Introduction
Pancreatic cancer is one of the leading causes of
cancer-related mortality worldwide, and the 5-year survival rate is
less than 9%; more than 50% of cases are diagnosed at an advanced
stage, which partially explains the low survival rate (1). The high mortality rate among advanced
pancreatic cancer patients is linked to their limited response to
chemotherapy and radiotherapy. The epithelial-mesenchymal
transition (EMT) phenotype is a critical mediator of drug
resistance in pancreatic cancer patients (2). The activation of the EMT process has
been proposed as the critical mechanism responsible for the
acquisition of drug resistance in pancreatic cancer cells (3,4).
Furthermore, EMT has been revealed to contribute to the invasion
and metastasis of various types of carcinomas, including pancreatic
cancer (5–7). However, the vital role of EMT in
cancer invasion and metastasis has been recently challenged based
on murine studies (8,9).
Amphiregulin (AREG) is a member of the epidermal
growth factor (EGF) family and is expressed in a plethora of
neoplasms, including ovarian, breast, bladder, colon, lung, major
salivary glands and pancreatic cancers (10–16).
AREG interacts with epidermal growth factor receptor (EGFR) to
trigger numerous signalling cascades that mediate cell survival,
proliferation, and motility. According to studies investigating
ovarian cancer (10), and breast
cancer (11), AREG plays a crucial
role in cell invasion and metastasis. AREG was linked to a reduced
life span in bladder cancer (12)
and lung cancer patients (13).
However, AREG overexpression was associated with a longer
disease-free survival in colorectal cancer (14) and mucoepidermoid carcinoma patients
(15). In our previous study, it
was revealed that AREG expression was correlated with a poor
prognosis in pancreatic cancer patients (16). To date, knowledge regarding the
molecular mechanisms underlying AREG-induced migration, invasion
and EMT in pancreatic cancer cells is limited. The aim of this
study was to detect the molecular mechanism of AREG mediating EMT
in pancreatic cancer cells in vitro and in vivo.
Materials and methods
Cell culture
Four human pancreatic cancer cell lines, PANC-1
(cat. no. CRL-1469), AsPC-1 (cat. no. CRL-1682), BxPC-3 (cat. no.
CRL-1687) and MIA PaCa-2 (cat. no. CRL-1420) were purchased from
American Type Culture Collection (ATCC). The cell lines (PANC-1,
AsPC-1, BxPC-3 and MIA PaCa-2) had been identified by specialized
STR profiling and tested for mycoplasma contamination. PANC-1 and
MIA PaCa-2 were maintained in Dulbecco's Modified Eagle's Medium
(DMEM), while AsPC-1 and BxPC-2 were maintained in RPMI-1640 medium
(both from Corning Incorporated). The medium was supplemented with
10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific,
Inc.), 1% penicillin/streptomycin and 1 mM glutamine (Invitrogen;
Thermo Fisher Scientific, Inc.). The cells were grown in 5%
CO2 at 37°C in a humidified atmosphere.
Antibodies and reagents
EMT Antibody Sampler Kit (cat. no. 9782), NF-κB
pathway Sampler Kit (cat. no. 9936), Phospho-EGF Receptor Antibody
Sampler Kit (cat. no. 9922) and anti-phospho-STAT3 (cat. no. 9145)
antibody were purchased from Cell Signaling Technology, Inc.,
except for anti-ERK (cat. no. sc-154) and anti-phospho-ERK (cat.
no. sc-7976-R), anti-AKT (cat. no. sc-81434) and anti-phospho-AKT
(cat. no. sc-81433) and anti-STAT3 (cat. no. sc-482) antibodies
(Santa Cruz Biotechnology, Inc.). The recombinant human AREG
protein (cat. no. 262-AR-100) and normal goat IgG control (cat. no.
AB-108-C) were purchased from R&D Systems. PD153035 (EGFR
tyrosine kinase inhibitor), U0126 (MEK1/2 inhibitor) and QNZ (NF-κB
inhibitor) were purchased from Selleckchem. PD153035, U0126 and QNZ
were dissolved in dimethyl sulphoxide (DMSO) and maintained at
−20°C until use. AREG and IgG were dissolved in sterile PBS
containing 0.1% FBS just before use.
Preparation of conditioned medium
The cells were grown to 80–90% confluence in
25-cm2 flasks and then rinsed three times with
serum-free medium. Then, the cells were incubated with serum-free
medium for 48 h. The conditioned medium was harvested and
centrifuged (335 × g for 10 min, 4°C). After centrifugation, the
supernatant was collected and stored at −80°C for the subsequent
enzyme-linked immunosorbent assay (ELISA).
Quantitative real-time PCR
Detailed procedures for total RNA extraction and
RT-qPCR have been previously described (17). The primer sequences were as follows:
AREG, 5′-TGAGATGTCTTCAGGGAGTG-3′ (sense) and
5′-AGCCAGGTATTTGTGGTTCG-3′ (antisense); EGFR,
5′-GGATGCCGACGAGTACCTC-3′ (sense) and 5′-GCTTTGCAGCCCATTTCTAT-3′
(antisense); and glyceraldehyde-3-phosphate dehydrogenase (GAPDH),
5′-GGCATCCTGGGCTACACTG-3′ (sense) and 5′-GTGGTCGTTGAGGGCAAT-3′
(antisense). GAPDH was used as a control. The mRNA levels were
analysed using the comparative quantification cycle (Cq) method
(2−ΔΔCq) (18).
Western blot analysis
Western blot analysis was performed as previously
described (17). Dilution rates for
all primary and secondary antibodies were 1:1,000 and 1:10,000,
respectively. The blots were probed with ECL Plus Western Blotting
Detection kit (Pierce Biotechnology; Thermo Fisher Scientific,
Inc.). The densities of the bands were scanned and analysed using
ImageJ software (version 1.48v; National Institutes of Health).
ELISA
The secretion level of AREG was assessed using an
AREG ELISA kit (cat. no. DAR00; R&D systems) according to the
manufacturer's instructions.
RNA interference
The siRNAs targeting AREG were as follows:
AREG-Homo-390 (siAR-390), 5′-GGAUUUGAGGUUACCUCAATT-3′ (sense) and
5′-UUGAGGUAACCUCAAAUCCTT-3′ (antisense); AREG-Homo-817 (siAR-817),
5′-GCAUGAUUGACAGUAGUUUTT-3′ (sense) and 5′-AAACUACUGUCAAUCAUGCTT-3′
(antisense); and AREG-Homo-348 (siAR-348),
5′-UCUGGGAAGCGUGAACCAUTT-3′ (sense) and 5′-AUGGUUCACGCUUCCCAGATT-3′
(antisense). The siRNA control sequences were
5′-UUCUCCGAACGUGUCACGUTT-3′ (sense) and 5′-ACGUGACACGUUCGGAGAATT-3′
(antisense). All siRNAs were purchased from Shanghai GenePharma
Co., Ltd. AREG was transfected into AsPC-1 cells in 6-well plates
using Lipofectamine RNAiMAX Reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). Total protein and RNA were extracted, and the
knockdown efficiency of AREG was verified by western blotting and
RT-qPCR analyses, respectively.
Immunofluorescence
AsPC-1 cells were transfected with AREG siRNA
(siAR-390) or control siRNA (NC) for 48 h. PANC-1 cells were
treated with AREG (100 ng/ml, AREG), or normal IgG (100 ng/ml, NC)
for 48 h. A total of 2×105 cells were seeded on
coverslips. After 24 h, serum-free culture medium was removed from
each well and replaced with 4% paraformaldehyde, and the plates
were incubated for 15 min at room temperature for fixation. Then,
the cells were washed with PBS three times. After blockade of
non-specific epitopes by 10% normal goat serum for 1 h at room
temperature, primary antibodies against E-cadherin (1:100 dilution;
cat. no. 3195; Cell Signaling Technology, Inc.) or vimentin (1:200
dilution; cat. no. 5741; Cell Signaling Technology, Inc.) were
added. After overnight incubation at 4°C, the primary antibody was
washed by rinsing three times with PBS, followed by incubation of
secondary antibodies (Alexa Fluor®488 Conjugate, cat.
no. 4412 or Alexa Fluor®594 Conjugate, cat. no. 8889,
Cell Signaling Technology, Inc., 1:1,000 dilution) for 1 h at room
temperature. The cells were stained with DAPI and observed under a
fluorescence microscope (Olympus Corp.).
Transwell assay
Transwell chambers (8-µm-pore; Corning Inc.) were
used to evaluate cell migration and invasion. Matrigel was applied
in the invasion assay. Briefly, after transfection or different
treatment conditions for 48 h, 2×104 cells were
resuspended in 200 µl of serum-free medium and added to the upper
chamber. Then, medium with 10% FBS was added to the opposite
(lower) chamber. After 24 h, the cells remaining in the upper
chamber were wiped away. The cells on the Transwell membrane were
fixed and stained (by 4% paraformaldehyde and 0.1% crystal violet,
respectively, for 15 min each at room temperature). Finally, the
number of cells was counted in five random fields (magnification,
×100; BX53 upright microscope; Olympus Corp.).
Wound-healing assay
PANC-1 cells were treated with AREG (100 ng/ml)
alone (AREG), AREG combined with QNZ (20 µM, AREG+QNZ), or normal
IgG (100 ng/ml, NC) for 48 h. A total of 5×105 cells
were seeded in a 6-well plate and grown to 90% confluence. A
sterile 200-µl pipette tip was used to form a constant width gap.
After removal of cell debris, 1 ml of serum-free medium was added
to each well. The scratched areas were imaged at 0, 24, 48, and 72
h using a DP-73 scientific camera on a phase-contrast microscope
(Olympus Corp.). The cell-free area was calculated using ImageJ
software (version 1.48v; National Institutes of Health), and the
percentage of the original cell-free zone at each time-point was
used to evaluate the speed of wound healing. The values were the
means of three independent experiments.
Lentivirus construction and
transduction
Short hairpin RNA (shRNA) targeting AREG was
designed and synthesized by Shanghai GeneChem Co., Ltd., and used
to construct the experimental plasmids (pGV248/AREG-shRNA); the
shRNA was inserted into an AgeI- and EcoRI-linearized
pGV248. The negative control vector (pGV248/control-shRNA) was
constructed similarly by inserting an unrelated shRNA sequence. All
inserted sequences were verified by sequencing. To generate stable
cell lines, recombinant lentiviruses (namely, LV-AREG and LV-con)
were generated and subsequently used to infect AsPC-1 cells.
Orthotopic model of pancreatic
cancer
Twelve six- to seven-week-old female athymic nude
mice (BALB/c nu/nu) were randomly assigned to 2 groups. Six mice
with LV-AREG cells were distributed to the experimental group,
while the other 6 mice with LV-con cells were in the control group.
In detail, the mice were anaesthetized and placed in a supine
position; the abdomen was disinfected with 70% alcohol, and then,
an incision of <1 cm was performed to expose the pancreas.
AsPC-1 cells (2×106) were injected into the tail of the
pancreas, and the wound was sutured. The mice were fed in SPF
conditions and weighed twice weekly. The mice were sacrificed by
CO2 asphyxiation on the 39th day after the injection.
The pancreas was removed, and the tumours were weighed. The
stomach, spleen, liver, intestine and kidney were examined for the
presence of metastases. This study was conducted with the approval
of the Institutional Animal Care and Use Committee of Peking Union
Medical College Hospital and was in accordance with the National
Policy on Use of Laboratory Animals.
Statistical analysis
All data were sorted and analysed using SPSS
software for Windows, version 17.0 (SPSS, Inc.). The results are
expressed as the mean ± SD. Significance was evaluated by
performing Student's t-tests or one-way analysis of variance
(ANOVA) with Tukey's post hoc test. A P-value <0.05 was
considered to indicate a statistically significant difference.
Results
EGFR and AREG expression in pancreatic
cancer cells
To investigate EGFR and AREG expression in
pancreatic cancer cell lines, RT-qPCR, western blot analysis and
ELISA were performed. These analyses revealed that EGFR expression
was significantly high in the AsPC-1 cells and BxPC-3 cells
compared with that in MIA PaCa-2 cells (Fig. 1A and B). AREG expression was
significantly high in the AsPC-1 cells, BxPC-3 cells and MIA PaCa-2
cells, compared with that in PANC-1 cells (Fig. 1C and D). AsPC-1 (high EGFR
expression and high AREG expression) and PANC-1 cells (high EGFR
expression and low AREG expression) were selected for the
subsequent loss-of-function and gain-of-function experiments. To
characterize the biological effects of AREG, AREG siRNAs were used
to suppress the endogenous expression of AREG in AsPC-1 cells.
Three different AREG siRNAs and 1 control siRNA were used. The
results revealed that siAR-390 was most effective in inhibiting
AREG mRNA (65% reduction) and protein (64% reduction) expression in
AsPC-1 cells (Fig. S1). Therefore,
siAR-390 was used in the subsequent experiments.
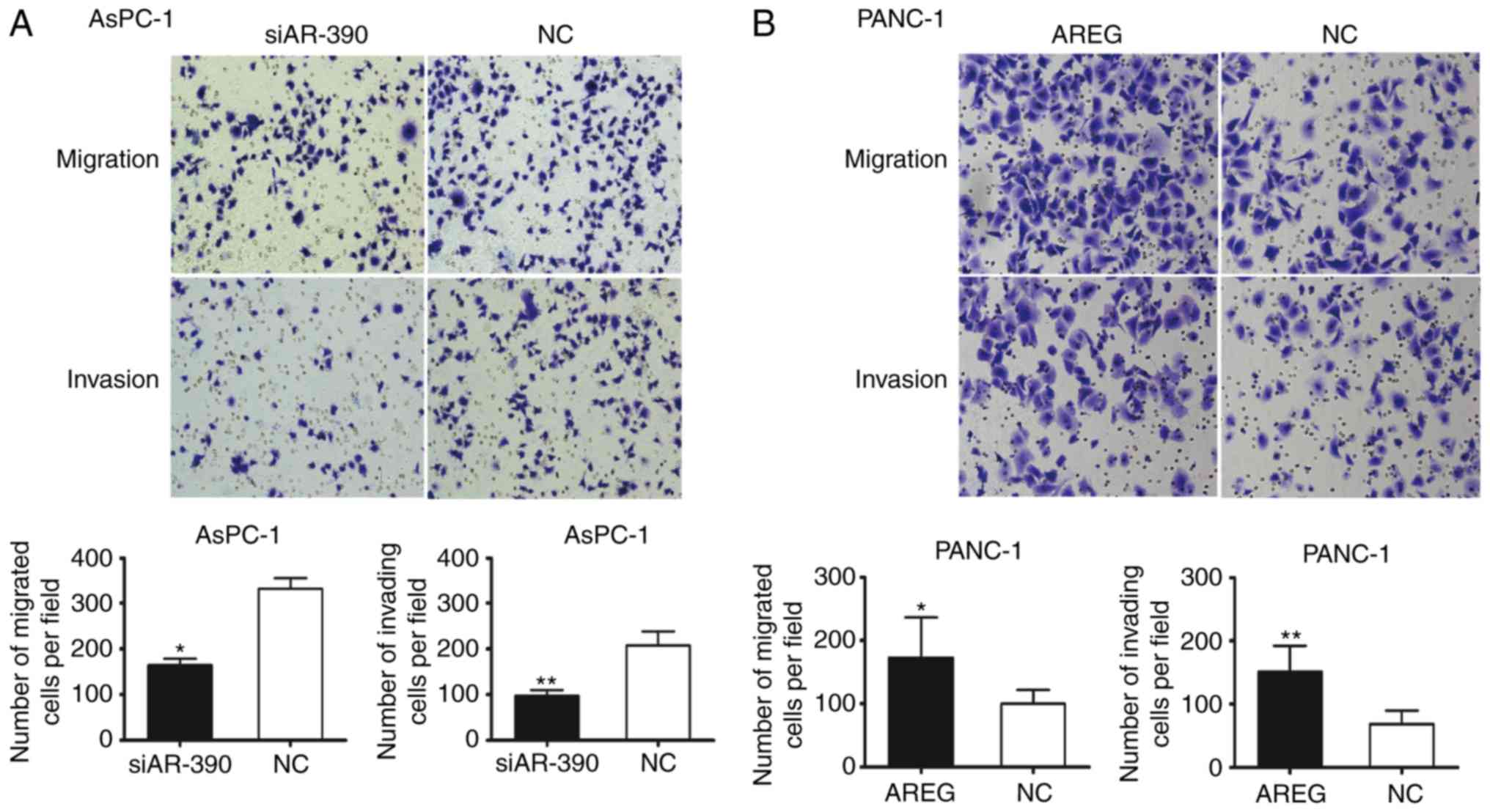
AREG facilitates the migration and
invasion of pancreatic cancer cells
Transwell assays were performed to monitor whether
AREG could affect pancreatic cancer cell migration and invasion.
The results revealed a nearly 50% reduction in cell migration and
47% reduction in cell invasion in AREG siRNA-transfected AsPC-1
cells (P<0.05 and P<0.01, Fig.
2A). Knockdown of AREG significantly decreased the migration
ability of BxPC-3 cells (P<0.01, Fig. S2). To confirm the involvement of
AREG in pancreatic cancer cell migration and invasion, recombinant
human AREG treatment was used. Exposure to exogenous AREG (100
ng/ml) significantly promoted the migration and invasion of the
PANC-1 cells (P<0.05 and P<0.01, Fig. 2B). It was revealed that AREG was
expressed in activated pancreatic stellate cells (PSCs), and
co-culture with PSCs significantly increased the migration and
invasion of PANC-1 cells (Fig.
S3). The results revealed that AREG facilitated the migration
and invasion of pancreatic cancer cells.
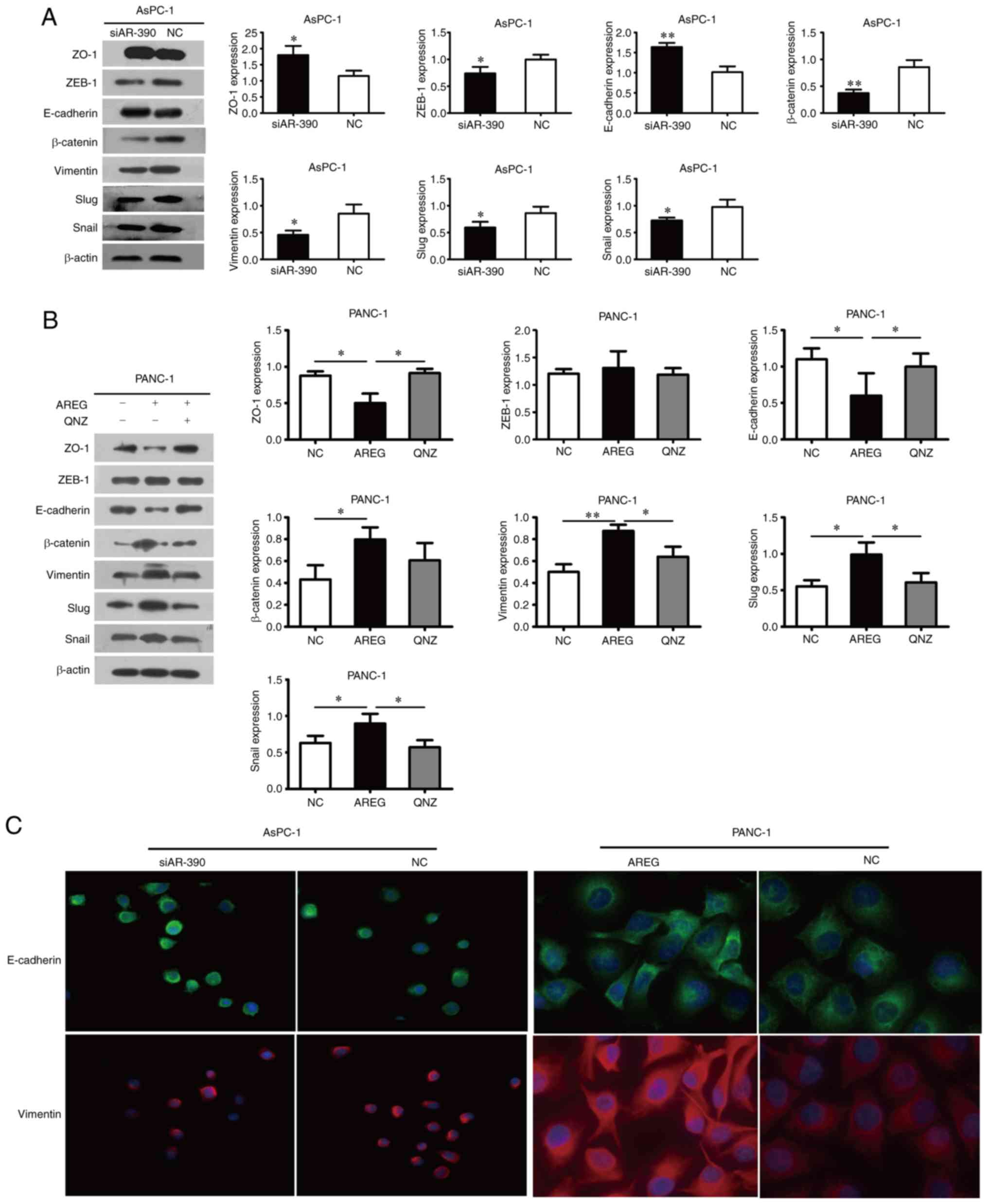
AREG promotes EMT in pancreatic cancer
cells
E-cadherin and ZO-1 expression was upregulated,
however, vimentin, β-catenin, Snail, Slug, and ZEB-1 levels were
downregulated in the AREG siRNA-transfected AsPC-1 cells (Fig. 3A). Knockdown of AREG upregulated the
expression of E-cadherin and downregulated the expression of
β-catenin and vimentin in BxPC-3 cells (Fig. S2). As anticipated, these proteins
displayed the opposite expression profiles in the AREG-treated (100
ng/ml) PANC-1 cells, whereas ZEB-1 expression exhibited no evident
change (Fig. 3B). The changes in
the expression of E-cadherin and vimentin induced by AREG were also
examined by immunofluorescence. The results revealed the
upregulated expression of E-cadherin and the downregulated
expression of vimentin in AREG siRNA-transfected AsPC-1 cells. In
contrast, the downregulated expression of E-cadherin and the
upregulated expression of vimentin were observed in AREG-treated
PANC-1 cells (Fig. 3C).
AREG activates the EGFR/ERK signalling
pathway in pancreatic cancer cells
To further explore the upstream signalling that may
be responsible for AREG-induced EMT, the phosphorylation of EGFR,
ERK, AKT and STAT3 was evaluated by performing western blot
analyses. The phosphorylation levels of EGFR, ERK, AKT and STAT3
levels were downregulated in the AREG siRNA-transfected AsPC-1
cells (Fig. 4A). As anticipated,
the exogenous treatment of AREG upregulated the phosphorylation
levels of EGFR, ERK, AKT and STAT3 to varying degrees in PANC-1
cells. It was revealed that the small molecule inhibitors of EGFR
(PD153035) suppressed the AREG-induced ERK, AKT and STAT3
phosphorylation (Fig. 4B). The
expression levels of total EGFR, ERK, AKT, and STAT3 were not
significantly altered in pancreatic cancer cells (Fig. 4A and B). The results revealed a
significant decrease in phospho-ERK in siRNA-transfected AsPC-1
cells (P<0.01) and a significant increase in phospho-ERK in
exogenous AREG-treated PANC-1 cells (P<0.05). To further
characterize the effect of AREG on ERK activation, ERK
phosphorylation was examined at various time-points after AREG
stimulation; the peak phosphorylated ERK level occurred 30 min
after AREG stimulation (Fig. 4C).
The small molecule inhibitor of MEK1/2 (U0126) significantly
reduced the phosphorylation level of ERK in exogenous AREG-treated
PANC-1 cells.
 | Figure 4.AREG activates the EGFR/ERK
signalling pathway in pancreatic cancer cells. (A) Phosphorylation
and total protein expression levels of EGFR, STAT3, AKT, and ERK in
AsPC-1 cells transfected with AREG siRNA (siAR-390) or control
siRNA (NC). (B) In PANC-1 cells, the exogenous treatment of AREG
induced EGFR signalling as revealed by the varying degrees of
activation of the p-ERK, p-AKT and p-STAT3 levels. The enhancement
effect was blocked by PD153035 (an inhibitor of EGF receptor
tyrosine kinase, 5 µM) or U0126 (an inhibitor of MEK1/2, 20 µM).
(C) Activation of ERK in response to AREG in PANC-1 cells. AREG
stimulation resulted in a significant induction of p-ERK, which
stabilized at 30 min. Graphs are presented as the relative density
of the phosphoprotein vs. the total protein. *P<0.05 compared
with the control untreated cells at 0 min. *P<0.05, **P<0.01.
AREG, amphiregulin; EGFR, epidermal growth factor receptor. |
Inhibition of the NF-κB signalling
pathway suppresses AREG-induced cell migration, invasion and EMT in
pancreatic cancer cells
Previous studies have suggested that NF-κB plays an
essential role in the induction and maintenance of EMT (19,20).
We proposed that the role of AREG-induced pancreatic cancer cell
migration, invasion and EMT may be through regulation of the NF-κB
signalling pathway. To evaluate this possibility, the nuclear
accumulation level of NF-κB/p65 and the phosphorylation level of
IκBα were analysed by western blot analysis. It was revealed that
the nuclear accumulation level of NF-κB/p65 and the phosphorylation
level of IκBα were significantly decreased in AREG
siRNA-transfected AsPC-1 cells compared with the siRNA control
group (P<0.05, Fig. 5A). In
contrast, the nuclear accumulation level of NF-κB/p65 and the
phosphorylation level of IκBα were significantly increased in
AREG-treated PANC-1 cells. U0126 attenuated the AREG-promoted
nuclear accumulation of NF-κB/p65 in PANC-1 cells (P<0.05,
Fig. 5B). Furthermore, the role of
the NF-κB signalling pathway in AREG-induced cell migration,
invasion and EMT in pancreatic cancer cells was investigated using
the selective NF-κB inhibitor QNZ. PANC-1 cells were pre-treated
with 20 µM QNZ for 1 h, followed by incubation with 100 ng/ml AREG
for 48 h. Based on the wound-healing assay, QNZ significantly
inhibited the migratory ability of PANC-1 cells induced by AREG at
72 h (P<0.01, Fig. 5C).
Transwell assay results revealed that QNZ abolished the invasive
properties of PANC-1 cells induced by AREG (P<0.01, Fig. 5D). According to the western blot
analysis, pre-treatment of PANC-1 cells with QNZ resulted in the
upregulation of E-cadherin and ZO-1 and the downregulation of
vimentin, Slug, and Snail (Fig.
3B). All of the aforementioned results indicated that
inhibition of the NF-κB signalling pathway suppressed AREG-induced
cell migration, invasion and EMT in pancreatic cancer cells.
 | Figure 5.The NF-κB pathway is involved in
AREG-induced migration and invasion of pancreatic cancer cells. (A)
Representative images and analysis of western blots for IκBα,
p-IκBα, nuclear p65 in AsPC-1 cells transfected with AREG siRNA
(siAR-390) or control siRNA (NC). (B) Representative images and
analysis of western blots for IκBα, p-IκBα, nuclear p65 in PANC-1
cells treated with AREG (100 ng/ml), U0126 (20 µM), PD153035 (5
µM), and normal IgG (100 ng/ml, NC). (C) Wound-healing assay. QNZ
inhibited the migratory ability of PANC-1 cells induced by AREG.
(D) Transwell assay. QNZ abolished the invasive properties of
PANC-1 cells induced by AREG. PANC-1 cells treated with AREG (100
ng/ml) alone (AREG), AREG combined with QNZ (20 µM, AREG+QNZ), or
normal IgG (100 ng/ml, NC). The data represent the mean ± SD of at
least three independent experiments, *P<0.05, **P<0.01. AREG,
amphiregulin. |
Orthotopic model of pancreatic
cancer
The survival rate in our model was 91.7%; one mouse
in the LV-AREG group died the day after the injection. All mice in
the LV-AREG and LV-con groups exhibited a tumour at the site of the
injection of cells into the pancreas tail. The incidence of
regional and distant metastasis was lower in the LV-AREG group
compared with the LV-con group (Table
I; Fig. 6A). Macroscopically,
the tumours appeared as firm, white, irregular masses. There had
been a downward trend in tumour growth with AREG silencing in the
orthotopic model of pancreatic cancer (0.74±0.31 g vs. 1.04±0.17
g); however, no significant differences were revealed between the
LV-AREG group and LV-con groups (P>0.05, Fig. 6B). H&E staining confirmed
pancreatic tumour development in mice (Fig. 6C).
 | Table I.Incidence of metastasis in a
pancreatic cancer orthotopic model. |
Table I.
Incidence of metastasis in a
pancreatic cancer orthotopic model.
| Metastatic
sites | LV-AREG (n=5) | LV-con (n=6) |
|---|
| Liver | 0 | 3 |
| Spleen | 1 | 2 |
| Intestine | 0 | 1 |
| Stomach | 0 | 1 |
| Kidney | 1 | 1 |
Discussion
Pancreatic cancer is one of the leading causes of
cancer-related mortality worldwide. The drug-resistant
characteristics of pancreatic cancer cells result in their partial
responses to traditional cytotoxic chemotherapy. The EMT phenotype
is emerging as an important feature of drug-resistant pancreatic
cancer cells (2–4). EMT modifies the adhesion molecules
expressed by the epithelial cells, allowing the cells to adopt
migratory and invasive behaviours (21).
AREG gene overexpression has been observed in a wide
variety of human cancer tissues (22). Willmarth et al (11) reported that AREG upregulated
numerous genes involved in cell motility and invasion in the human
mammary epithelial cell line MCF10A. Liu et al (23) revealed that AREG promoted cancer
cell motility in osteosarcoma and upregulated the expression of
ICAM-1 through the EGFR/PI3K/Akt/NF-κB signalling pathway.
Loss-of-function and gain-of-function experiments have revealed
that AREG modulates the cell migration and invasion of pancreatic
cancer cells in vitro. In an orthotopic model of pancreatic
cancer, AREG silencing was revealed to be related to smaller tumour
sizes and reduced metastatic ability. The present data are in line
with the results of previous studies, suggesting that AREG plays a
critical role in the motility and metastasis of pancreatic cancer
cells.
EMT may be a key process for the acquisition of
capabilities required for metastasis (7,21). The
functional decrease in E-cadherin is among the hallmarks of EMT.
AREG has been reported to stimulate ovarian cancer cell invasion by
downregulating the expression level of E-cadherin (10). AREG was revealed to markedly
decrease E-cadherin expression in psoriatic lesions from both
AREG-transgenic mice and individuals with psoriasis (24). Consistent with these studies, in the
present study, knockdown of AREG upregulated the expression of
E-cadherin and downregulated the expression of β-catenin and
vimentin in AsPC-1 and BxPC-3 cells. The loss of cell polarity
complexes and adhesion complexes leads to the failure to maintain
epithelial cell polarity, induces EMT and enhances cell invasive
potential (25). Kleeff et
al have revealed that ZO-1 was significantly increased in
pancreatic cancer (26). In the
present study, the inhibition of AREG induced ZO-1 expression in
pancreatic cancer cells. Transcriptional repression mediated by
factors from the Snail, ZEB and basic helix-loop-helix families is
a basic mechanism of the dynamic silencing of the E-cadherin
promoter CDH1 (27). Snail was
revealed to suppress the expression of E-cadherin and induce the
expression of the mesenchymal markers fibronectin and ZEB-1 in
tumour cells (28,29). The knockdown of Slug in glioblastoma
cells decreased invasion and increased survival in a mouse
intracranial human glioblastoma transplantation model (30). Consistently, in the present study,
AREG increased the levels of Snail and Slug. Thus, AREG may be
involved in EMT in pancreatic cancer cells.
Numerous cellular pathways have been implicated in
the regulation of the EMT process in pancreatic cancer (6,31). The
present results revealed that AREG induced the activation of the
ERK, AKT and STAT3 pathways, although the activation of the ERK
pathway was the most marked. NF-κB is constitutively active in
different cancers. It was revealed that AREG induced the activation
of IκBα and NF-κB in pancreatic cancer cells. The pharmacological
inhibition of EGFR or ERK attenuated the effects of the
AREG-induced nuclear accumulation of NF-κB. Thus, the
EGFR/ERK/NF-κB axis was likely involved in AREG-mediated pancreatic
cancer cell migration, invasion and EMT.
The stroma plays a dynamic role in tumour cell
proliferation, invasion and metastasis in pancreatic cancer
(32). PSCs play a critical role in
the tumour desmoplasticity of pancreatic cancer (33,34).
PSCs were revealed to promote EMT in pancreatic cancer cells and
facilitate BxPC-3 cell invasion (35,36).
The expression of AREG by PSCs and its roles in the tumour
microenvironment remains elusive. In the present study, it was
revealed that AREG was expressed in activated PSCs, and co-culture
with PSCs significantly increased the migration and invasion of
PANC-1 cells. However, using an anti-human AREG-neutralizing
antibody did not decrease the migration and invasion abilities of
PANC-1 cells, indicating that AREG was not the main inducer of
PANC-1 cell migration in PSC secretions. In future research, the
role of how AREG affects the biological characteristics of PSCs
requires further thorough investigation. The animal experiment was
originally designed to verify whether knockdown of AREG would
affect tumorigenicity in vivo. Although there was a trend in
reduced tumour burden with AREG silencing, no significant
differences were revealed between the LV-AREG group and LV-con
groups (P>0.05). In a future experimental design, appropriate
increasing of the sample size of animal experiments will be
considered in order to obtain more sample sizes for subsequent
experiments and render the research results more complete.
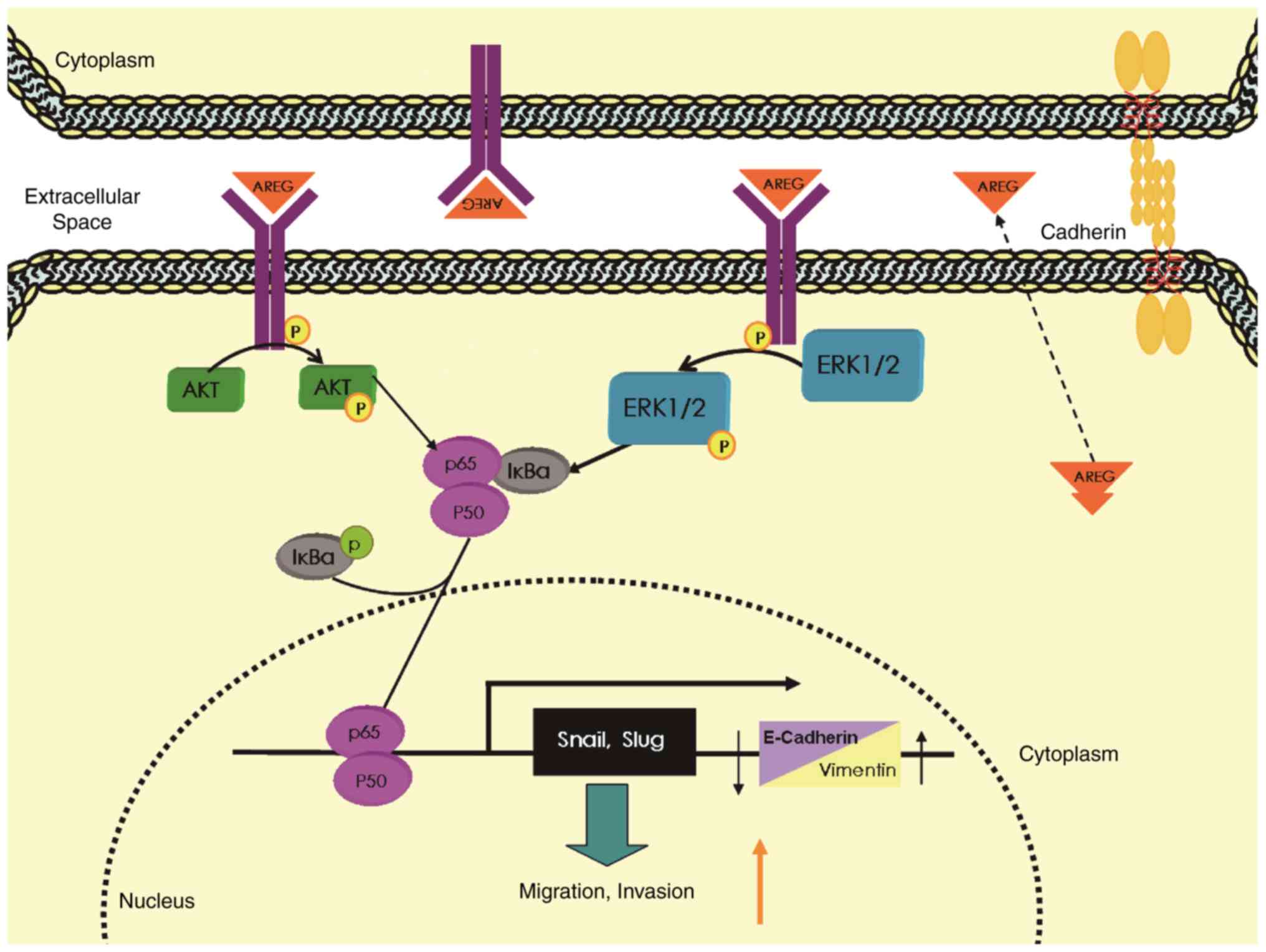
In conclusion, the EGFR ligand AREG plays an
important role in NF-κB-mediated migration and invasion as well as
EMT in pancreatic cancer cells by activating the EGFR/ERK/NF-κB
signalling pathway (Fig. 7). AREG
may be a promising target for the treatment of pancreatic
cancer.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (31471366), the Chinese Academy of
Medical Sciences (CAMS) Initiative for Innovative Medicine
(CAMS-I2M) 2016-I2M-1-002 and the National Natural Science
Foundation of China (81502625). The study sponsors did not play any
role in the study design, collection, analysis, interpretation of
data and in the writing of the manuscript.
Availability of data and material
The datasets used during and/or analysed during the
present study are available from the corresponding author on
reasonable request.
Authors' contributions
LiW was mainly responsible for the research and
writing the manuscript. LilW assisted in selecting the specimens
and performing quantitative real-time PCR and RNA interference
assays and the orthotopic model of pancreatic cancer. HZ
contributed to the immunofluorescence and Transwell assays and
lentivirus construction and transduction. JL assisted with the
western blot assays and ELISA. ZZ performed the statistical
analyses. HW supplied the technical analysis and performed the
manuscript revisions. ZL conceived and designed the whole study.
All authors read and approved the final manuscript and agree to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
The present study was conducted with the approval of
the Institutional Animal Care and Use Committee of Peking Union
Medical College Hospital and was in accordance with the National
Policy on Use of Laboratory Animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2019. CA Cancer J Clin. 69:7–34. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Amrutkar M and Gladhaug I: Pancreatic
cancer chemoresistance to gemcitabine. Cancers. 9:1572017.
View Article : Google Scholar :
|
|
3
|
Du B and Shim JS: Targeting
Epithelial-mesenchymal transition (EMT) to overcome drug resistance
in cancer. Molecules. 21(pii): E9652016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Arumugam T, Ramachandran V, Fournier KF,
Wang H, Marquis L, Abbruzzese JL, Gallick GE, Logsdon CD, McConkey
DJ and Choi W: Epithelial to mesenchymal transition contributes to
drug resistance in pancreatic cancer. Cancer Res. 69:5820–5828.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bronsert P, Enderle-Ammour K, Bader M,
Timme S, Kuehs M, Csanadi A, Kayser G, Kohler I, Bausch D, Hoeppner
J, et al: Cancer cell invasion and EMT marker expression: A
three-dimensional study of the human cancer-host interface. J
Pathol. 234:410–422. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang L, Wu H, Wang L, Zhang H, Lu J, Liang
Z and Liu T: Asporin promotes pancreatic cancer cell invasion and
migration by regulating the epithelial-to-mesenchymal transition
(EMT) through both autocrine and paracrine mechanisms. Cancer Lett.
398:24–36. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rhim AD, Mirek ET, Aiello NM, Maitra A,
Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK,
Vonderheide RH, et al: EMT and dissemination precede pancreatic
tumor formation. Cell. 148:349–361. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fischer KR, Durrans A, Lee S, Sheng J, Li
F, Wong ST, Choi H, El Rayes T, Ryu S, Troeger J, et al:
Epithelial-to-mesenchymal transition is not required for lung
metastasis but contributes to chemoresistance. Nature. 527:472–476.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zheng X, Carstens JL, Kim J, Scheible M,
Kaye J, Sugimoto H, Wu CC, LeBleu VS and Kalluri R:
Epithelial-to-mesenchymal transition is dispensable for metastasis
but induces chemoresistance in pancreatic cancer. Nature.
527:525–530. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
So WK, Fan Q, Lau MT, Qiu X, Cheng JC and
Leung PC: Amphiregulin induces human ovarian cancer cell invasion
by down-regulating E-cadherin expression. FEBS Lett. 588:3998–4007.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Willmarth NE and Ethier SP: Autocrine and
juxtacrine effects of amphiregulin on the proliferative, invasive,
and migratory properties of normal and neoplastic human mammary
epithelial cells. J Biol Chem. 281:37728–37737. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Thøgersen VB, Sorensen BS, Poulsen SS,
Orntoft TF, Wolf H and Nexø E: A subclass of HER1 ligands is a
prognostic marker for survival in bladder cancer patients. Cancer
Res. 61:6227–6233. 2001.PubMed/NCBI
|
|
13
|
Ishikawa N, Daigo Y, Takano A, Taniwaki M,
Kato T, Hayama S, Murakami H, Takeshima Y, Inai K, Nishimura H, et
al: Increases of amphiregulin and transforming growth factor-alpha
in serum as predictors of poor response to gefitinib among patients
with advanced non-small cell lung cancers. Cancer Res.
65:9176–9184. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jing C, Jin YH, You Z, Qiong Q and Jun Z:
Prognostic value of amphiregulin and epiregulin mRNA expression in
metastatic colorectal cancer patients. Oncotarget. 7:55890–55899.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shinomiya H, Ito Y, Kubo M, Yonezawa K,
Otsuki N, Iwae S, Inagaki H and Nibu KI: Expression of amphiregulin
in mucoepidermoid carcinoma of the major salivary glands: A
molecular and clinicopathological study. Hum Pathol. 57:37–44.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang L, Wu H, Wang L, Lu J, Duan H, Liu X
and Liang Z: Expression of amphiregulin predicts poor outcome in
patients with pancreatic ductal adenocarcinoma. Diagn Pathol.
11:602016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang H, Wu H, Guan J, Wang L, Ren X, Shi
X, Liang Z and Liu T: Paracrine SDF-1α signaling mediates the
effects of PSCs on GEM chemoresistance through an IL-6 autocrine
loop in pancreatic cancer cells. Oncotarget. 6:3085–3097.
2015.PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gao S, Sun Y, Zhang X, Hu L, Liu Y, Chua
CY, Phillips LM, Ren H, Fleming JB, Wang H, et al: IGFBP2 Activates
the NF-κB pathway to drive epithelial-mesenchymal transition and
invasive character in pancreatic ductal adenocarcinoma. Cancer Res.
76:6543–6554. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nomura A, Majumder K, Giri B, Dauer P,
Dudeja V, Roy S, Banerjee S and Saluja AK: Inhibition of NF-kappa B
pathway leads to deregulation of epithelial-mesenchymal transition
and neural invasion in pancreatic cancer. Lab Invest. 96:1268–1278.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nieto MA, Huang RY, Jackson RA and Thiery
JP: EMT: 2016. Cell. 166:21–45. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Busser B, Sancey L, Brambilla E, Coll JL
and Hurbin A: The multiple roles of amphiregulin in human cancer.
Biochim Biophys Acta. 1816:119–131. 2011.PubMed/NCBI
|
|
23
|
Liu JF, Tsao YT and Hou CH: Amphiregulin
enhances intercellular adhesion molecule-1 expression and promotes
tumor metastasis in human osteosarcoma. Oncotarget. 6:40880–40895.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chung E, Cook PW, Parkos CA, Park YK,
Pittelkow MR and Coffey RJ: Amphiregulin causes functional
downregulation of adherens junctions in psoriasis. J Invest
Dermatol. 124:1134–1140. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Coradini D, Casarsa C and Oriana S:
Epithelial cell polarity and tumorigenesis: New perspectives for
cancer detection and treatment. Acta Pharmacol Sin. 32:552–564.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kleeff J, Shi X, Bode HP, Hoover K,
Shrikhande S, Bryant PJ, Korc M, Büchler MW and Friess H: Altered
expression and localization of the tight junction protein ZO-1 in
primary and metastatic pancreatic cancer. Pancreas. 23:259–265.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Peinado H, Olmeda D and Cano A: Snail, Zeb
and bHLH factors in tumour progression: An alliance against the
epithelial phenotype? Nat Rev Cancer. 7:415–428. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Batlle E, Sancho E, Francí C, Domínguez D,
Monfar M, Baulida J and García De Herreros A: The transcription
factor snail is a repressor of E-cadherin gene expression in
epithelial tumour cells. Nat Cell Biol. 2:84–89. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Guaita S, Puig I, Francı́ C, Garrido M,
Dominguez D, Batlle E, Sancho E, Dedhar S, De Herreros AG and
Baulida J: Snail induction of epithelial to mesenchymal transition
in tumor cells is accompanied by MUC1 repression and ZEB1
expression. J Biol Chem. 277:39209–39216. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang HW, Menon LG, Black PM, Carroll RS
and Johnson MD: SNAI2/Slug promotes growth and invasion in human
gliomas. BMC Cancer. 10:3012010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liang C, Wang Z, Li YY, Yu BH, Zhang F and
Li HY: miR-33a suppresses the nuclear translocation of β-catenin to
enhance gemcitabine sensitivity in human pancreatic cancer cells.
Tumor Biol. 36:9395–9403. 2015. View Article : Google Scholar
|
|
32
|
von Ahrens D, Bhagat TD, Nagrath D, Maitra
A and Verma A: The role of stromal cancer-associated fibroblasts in
pancreatic cancer. J Hematol Oncol. 10:762017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Duner S, Lopatko LJ, Ansari D, Gundewar C
and Andersson R: Pancreatic cancer: The role of pancreatic stellate
cells in tumor progression. Pancreatology. 10:673–681. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vonlaufen A, Joshi S, Qu C, Phillips PA,
Xu Z, Parker NR, Toi CS, Pirola RC, Wilson JS, Goldstein D and Apte
MV: Pancreatic stellate cells: Partners in crime with pancreatic
cancer cells. Cancer Res. 68:2085–2093. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kikuta K, Masamune A, Watanabe T, Ariga H,
Itoh H, Hamada S, Satoh K, Egawa S, Unno M and Shimosegawa T:
Pancreatic stellate cells promote epithelial-mesenchymal transition
in pancreatic cancer cells. Biochem Biophys Res Commun.
403:380–384. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Saito K, Sakaguchi M, Maruyama S, Iioka H,
Putranto EW, Sumardika IW, Tomonobu N, Kawasaki T, Homma K and
Kondo E: Stromal mesenchymal stem cells facilitate pancreatic
cancer progression by regulating specific secretory molecules
through mutual cellular interaction. J Cancer. 9:2916–2929. 2018.
View Article : Google Scholar : PubMed/NCBI
|