Introduction
Lung cancer is the leading cause of cancer-related
death worldwide, and approximately 85% of all diagnosed lung cancer
cases are non-small cell lung cancer (NSCLC) (1,2). Great
progress in therapeutic strategies has been made over the past
decades including the discovery of epidermal growth factor receptor
(EGFR)-tyrosine kinase inhibitors (TKIs), which have dramatically
changed the prognosis of advanced NSCLC patients harboring specific
EGFR-activating mutations (3,4).
However, patients who initially respond to EGFR-TKIs eventually
acquire resistance, resulting in progression and relapse (5,6).
Several mechanisms are believed to be responsible for acquired
EGFR-TKI resistance. The secondary EGFR T790M mutation that
can eliminate inhibition of the respective TKIs accounts for half
of the potential mechanisms (7).
The remaining resistance mechanisms under non-T790M mutation status
can be classified into three types. Phenotypic or histological
changes include small cell lung cancer (SCLC) transformation and
epithelial to mesenchymal transition (EMT) process. Accumulating
studies point to a molecular association between EMT and TKI
resistance. Tissue samples of lung cancer patients who develop
acquired resistance to erlotinib were found to consist of EMT
features (8). Activation of AXL
receptor tyrosine kinase (AXL) and transforming growth
factor-β (TGF-β) is associated with the EMT process in
EGFR-TKI-resistant NSCLC (9). In
terms of compensatory bypass signaling pathways, insulin-like
growth factor 1 receptor (IGF1R)/MEK, AXL/PI3K/AKT,
hepatocyte growth factor receptor (HGFR)/MAPK pathways were found
to be involved in TKI resistance (10). It has been reported that the
InsR/IGF1R pathway confers resistance to gefitnib resistance in
glioblastoma through AKT regulation (11). Point mutations of target genes also
contribute to non-T790M mutations such as HER2
amplification, MET amplification, BRAF mutation and
PIK3CA mutation (12).
Osimertinib is a third-generation EGFR-TKI used for the treatment
of patients with the T790M mutation; however no special treatment
has been discovered for patients harboring non-T790M mutations
(13,14). Therefore, further elucidation of
other potential mechanisms that are critical for the development of
effective therapeutic strategies targeting patients without the
T790M mutation is urgent.
MicroRNAs are a class of small non-coding RNAs that
play essential roles in tumor development and progression via the
regulation of various networks that are associated with multiple
cellular functions, such as proliferation, migration, and
metabolism (15). Accumulating
evidence has shown that a number of microRNAs may have a specific
role in lung cancer pathogenesis and biological and pathological
behaviors as well as in modulating the response to anticancer
treatments, particularly EGFR-TKIs (16,17).
It is reported that circulating miR-21 expression in the peripheral
blood of patients significantly increased from the baseline to high
levels with the progression of disease following treatment with
EGFR-TKI. Mechanically, miR-21 was found to induce EGFR-TKI
resistance via downregulating PTEN and PDCD4 and
activating the PI3K/AKT pathway (18). MicroRNAs have also been reported to
reverse drug resistance in addition to contributing to gefitinib
resistance in tumor cells. miR-506-3p was identified to reverse
gefitinib resistance by targeting Yes-associated protein 1 in the
PC9GR cell line (19). miR-497 was
reported to enhance the sensitivity of NSCLC cells to gefitinib by
targeting IGFR-1 (20).
In the present study, we mainly focused on the
identification of new microRNAs underlying non-T790M
mutation-induced gefitinib resistance. Here, we found that the
PC9GR cell line acquired a secondary T790M mutation, herein the
non-T790M mutated HCC827GR cell line was selected for our
experiments. Our results showed that miRNA-625-3p was significantly
downregulated in HCC827GR cells compared to that noted in the
HCC827 cells. Overexpression of miRNA-625-3p was found to enhance
sensitivity to gefitinib and inhibit the migratory and invasive
abilities of HCC827GR cells. Furthermore, a functional assay also
indicated that miRNA-625-3p could directly target AXL to
reverse the EMT process. Taken together, these results suggest that
the modulation of miRNA-625-3p may be a potential strategy to
overcome gefitinib acquired resistance in NSCLC.
Materials and methods
Cell culture and reagents
The NSCLC cell line HCC827 and 293T cells were
purchased from the Cell Bank of the Chinese Academy of Sciences
(Shanghai, China). To establish the gefitinib-resistant cell strain
HCC827GR, HCC827 cells were exposed to gefitinib as previously
described (21). The NSCLC cell
line PC9 and PC9 gefitinib-resistant (PC9GR) cell line were
obtained from Professor Caicun Zhou (Shanghai Pulmonary Hospital)
as a gift and were maintained in Dulbecco's modified Eagle's medium
(DMEM; Gibco, Carlsbad, CA, USA) supplemented with 10% foetal
bovine serum (FBS) (Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin/streptomycin (Gibco; Thermo Fisher Scientific, Inc.).
All cell lines were cultured at 37°C in a humidified atmosphere
containing 5% CO2. Among all cell lines, both HCC827 and
PC9 cell lines contain exon 19 deletions (del 19). PC9GR cells
contain the T790M mutation while HCC827 do not. Detailed mutation
information is documented in Table
SII. The EGFR inhibitor gefitinib was purchased from Selleck,
at doses of 0–40 µM (Selleck Chemicals).
Next-generation DNA sequencing
The DNAseq was performed by Geneseeq Co. DNA from
cell lines was profiled and then analyzed using a capture-based
targeted sequencing panel. Human genomic regions totalling 1.4
megabases in size, including selected exons and introns of 357
genes, were captured using 120-bp probes. DNA was fragmented into
segments 200 to 250 bp in length, captured by the 120-bp probes,
and sequenced by obtaining paired 2×150-bp reads. After DNA
extraction with the QIAamp DNA Mini Kit (Qiagen), DNA
concentrations were measured using the Qubit dsDNA assay
(Invitrogen; Thermo Fisher Scientific, Inc.). The DNA quality was
confirmed by checking that the A260/A280 ratio was 1.8:2.0. The
appropriate concentration value for samples was higher than 100
ng/µl and 4 mg DNA was needed for each sample. DNA was hybridized
with the capture probes (the bait), selected using magnetic beads,
and polymerase chain reaction (PCR)-amplified. Then, a Qubit and
Agilent 2100 bioanalyzer (Agilent Technologies) was used to perform
high-sensitivity assays assessing DNA quality and size range. All
samples were sequenced on a HiSeq 4000 platform (Illumina, Inc.)
and pair-end reads were obtained. For tissue samples, we aimed to
achieve an average sequencing depth of 2,000× for all targeted
regions.
miRNA library construction and RNA
sequencing
Sample preparation
Total RNA was extracted from HCC827 and HCC827GR
cells by TRIzol reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) separately. The RNA quality was checked by a Bioanalyzer 2200
(Agilent Technologies) and stored at −80°C. RNA with RNA integrity
number (RIN) >6.0 was appropriate for miRNA purification. miRNA
was purified by the miRNeasy Mini Kit (Qiagen), and the purity was
validated by gel electrophoresis.
The complementary DNA (cDNA) libraries for
single-end sequencing were prepared using the Ion Total RNA-Seq Kit
v2.0 (Life Technologies; Thermo Fisher Scientific, Inc.) according
to the manufacturer's instructions. The cDNA library was size
selected by PAGE gel electrophoresis for miRNA sequencing. The cDNA
libraries were then processed for the proton sequencing process
according to commercially available protocols. Samples were diluted
and mixed, and the mixture was processed on a OneTouch 2 Instrument
(Life Technologies; Thermo Fisher Scientific, Inc.) and enriched on
a OneTouch 2 ES station (Life Technologies; Thermo Fisher
Scientific, Inc.) to prepare the template-positive Ion PI™ Ion
Sphere™ Particles (Life Technologies; Thermo Fisher Scientific,
Inc.) according to the instructions for the Ion PI™ Hi-Q OT2 200
Kit (Life Technologies; Thermo Fisher Scientific, Inc.). After
enrichment, the mixed template-positive Ion PI™ Ion Sphere™
Particles of the samples were loaded onto 1 v3 Proton Chip (Life
Technologies; Thermo Fisher Scientific, Inc.) and sequenced on
Proton Sequencers according to the protocol for the Ion PI Hi-Q
Sequencing 200 Kit (Life Technologies; Thermo Fisher Scientific,
Inc.) by NovelBio Corp. Laboratory, Shanghai.
Filtering and miRNA mapping
We performed filtering of the raw reads after
sequencing to obtain clean data using the following criteria: i)
30% base quality <20, ii) the sequences shorter than 15 bp or
longer than 33 bp were discarded, and iii) presence of the adaptor
sequence. Utilizing the BWA software BSA-backtrack) (22), we mapped the clean data to the human
miRNA database (miRBase v21.0) (23) and the human genome (GRCh38, NCBI)
(24).
RNA sequencing mapping/mapping of pair-end
reads
Before read mapping, clean reads were obtained from
the raw reads by removing the adaptor sequences, reads with >5%
ambiguous bases (noted as N) and low-quality reads containing more
than 20% of bases with quality of <20. The clean reads were then
aligned to the human genome (version: GRCh38) using the HISAT2
programme (25). Dif-Gene-Finder
(26) and Target Analysis were
described in our previous study (27).
Cell transfection
Transient transfection of miR-625-3p mimics, miR-NC,
control siRNA and AXL siRNA was carried out using
Lipofectamine 2000 Reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocols. The target siRNA
sequences are: si-NC, 5′-UUCUCCGAACGUGUCACGUTT-3′; si-AXL:
5′-ACAGCGAGAUUUAUGACUATT-3′. The sequences of miR-625-3p are as
follows: Sense, GACUAUAGAACUUUCCCCCUC A and antisense,
UGAGGGGGAAAGUUCUAUAGUC; for miR-NC, sense, UUUGUACUACACAAAAGUACUG
and antisense, CAGUACUUUUGUGUAGUACAAA. All mimics were synthesized
by Guangzhou Ribo BioCompany (Guangzhou, China). siRNAs were
provided by GenePharma Co. Cells were plated in 6-well plates
(2×105 cells/well) in advance. At 50% confluence, the
cells were transfected with 0.1 nmol miR-625-3p mimics, miR-NC or
100 pmol siRNAs using Lipofectamine 2000 transfection reagent in
medium without serum. After 6 h, the medium was changed to
RPMI-1640 supplemented with 10% FBS. After 48 h, the cells were
collected for subsequent experiments.
Cell growth inhibition assay
After transfection with miR-625-3p mimics, miR-NC or
siRNAs, the cells were seeded in 96-well plates (3×103
cells/well) overnight. Gefitinib was added in a dose-dependent
manner, and the cells were incubated for 72 h. Then, we used the
Cell Counting Kit-8 assay kit (CCK-8, Boster) to assess cell
proliferation according to the manufacturer's instructions. Each
sample was plated in triplicate, and three independent experiments
were performed. The half maximal inhibitory concentration
(IC50) was defined as the concentration needed for a 50%
reduction in the absorbance.
Wound healing assay
The wound healing assay was used to determine the
cell migration ability. After cells formed a monolayer in 6-well
plates, cells were washed with PBS and then cultured in RPMI-1640
medium with 1% FBS overnight. Then the wound assay was performed. A
straight scratch was made gently through the cell monolayer using a
10-µl pipette tip. Detached cells were washed away with PBS, and
then fresh RPMI-1640 medium with 10% FBS was added. The cells were
imaged at 0 and 24 h after wounding using a light microscope
(CKX41; Olympus, magnification, ×100).
Transwell assays
For the migration assay, 800 µl medium supplemented
with 10% FBS was added to the lower well, and 4×104
cells in 200 µl medium supplemented with 1% FBS were seeded into
the upper chamber of a Transwell insert. The cells were incubated
for 24 h at 37°C in 5% CO2. For the invasion assay, the
inserts were coated with a Matrigel matrix (BD Science), which was
diluted in serum-free medium and incubated at 37°C for 2 h before
cell plating. In both assays, the cells were then imaged under a
light microscope (IX73; Olympus). The detailed cell numbers in each
field were counted via ImageJ software (National Institutes of
Health). Three independent experiments were performed.
Plasmid construction and dual-luciferase reporter
assay
To construct a plasmid containing the AXL
3′-UTR (3′ untranslated region) fused to the 3′ end of a luciferase
reporter, a 221-bp fragment containing the predicted miR-625-3p
target site (positions 905–911) was inserted into the psiCHECK-2
dual-luciferase vector (Promega). The potential binding sites were
predicted by TargetScan (http://www.targetscan.org/). The
psiCHECK-2-AXL−3′-UTR wild-type and mutated fragments were
synthesized and subcloned directly (Genewiz). Cells were plated in
a 24-well plate overnight and then co-transfected with the
wild-type or mutated plasmid, control pRL-TK plasmid and with
either miR-625-3p mimics or miR-NC using Lipofectamine 2000 (Life
Technologies; Thermo Fisher Scientific, Inc.). Then, the cells were
harvested, and luciferase activities were evaluated using a
Dual-Luciferase Reporter Assay Kit (Promega). Each experiment was
performed in triplicate.
RNA isolation and RT-qPCR
Trizol reagent was used to extract RNA (Takara). In
our study, we extracted RNA from HCC827 and HCC827GR cells to
detect miR-625-3p and AXL mRNA expression to verify the
sequencing results. After transfection of the HCC827GR cells with
miR-NC and miR-625-3p, RNA was extracted to examine miR-625-3p and
AXL expression to confirm whether miR-625-3p was upregulated
and AXL was downregulated in the cells. We also extracted
RNA from HCC827GR cells transfected with si-NC and si-AXL to
confirm whether AXL was downregulated.
Reverse transcription was performed using reverse
transcriptase M-MLV (Takara) according to the manufacturer's
protocol. miR-625-3p and U6-specific cDNA were synthesized using
gene-specific primers designed and synthesized by Guangzhou RiboBio
Co. (cat. no. MQPS0001992-1-100 and MQPS0001992-1-100). Reverse
transcription polymerase chain reaction (RT-qPCR) was performed
using SYBR Premix ExTaq™ (Takara) according to the manufacturer's
instructions. The primer sequences for RT-qPCR of AXL and
GAPDH were as follows: AXL sense, 5′-GGTGGCTGTGAAGACGATGA-3′
and antisense, 5′-CTCAGATACTCCATGCCA-3′; GAPDH sense,
5′-TGCACCACCAACTGCTTAGC-3′ and antisense,
5′-GGCATGGACTGTGGTCATGG-3′. The PCR programme was as follows: 50°C
for 2 min; 95°C for 10 min; and 45 cycles of 95°C for 15 sec and
60°C for 1 min. The expression values of AXL and miR-625-3p
were normalized to the values of the internal controls GAPDH and
U6, respectively. Relative expression was calculated using the
2−ΔΔCt method (28).
Western blot analysis
In the present study, HCC827, HCC827GR, HCC82GR cell
lines transfected with miR-625-3p, si-AXL or negative
control were proceeded to extract proteins to identify the changes
in the SMAD pathway and EMT-related signaling. Generally, the cells
were harvested and lysed on ice for 30 min in RIPA lysis buffer
(cat. no. 9806, Cell Signaling Technology, Inc.) to extract protein
fractions. The lysates were collected by high-speed centrifugation
at 11,000 × g for 15 min at 4°C. Lysates were subjected to western
blot analysis as previously described (29). The antibodies used in our study
included anti-vimentin (RV202, dilution 1:1,000, cat. no. 550513,
BD Biosciences), anti-pAXL (Y779, dilution 1:1,000, cat. no.
AF2228, R&D Systems), and anti-AXL (C89E7, dilution 1:1,000,
cat. no. 8661), anti-Smad3 (C67H9, dilution 1:1,000, cat. no.
9523), anti-pSmad3 (Ser423/425, dilution 1:1,000, cat. no. 9520),
anti-ZEB1 (D80D3, dilution 1:1,000, cat. no. 3396), anti-Snail
(C15D3, dilution 1:1,000, cat. no. 3895s), anti-MMP2 (D8N9Y,
dilution 1:1,000, cat. no. 13132), and anti-MMP9 (603H, dilution
1:1,000, cat. no. 13667), anti-N-cadherin (D4R1H, dilution 1:1,000,
cat. no. 13116) and anti-E-cadherin (4A2, dilution 1:1,000, cat.
no. 14472) (all from Cell Signaling Technology, Inc.), In addition,
anti-β-actin (13E5, dilution 1:1,000, cat. no. 4970S) and
anti-mouse (dilution 1:2,000, cat. no. 7076S) or anti-rabbit
(dilution 1:2,000, cat. no. 7074S) constituted the secondary
antibodies and were also purchased from Cell Signaling Technology,
Inc.
Statistical analysis
GraphPad Prism 5.02 (GraphPad Software, Inc.) and
SPSS 16.0 software (SPSS, Inc.) were used to perform the
statistical analysis. Significant differences between two groups
were assessed by a non-paired Student's t-test. Significant
differences between three groups were analyzed using one-way ANOVA
followed by Dunnett's post-hoc test. All statistical tests were
two-tailed, and statistical significance was defined as
P<0.05.
Results
EMT features are detected in HCC827
gefitinib-resistant (HCC827GR) cells without the EGFR T790M
mutation
First, we established a gefitinib-resistant subline
from the parental HCC827 and PC9 cells harboring EGFR-activating
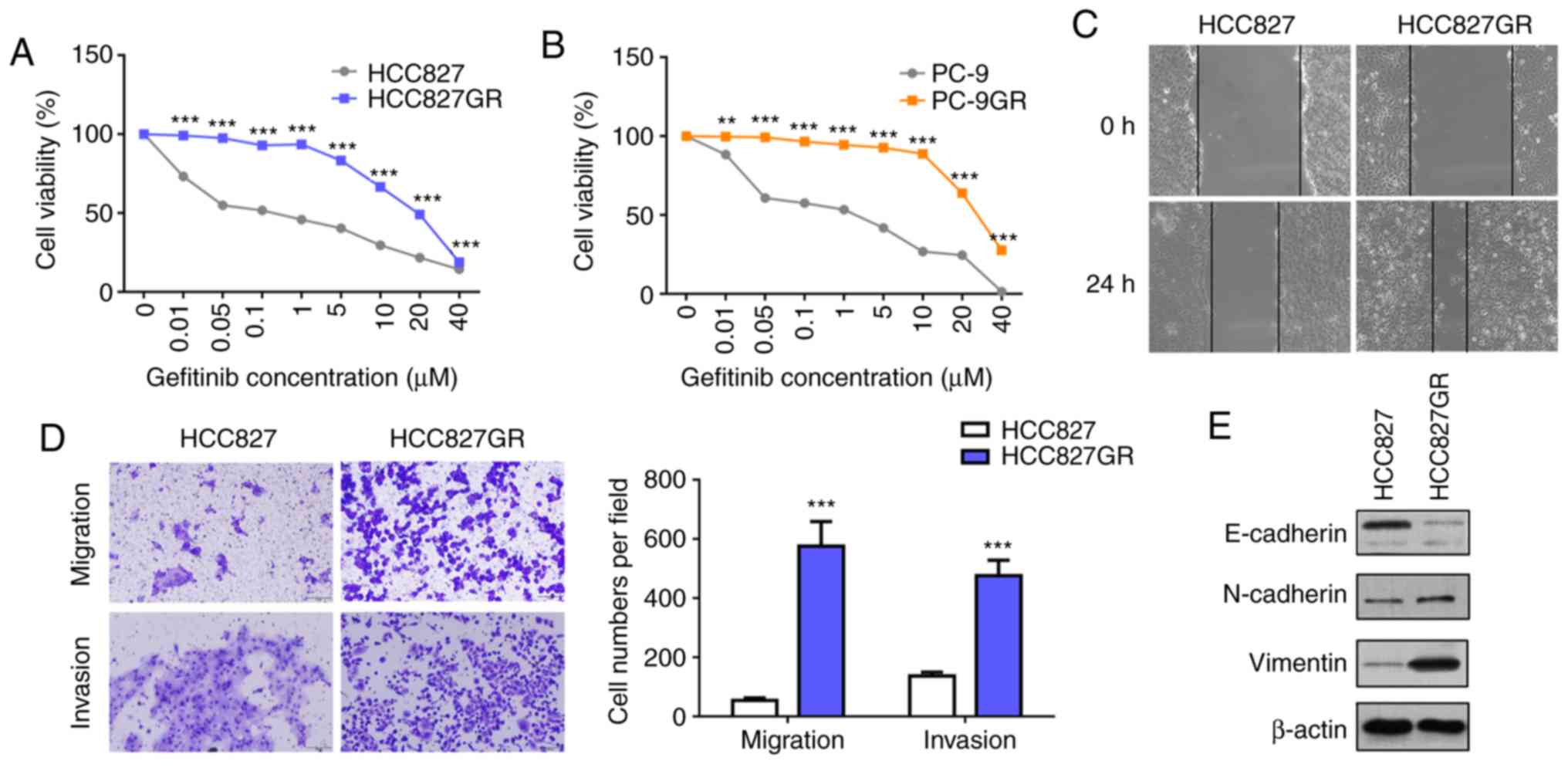
mutations. As shown in Fig. 1A and
B, the gefitinib-resistant sublines HCC827GR (IC50:
0.07 µM vs. 23.64 µM) and PC9GR (IC50: 2.06 µM vs. 28.45
µM ) manifested significant resistance to gefitinib. The T790M
mutation and MET gene amplification are the most clearly
defined mechanisms underlying acquired resistance to gefitinib.
Therefore, we profiled DNA from the cell lines and analyzed the
gene status using a capture-based targeted sequencing panel that
included the EGFR and MET genes (Table SI). The results revealed that PC9GR
cells acquired a secondary T790M mutation compared with HCC827GR
cells, and MET gene amplification was not detected (Table SII). Growing evidence has shown
that in addition to the T790M mutation and MET
amplification, EMT plays an important role in acquired resistance
to EGFR-TKIs. EMT is an important process during malignant cancer
progression, accompanied by upregulation of N-cadherin and vimentin
and downregulation of E-cadherin (30). Furthermore, wound healing and
Transwell assays were performed, and the results showed that
HCC827GR cells contained high migratory and invasive ability
compared with HCC827 cells (Fig. 1C and
D). Moreover, western blot analysis revealed decreased protein
levels of E-cadherin and increased protein levels of N-cadherin and
vimentin in the HCC827GR cells (Fig.
1E).
miR-625-3p is decreased in HCC827GR
cells, and overexpression of miR-625-3p attenuates gefitinib
resistance via regulation of EMT phenotypes
MicroRNAs can regulate multiple signaling cascades
via transcriptional silencing effects. First, microRNA library
construction and RNA sequencing were performed. We found 16
upregulated microRNAs and 27 downregulated microRNAs in HCC827GR
cells when compared to the parental cells [Fig. 2A, >1.5-fold change, false
discovery rate (FDR) <0.05, P<0.05]. To verify the results
above, we performed RT-qPCR analysis and found that miR-625-3p
expression was significantly reduced in HCC827GR cells compared
with HCC827 cells (Fig. 2B,
P<0.001). In addition, data showed similar downregulated
miR-625-3p expression in PC9GR cells when compared to PC9 cells
(Fig. S1A).
As microRNA interventions can attenuate
EGFR-TKI-resistant phenotypes, we next verified whether miR-625-3p
overexpression could reverse the acquired resistance to EGFR-TKI.
HCC827GR cells were transfected with miR-625-3p mimics (Fig. 3A). As shown in Fig. 3B, our results demonstrated that
overexpression of miR-625-3p could partly reverse gefitinib
resistance in the HCC827GR cells. In addition, the IC50
value declined to 12.26 µM after miR-625-3p transfection.
Considering the findings that EMT is a mechanism of acquired
resistance to EGFR-TKI, wound healing and Transwell assays were
performed. The results showed that overexpression of miR-625-3p
inhibited the migratory and invasive ability of the HCC827GR cells
(Fig. 3C and D). Moreover, western
blot analysis showed increased E-cadherin expression and decreased
N-cadherin and vimentin expression after miR-625-3p overexpression
(Fig. 3E).
AXL is upregulated in
EGFR-TKI-resistant lung cancer cell lines and is a direct target of
miR-625-3p
It is well accepted that microRNAs execute their
function by attenuating the stability and translation of downstream
target genes. The miRanda and RNAhybrid databases were used to
predict the potential mRNAs targeted by miR-625-3p. Among all mRNAs
presented in Fig. 4A, AXL
has been shown to be associated with EMT and drug resistance
(31,32). Therefore, we focused on AXL
as a new post-transcriptional mechanism of miR-625-3p. RNA
sequencing showed that AXL was upregulated in the HCC827GR
cells (Fig. 4B). Additionally,
upregulated AXL expression was also found in HCC827
erlotinib-resistant cells based on the data extracted from the
public datasets GSE38121 and GSE71587 (Fig. 4C and D). To validate that AXL
is a direct target of miR-625-3p, the AXL wild-type (WT)
3′-UTR (containing the miR-625-3p binding sequence) and the
AXL MUT 3′-UTR were constructed (Fig. 5A). The dual-luciferase reporter
assay conducted in 293T cells showed that miR-625-3p significantly
inhibited the luciferase activity in cells transfected with the
wild-type AXL 3′-UTR but did not in cells transfected with
the mutant construct (Fig. 5B).
Moreover, we also performed the luciferase assay in HCC827 and
HCC827GR cells and got similar results (Fig. S2). In addition, we found that
AXL expression was downregulated in the HCC827GR cells at
both the mRNA and protein levels after transfection with miR-625-3p
mimics (Fig. 5C and D).
Collectively, our data showed that AXL is a target of
miR-625-3p in NSCLC cells.
AXL knockdown attenuates gefitinib
resistance in HCC827GR cells by reversing EMT phenotypes
The role of AXL-mediated EMT and drug
resistance has been documented in many types of cancers, especially
lung cancer. First, we performed RT-qPCR analysis to confirm the
results from RNA sequencing and found that the AXL level was
significantly increased in HCC827GR cells compared with that noted
in the HCC827 cells (Fig. 6A,
P<0.01). In contrary, PC9GR cells were found to contain lower
AXL mRNA expression when compared to PC9 cells (Fig. S1B). Next, we inhibited AXL
using specific small interfering RNA (siRNA) and found that
knockdown of AXL enhanced gefitinib sensitivity in the
HCC827GR cells. Similarly, the IC50 value decreased to
18.02 µM after AXL knockdown (Fig. 6B and C). To investigate whether
knockdown of AXL enhanced gefitinib sensitivity by reversing
EMT, wound healing and Transwell assays were performed. The results
showed that AXL knockdown significantly inhibited the
migratory and invasive ability of HCC827GR cells (Fig. 6D and E). Western blot analysis
demonstrated increased protein levels of E-cadherin and decreased
protein levels of N-cadherin and vimentin in the HCC827GR cells
following AXL knockdown (Fig.
6F).
miR-625-3p/AXL axis contributes to
gefitinib resistance via the SMAD pathway
From previous results we know that overexpression of
miR-625-3p or knockdown of AXL can attenuate gefitinib
resistance by reversing the EMT phenotype. However, the underlying
mechanism remains unclear. Given that the transforming growth
factor (TGF)-β1 signaling pathway can contribute to the EMT
phenotype in the progression of various cancers (33,34),
and that Smad3 functions as a canonical downstream player involved
in the TGF-β1 signaling pathway (35,36),
we hypothesized that Smad3 may participate in AXL-mediated
acquired resistance to gefitinib. As shown in Fig. 7A, western blot analysis demonstrated
that the expression level of phosphorylated Smad3 was increased in
the HCC827GR cell line compared to that found in the HCC827 cells.
In addition, the expression of the transcription factors Snail and
ZEB1 was increased in the HCC827GR cells. MMP family members, such
as MMP2 and MMP9, showed similar upregulated tendencies. Moreover,
knockdown of AXL and overexpression of miR-635-3p decreased
the protein levels of p-Smad3, ZEB1, Snail, MMP2 and MMP9 in the
HCC827GR cells. All these changes confirmed that the TGF-β1/Smad3
pathway was indeed involved in the miR-625-3p/AXL
axis-mediated gefitinib resistance (Fig. 7A).
 | Figure 7.The miR-625-3p/AXL axis
contributes to gefitinib resistance via the SMAD pathway. (A)
Western blot analysis of AXL, phosphorylated (p)-AXL,
Smad3, p-Smad3, ZEB1, Snail, MMP2, and MMP9 expression in the
parental HCC827GR cells compared to HCC827 cells or in cells after
transfection of miR-625-3p mimic or knockdown of AXL. (B) A
diagram of the mechanism of miR-625-3p/AXL-mediated
gefitinib resistance. Gas6, growth arrest specific 6; TGFβ,
transforming growth factor β; AXL, AXL receptor tyrosine
kinase;; ZEB1, zinc finger E-box binding homeobox 1; MMP, matrix
metalloproteinases; Snail, Snail family transcriptional repressor
1; p, phorphorylation; EMT, epithelial to mesenchymal
transition. |
Discussion
Recently, the identification of epidermal growth
factor receptor (EGFR) mutations as oncogenic drivers has
launched the era of precision medicine in the treatment of
non-small cell lung cancer (NSCLC). A majority of patients who
carry EGFR-sensitive mutations, such as exon 19 deletions
(del 19) and exon 21 L858R substitutions, have benefited from the
clinical application of gefitinib (37,38).
However, some patients eventually develop acquired drug resistance
during treatment, and thus, the efficacy is limited. It has been
reported that the T790M mutation accounts for more than 50% of the
cases of known acquired resistance. However, the mechanism for
acquired resistance in the remaining patients without the T790M
mutation still needs to be explored. After profiling DNA from cell
lines using a capture-based targeted sequencing panel, we found
that PC9GR cells contained the T790M mutation compared to HCC827GR
cells. Therefore, in the next step, we mainly focused on the
mechanism of resistance to gefitinib in the HCC827GR cell line.
MicroRNAs are important regulators in tumor
development and progression. A vast variety of evidence also
indicates that microRNAs can participate in the acquired resistance
to epidermal growth factor receptor-tyrosine kinase inhibitors
(EGFR-TKIs). It has been reported that microRNA-147 overexpression
can induce colon cancer cells to undergo a
mesenchymal-to-epithelial transition phenotype to reverse gefitinib
resistance (39). In NSCLC, miR-124
modulates gefitinib resistance through SNAI2 and
STAT3, providing a therapeutic strategy for reversing
acquired gefitinib resistance in patients (40). Our microarray data showed that
miR-625-3p was significantly downregulated in HCC827GR cells
compared to HCC827 cells, implying that miR-625-5p may play an
important role in the development of resistance to EGFR-TKIs.
Importantly, we found that overexpression of
miR-625-3p decreased the IC50 value in HCC827GR cells
and thus partly reversed gefitinib resistance. The EMT process has
been demonstrated to contribute to acquired resistance to
gefitinib. Consequently, next we mainly focused on whether
overexpression of miR-625-5p could induce mesenchymal-to-epithelial
transition to reverse gefitinib resistance. The Transwell and wound
healing assays both showed that overexpression of miR-625-3p
inhibited the migratory and invasive ability of HCC827GR cells.
Western blot assays further revealed increased expression of
E-cadherin and decreased expression of N-cadherin and vimentin in
HCC827GR cells transfected with miR-625-3p. All of these findings
confirmed that miR-625-3p overexpression indeed induced NSCLC cells
to undergo mesenchymal-to-epithelial transition to reverse
gefitinib resistance.
After integrating the data obtained from Targetscan
analysis, we were surprised to find that AXL receptor tyrosine
kinase (AXL) was among the potential targets of miR-625-3p.
It is known that AXL is associated with EMT and TKI
resistance. We found higher AXL expression in HCC827GR cells
based on RNA sequencing analysis. Analysis of data from public
databases also demonstrated that AXL is highly expressed in
both HCC827GR and HCC827 erlotinib-resistant (HCC827ER) cells.
Dual-luciferase reporter assays were performed to prove that
AXL is a direct target of miR-625-3p. After knockdown of
AXL expression, the IC50 value was decreased, and
the migratory and invasive ability were decreased in HCC827GR
cells. Western blot analysis showed similar results that the
expression of E-cadherin was increased. Moreover, the expression of
N-cadherin and vimentin were decreased, implying that the cells had
acquired a mesenchymal-to-epithelial transition phenotype. We
should not ignore the fact that miR-625-3p and AXL mRNA expression
were all showed to be downregulated in PC9GR cells when compared to
the PC9 cells, indicating that the miR-625-3p/AXL axis may
be responsible for the mechanism underlying non-T790M mutation.
However the hypothesis needs to be further explored.
Tumor growth factor-β1 (TGF-β1)-induced epithelial
to mesenchymal transition (EMT) is recognized to contribute to the
reduction in drug sensitivity and acquisition of resistance to
EGFR-TKIs (41). Among all
downstream signaling molecules, the Smad pathway is a major
transducer in response to TGF-β1 stimulation. Western blot analysis
showed that the expression level of phosphorylated Smad3 was
upregulated in the HCC827GR cells. The expression level of p-Smad3
was downregulated after transfection with miR-625-3p or
si-AXL. In addition to the change in EMT features, ZEB1,
Snail, MMP2 and MMP9 levels were also altered. Collectively, these
data indicate that the miR-625-3p/AXL axis contributes to
gefitinib resistance via the SMAD pathway in NSCLC cells.
However, the present study also consisted of various
limitations. We did not co-transfect the AXL plasmid and miR-625-3p
mimics to assess gefitinib resistance and EMT phenotypes. This
rescue experiment can be regarded as another evidence of the
miR-625-3p/AXL axis. In addition, when we performed the
wound healing assay, cells were starved before we scratched the
cellular layer. However in various research studies, to prevent
cells from proliferating, cells were cultured in serum-free medium
or in medium with lower concentrations of FBS after the scratch was
made (42,43). Thus, we can repeat the wound healing
assay under the latter condition to ensure the accuracy of the
results in the future.
In conclusion, this is the first report showing that
miR-625-3p overexpression reversed TGF-β1-induced EMT and enhanced
gefitinib sensitivity by directly targeting AXL in lung
cancer cells (Fig. 7B). The
miR-625-3p/AXL axis was identified as a mechanism for the
development of drug resistance in HCC827GR cells without the T790M
mutation. Our data may provide therapeutic approaches to increase
drug sensitivity and combat resistance to EGFR-TKIs. However,
further studies are needed to clarify the underlying mechanism of
the miR-625-3p/AXL axis and TGF-β1-induced EMT in NSCLC.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the Jiangsu
Provincial Medical Youth Talent (no. QNRC2016746), the Postgraduate
Research and Practice Innovation Program of Jiangsu Province (no.
KYCX18_2525), the Suzhou Key Laboratory for Respiratory Medicine
(no. SZS201617), the Clinical Medical Center of Suzhou (no.
Szzx201502), the Jiangsu Provincial Key Medical Discipline (no.
ZDXKB2016007), and the National Natural Science Foundation of China
(no. 81702870).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
All authors contributed to this article
substantially. WD, LS, and TL performed the experiments. JZ and YZe
were responsible for the literature research and collected the cell
line data. YZh and XW analysed the data. ZL and JAH contributed to
the design of this study and draft of the manuscript. All authors
read and approved the manuscript and agree to be accountable for
all aspects of the research in ensuring that the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
NSCLC
|
non-small cell lung cancer
|
|
TGF-β1
|
transforming growth factor-β1
|
|
HCC827GR
|
HCC827 gefitinib resistant
|
|
EGFR-TKIs
|
epidermal growth factor
receptor-tyrosine kinase inhibitors
|
|
EMT
|
epithelial to mesenchymal
transition
|
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
Statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yoda S, Dagogo-Jack I and Hata AN:
Targeting oncogenic drivers in lung cancer: Recent progress,
current challenges and future opportunities. Pharmacol Ther.
193:20–30. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Remon J, Ahn MJ, Girard N, Johnson M, Kim
DW, Lopes G, Pillai RN, Solomon B, Villacampa G and Zhou Q:
Advanced stage non-small cell lung cancer: Advances in thoracic
oncology 2018. J Thorac Oncol. 14:1134–1155. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zheng L, Wang Y, Xu Z, Yang Q, Zhu G, Liao
XY, Chen X, Zhu B, Duan Y and Sun J: Concurrent EGFR-TKI and
thoracic radiotherapy as first-line treatment for stage IV
non-small cell lung cancer harboring EGFR active mutations.
Oncologist. 24:e1031–e1612. 2019. View Article : Google Scholar
|
|
5
|
Gao J, Li HR, Jin C, Jiang JH and Ding JY:
Strategies to overcome acquired resistance to EGFR TKI in the
treatment of non-small cell lung cancer. Clin Transl Oncol.
21:1287–1301. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee CK, Kim S, Lee JS, Lee JE, Kim SM,
Yang IS, Kim HR, Lee JH, Kim S and Cho BC: Next-generation
sequencing reveals novel resistance mechanisms and molecular
heterogeneity in EGFR-mutant non-small cell lung cancer with
acquired resistance to EGFR-TKIs. Lung Cancer. 113:106–114. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ahsan A: Mechanisms of resistance to EGFR
tyrosine kinase inhibitors and therapeutic approaches: An update.
Adv Exp Med Biol. 893:137–153. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhu X, Chen L, Liu L and Niu X:
EMT-mediated acquired EGFR-TKI resistance in NSCLC: Mechanisms and
strategies. Front Oncol. 9:10442019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sato H, Shien K, Tomida S, Okayasu K,
Suzawa K, Hashida S, Torigoe H, Watanabe M, Yamamoto H, Soh J, et
al: Targeting the miR-200c/LIN28B axis in acquired EGFR-TKI
resistance non-small cell lung cancer cells harboring EMT features.
Sci Rep. 7:408472017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shah R and Lester JF: Tyrosine kinase
inhibitors for the treatment of EGFR mutation-positive
non-small-cell lung cancer: A clash of the generations. Clin Lung
Cancer. Dec 20–2019.(Epub ahead of print). PubMed/NCBI
|
|
11
|
Ma Y, Tang N, Thompson RC, Mobley BC,
Clark SW, Sarkaria JN and Wang J: InsR/IGF1R pathway mediates
resistance to EGFR inhibitors in glioblastoma. Clin Cancer Res.
22:1767–1776. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu SG and Shih JY: Management of acquired
resistance to EGFR TKI-targeted therapy in advanced non-small cell
lung cancer. Mol Cancer. 17:382018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Murtuza A, Bulbul A, Shen JP, Keshavarzian
P, Woodward BD, Lopez-Diaz FJ, Lippman SM and Husain H: Novel
third-generation EGFR tyrosine kinase inhibitors and strategies to
overcome therapeutic resistance in lung cancer. Cancer Res.
79:689–698. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Carlisle JW and Ramalingam SS: Role of
osimertinib in the treatment of EGFR-mutation positive
non-small-cell lung cancer. Future Oncol. 15:805–816. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Frixa T, Donzelli S and Blandino G:
Oncogenic MicroRNAs: Key players in malignant transformation.
Cancers (Basel). 7:2466–2485. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sin TK, Wang F, Meng F, Wong SC, Cho WC,
Siu PM, Chan LW and Yung BY: Implications of MicroRNAs in the
treatment of gefitinib-resistant non-small cell lung cancer. Int J
Mol Sci. 17:2372016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zang H, Wang W and Fan S: The role of
microRNAs in resistance to targeted treatments of non-small cell
lung cancer. Cancer Chemother Pharmacol. 79:227–231. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li B, Ren S, Li X, Wang Y, Garfield D,
Zhou S, Chen X, Su C, Chen M, Kuang P, et al: MiR-21 overexpression
is associated with acquired resistance of EGFR-TKI in non-small
cell lung cancer. Lung Cancer. 83:146–153. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhu J, Tao L and Jin L: MicroRNA506-3p
reverses gefitinib resistance in non-small cell lung cancer by
targeting Yes-associated protein 1. Mol Med Rep. 19:1331–1339.
2019.PubMed/NCBI
|
|
20
|
Ma W, Feng W, Tan J, Xu A, Hu Y, Ning L,
Kang Y, Wang L and Zhao Z: miR-497 may enhance the sensitivity of
non-small cell lung cancer cells to gefitinib through targeting the
insulin-like growth factor-1 receptor. J Thorac Dis. 10:5889–5897.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Koizumi F, Shimoyama T, Taguchi F, Saijo N
and Nishio K: Establishment of a human non-small cell lung cancer
cell line resistant to gefitinib. Int J Cancer. 116:36–44. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kozomara A and Griffiths-Jones S: miRBase:
Annotating high confidence microRNAs using deep sequencing data.
Nucleic Acids Res. 42:D68–D73. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schneider VA, Graves-Lindsay T, Howe K,
Bouk N, Chen HC, Kitts PA, Murphy TD, Pruitt KD, Thibaud-Nissen F,
Albracht D, et al: Evaluation of GRCh38 and de novo haploid genome
assemblies demonstrates the enduring quality of the reference
assembly. Genome Res. 27:849–864. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim D, Langmead B and Salzberg SL: HISAT:
A fast spliced aligner with low memory requirements. Nat Methods.
12:357–360. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schaid DJ, Sinnwell JP and Thibodeau SN:
Robust multipoint identical-by-descent mapping for affected
relative pairs. Am J Hum Genet. 76:128–138. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhu J, Zeng Y, Xu C, Qin H, Lei Z, Shen D,
Liu Z and Huang J: Expression profile analysis of microRNAs and
downregulated miR-486-5p and miR-30a-5p in non-small cell lung
cancer. Oncol Rep. 34:1779–1786. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhu J, Zeng Y, Li W, Qin H, Lei Z, Shen D,
Gu D, Huang JA and Liu Z: CD73/NT5E is a target of miR-30a-5p and
plays an important role in the pathogenesis of non-small cell lung
cancer. Mol Cancer. 16:342017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Weng CH, Chen LY, Lin YC, Shih JY, Lin YC,
Tseng RY, Chiu AC, Yeh YH, Liu C, Lin YT, et al:
Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per
se is a common mechanism for acquired resistance to EGFR TKI.
Oncogene. 38:455–468. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Debruyne DN, Bhatnagar N, Sharma B, Luther
W, Moore NF, Cheung NK, Gray NS and George RE: ALK inhibitor
resistance in ALK(F1174L)-driven neuroblastoma is associated with
AXL activation and induction of EMT. Oncogene. 35:3681–3691. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wu F, Li J, Jang C, Wang J and Xiong J:
The role of Axl in drug resistance and epithelial-to-mesenchymal
transition of non-small cell lung carcinoma. Int J Clin Exp Pathol.
7:6653–6661. 2014.PubMed/NCBI
|
|
33
|
Lin RL and Zhao LJ: Mechanistic basis and
clinical relevance of the role of transforming growth factor-β in
cancer. Cancer Biol Med. 12:385–393. 2015.PubMed/NCBI
|
|
34
|
Tan EJ, Olsson AK and Moustakas A:
Reprogramming during epithelial to mesenchymal transition under the
control of TGFβ. Cell Adhes Migr. 9:233–246. 2015. View Article : Google Scholar
|
|
35
|
Jin Q, Gao G and Mulder KM: Requirement of
a dynein light chain in TGFbeta/Smad3 signaling. J Cell Physiol.
221:707–715. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shirai K, Saika S, Tanaka T, Okada Y,
Flanders KC, Ooshima A and Ohnishi Y: A new model of anterior
subcapsular cataract: Involvement of TGFbeta/Smad signaling. Mol
Vis. 12:681–691. 2006.PubMed/NCBI
|
|
37
|
Nukaga S, Yasuda H, Tsuchihara K, Hamamoto
J, Masuzawa K, Kawada I, Naoki K, Matsumoto S, Mimaki S, Ikemura S,
et al: Amplification of EGFR wild-type alleles in non-small cell
lung cancer cells confers acquired resistance to mutation-selective
EGFR tyrosine kinase inhibitors. Cancer Res. 77:2078–2089. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Corallo S, D'Argento E, Strippoli A, Basso
M, Monterisi S, Rossi S, Cassano A and Barone CM: Treatment options
for EGFR T790M-negative EGFR tyrosine kinase inhibitor-resistant
non-small cell lung cancer. Target Oncol. 12:153–161. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lee CG, McCarthy S, Gruidl M, Timme C and
Yeatman TJ: MicroRNA-147 induces a mesenchymal-to-epithelial
transition (MET) and reverses EGFR inhibitor resistance. PLoS One.
9:e845972014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hu FY, Cao XN, Xu QZ, Deng Y, Lai SY, Ma J
and Hu JB: miR-124 modulates gefitinib resistance through SNAI2 and
STAT3 in non-small cell lung cancer. J Huazhong Univ Sci Technolog
Med Sci. 36:839–845. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kitamura K, Seike M, Okano T, Matsuda K,
Miyanaga A, Mizutani H, Noro R, Minegishi Y, Kubota K and Gemma A:
MiR-134/487b/655 cluster regulates TGF-β-induced
epithelial-mesenchymal transition and drug resistance to gefitinib
by targeting MAGI2 in lung adenocarcinoma cells. Mol Cancer Ther.
13:444–453. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kang CW, Han YE, Kim J, Oh JH, Cho YH and
Lee EJ: 4-Hydroxybenzaldehyde accelerates acute wound healing
through activation of focal adhesion signalling in keratinocytes.
Sci Rep. 7:141922017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jiang J and Gu J:
Beta1,4-galactosyltransferase V A growth regulator in glioma.
Methods Enzymol. 479:3–23. 2010. View Article : Google Scholar : PubMed/NCBI
|