orf54. Nat Genet.
42:759–763. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang Y, Wu X, Ou L, Yang X, Wang X, Tang
M, Chen E and Luo C: PLCε knockdown inhibits prostate cancer cell
proliferation via suppression of Notch signalling and nuclear
translocation of the androgen receptor. Cancer Lett. 362:61–69.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li L, Du Z, Gao Y, Tang Y, Fan Y, Sun W,
Li T, Liu N, Yuan M, Fan J, et al: PLCε knockdown overcomes drug
resistance to androgen receptor antagonist in castration-resistant
prostate cancer by suppressing the wnt3a/β-catenin pathway. J Cell
Physiol. 234:15472–15486. 2019. View Article : Google Scholar
|
|
12
|
Fan J, Fan Y, Wang X, Niu L, Duan L, Yang
J, Li L, Gao Y, Wu X and Luo C: PLCε regulates prostate cancer
mitochondrial oxidative metabolism and migration via upregulation
of Twist1. J Exp Clin Cancer Res. 38:3372019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Culig Z and Santer FR: Androgen receptor
signaling in prostate cancer. Cancer Metastasis Rev. 33:413–427.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Coutinho I, Day TK, Tilley WD and Selth
LA: Androgen receptor signaling in castration-resistant prostate
cancer: A lesson in persistence. Endocr Relat Cancer. 23:T179–T197.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chin Y, Clegg NJ and Scher HI:
Anti-androgens and androgen-depleting therapies in prostate cancer:
New agents for an established target. Lancet Oncol. 10:981–991.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ta HQ and Gioeli D: The convergence of DNA
damage checkpoint pathways and androgen receptor signaling in
prostate cancer. Endocr Relat Cancer. 21:R395–R407. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Goodwin JF, Schiewer MJ, Dean JL,
Schrecengost RS, de Leeuw R, Han S, Ma T, Den RB, Dicker AP, Feng
FY and Knudsen KE: A hormone-DNA repair circuit governs the
response to genotoxic insult. Cancer Discov. 3:1254–1271. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Spratt DE, Evans MJ, Davis BJ, Doran MG,
Lee MX, Shah N, Wongvipat J, Carnazza KE, Klee GG, Polkinghorn W,
et al: Androgen receptor upregulation mediates radioresistance
after ionizing radiation. Cancer Res. 75:4688–4696. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang J: Poly (ADP-ribose) polymerase
inhibitor: An evolving paradigm in the treatment of prostate
cancer. Asian J Androl. 16:401–406. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schiewer MJ, Goodwin JF, Han S, Brenner
JC, Augello MA, Dean JL, Liu F, Planck JL, Ravindranathan P,
Chinnaiyan AM, et al: Dual roles of PARP-1 promote cancer growth
and progression. Cancer Discov. 2:1134–1149. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pu H, Horbinski C, Hensley PJ, Matuszak
EA, Atkinson T and Kyprianou N: PARP-1 regulates
epithelial-mesenchymal transition (EMT) in prostate tumorigenesis.
Carcinogenesis. 35:2592–2601. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
EAU Guidelines. Edn. presented at the EAU
Annual Congress Barcelona 2019. ISBN 978-94-92671-04-2. EAU
Guidelines Office, Arnhem. 2019.
|
|
23
|
Hunt CR, Ramnarain D, Horikoshi N, Iyengar
P, Pandita RK, Shay JW and Pandita TK: Histone modifications and
DNA double-strand break repair after exposure to ionizing
radiations. Radiat Res. 179:383–392. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Davey RA and Grossmann M: Androgen
receptor structure, function and biology: From bench to bedside.
Clin Biochem Rev. 37:3–15. 2016.PubMed/NCBI
|
|
25
|
Friedlander TW and Ryan CJ: Targeting the
androgen receptor. Urol Clin North Am. 39:453–464. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jones CU, Hunt D, Mcgowan DG, Amin MB,
Chetner MP, Bruner DW, Leibenhaut MH, Husain SM, Rotman M, Souhami
L, et al: Radiotherapy and short-term androgen deprivation for
localized prostate cancer. N Engl J Med. 365:107–118. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Roach M III, Bae K, Speight J, Wolkov HB,
Rubin P, Lee RJ, Lawton C, Valicenti R, Grignon D and Pilepich MV:
Short-term neoadjuvant androgen deprivation therapy and
external-beam radiotherapy for locally advanced prostate cancer:
Long-term results of RTOG 8610. J Clin Oncol. 26:585–591. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bolla M, Van Tienhoven G, Warde P, Dubois
JB, Mirimanoff RO, Storme G, Bernier J, Kuten A, Sternberg C,
Billiet I, et al: External irradiation with or without long-term
androgen suppression for prostate cancer with high metastatic risk:
10-year results of an EORTC randomised study. Lancet Oncol.
11:1066–1073. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pignot G, Maillet D, Gross E, Barthelemy
P, Beauval JB, Constans-Schlurmann F, Loriot Y, Ploussard G, Sargos
P, Timsit MO, et al: Systemic treatments for high-risk localized
prostate cancer. Nat Rev Urol. 15:498–510. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Polkinghorn WR, Parker JS, Lee MX, Kass
EM, Spratt DE, Iaquinta PJ, Arora VK, Yen WF, Cai L, Zheng D, et
al: Androgen receptor signaling regulates DNA repair in prostate
cancers. Cancer Discov. 3:1245–1253. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bunney TD and Katan M: Phospholipase C
epsilon: Linking second messengers and small GTPases. Trends Cell
Biol. 16:640–648. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ou L, Guo Y, Luo C, Wu X, Zhao Y and Cai
X: RNA interference suppressing PLCE1 gene expression decreases
invasive power of human bladder cancer T24 cell line. Cancer Genet
Cytogenet. 200:110–119. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Du HF, Ou LP, Yang X, Song XD, Fan YR, Tan
B, Luo CL and Wu XH: A new PKCα/β/TBX3/E-cadherin pathway is
involved in PLCε-regulated invasion and migration in human bladder
cancer cells. Cell Signal. 26:580–593. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cheng H, Luo C, Wu X, Zhang Y, He Y, Wu Q,
Xia Y and Zhang J: shRNA targeting PLCε inhibits bladder cancer
cell growth in vitro and in vivo. Urology. 78:474.e7–11. 2011.
View Article : Google Scholar
|
|
35
|
Ma H, Wang LE, Liu Z, Sturgis EM and Wei
Q: Association between novel PLCE1 variants identified in published
esophageal cancer genome-wide association studies and risk of
squamous cell carcinoma of the head and neck. BMC Cancer.
11:2582011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li M, Edamatsu H, Kitazawa R, Kitazawa S
and Kataoka T: Phospholipase cepsilon promotes intestinal
tumorigenesis of Apc(Min/+) mice through augmentation of
inflammation and angiogenesis. Carcinogenesis. 30:1424–1432. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Abnet CC, Freedman ND, Hu N, Wang Z, Yu K,
Shu XO, Yuan JM, Zheng W, Dawsey SM, Dong LM, et al: A shared
susceptibility locus in PLCE1 at 10q23 for gastric adenocarcinoma
and esophageal squamous cell carcinoma. Nat Genet. 42:764–767.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Luo J, Jin J, Yang F, Sun Z, Zhang W, Shi
Y, Xu J and Guan X: The correlation between PARP1 and BRCA1 in AR
positive triple-negative breast cancer. Int J Biol Sci.
12:1500–1510. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ray Chaudhuri A and Nussenzweig A: The
multifaceted roles of PARP1 in DNA repair and chromatin
remodelling. Nat Rev Mol Cell Biol. 18:610–621. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tessarz P and Kouzarides T: Histone core
modifications regulating nucleosome structure and dynamics. Nat Rev
Mol Cell Biol. 15:703–708. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gan ES, Xu Y and Ito T: Dynamics of
H3K27me3 methylation and demethylation in plant development. Plant
Signal Behav. 10:e10278512015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mentzel T and Kiss K: Reduced H3K27me3
expression in radiation-associated angiosarcoma of the breast.
Virchows Arch. 472:361–368. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Prieto-Granada CN, Wiesner T, Messina JL,
Jungbluth AA, Chi P and Antonescu CR: Loss of H3K27me3 expression
is a highly sensitive marker for sporadic and radiation-induced
MPNST. Am J Surg Pathol. 40:479–489. 2016. View Article : Google Scholar : PubMed/NCBI
|