Introduction
Cardiovascular diseases (CVDs) are major killers
that cause over 17.3 million deaths each year worldwide (1). Atherosclerosis (AS), which accounts
for a large proportion of CVDs, may lay a foundation for strokes
and heart attacks. AS, an inflammatory disease, is characterized by
fibrous cap generation, lipid assembly, and necrotic core formation
(2). Human aortic endothelial cells
(HAECs) are implicated in the generation of AS lesions (3). In addition, previous research has
indicated that the aberrant migration, proliferation, and apoptosis
of HAECs is closely related to AS advancement (4). HAEC dysfunction can be influenced by
oxidized low-density lipoprotein (ox-LDL), a pivotal risk factor in
AS advancement (5). Therefore, in
the present research, ox-LDL-induced HAECs were selected as AS cell
models to investigate the modulating mechanism implicated in
AS.
Long non-coding RNAs (lncRNAs), which are 200-nt
long, are mainstay modulators in AS and other complex diseases
(6). The significance of lncRNAs in
AS has received considerable attention since they can modulate
inflammatory responses, and lipid metabolism as well as cell
adhesion, migration, proliferation, and apoptosis (7). The expression of lncRNA H19 has
been revealed to be markedly increased in AS, and to upregulate
cell proliferation and suppress the apoptosis of HAECs (8). LncRNA RNCR3 was revealed to
accelerate the development of AS by impeding the migration and
proliferation and triggering the apoptosis of vascular smooth
muscle cells in vitro (9).
The lncRNA nuclear paraspeckle assembly transcript 1
(NEAT1) has been confirmed to be a pivotally blotted gene in cell
differentiation and growth (10).
NEAT1 modulates ox-LDL-triggered inflammation and lipid
uptake in macrophages via paraspeckle formation (11). In addition, NEAT1 blockade
was revealed to suppress inflammation response and lipid intake by
adjusting miR-342-3p in human macrophages (THP-1 cells) (12). However, the exact molecular
mechanism of NEAT1 in the growth of AS requires further
research.
miRNAs are long small ncRNAs with lengths of 22 nt
and post-transcriptionally modulate genes (13). miRNAs critically adjust AS
pathological processes, including cholesterol efflux and
lipoprotein metabolism, lipid and cholesterol biosynthesis,
endothelial cell biology, immune responses, and vascular function
(14). miR-638 was revealed to
suppress tumors in breast (15),
hepatocellular (16), and other
cancers. However, the roles of miR-638 in AS remain unclear.
The present research aimed to identify the
underlying treatment targets for AS by further investigating the
possible roles and molecular bases of NEAT1 and miR-638 in
the advancement of HAECs.
Materials and methods
Clinical specimens and ethics
statement
The present research was conducted with the approval
of the Ethics Committee of Tianjin Chest Hospital. A total of 10 ml
of blood specimens was harvested from 24 healthy volunteers without
malignant tumors, recent infection, AS disease, autoimmune
diseases, or inflammatory diseases (<1 month) and from 24
patients with AS. The samples were placed in centrifuge tubes
without anticoagulant. Subsequently, the specimens were maintained
for approximately 1 h at room temperature, and serum was collected
through 50 min of centrifugation at 1,006 × g. All the patients and
healthy volunteers signed informed consent to participate in this
study.
Cell culture
HAECs were obtained from the American Type Culture
Collection and cultured in DMEM containing 10% fetal bovine serum,
100 U/ml penicillin, and streptomycin in an incubator with 5%
CO2 at 37°C.
Cell transfection and treatment
The full-length NEAT1 sequence was amplified
through polymerase chain reaction (PCR), and pcDNA-NEAT1
overexpression plasmids were constructed following the subcloning
of the sequence into pcDNA3.1 vectors (Invitrogen; Thermo Fisher
Scientific, Inc.). Shanghai GenePharma Co., Ltd. synthesized
miR-638 mimics, the miR-638 inhibitor, and their negative control
(miR-con), as well as small interference RNA (siRNA) specific for
NEAT1 (si-NEAT1#1 and si-NEAT1#2) and its
negative control (si-con). Cells were transfected with
Lipofectamine 2000 reagent acquired from Invitrogen in accordance
with the manufacturer's instructions. HAECs were treated with
different concentrations of ox-LDL (0, 25, 50 and 75 µg/ml) for 24
h to elucidate the effects of ox-LDL, which was provided by
Biosynthesis Biotechnology, on NEAT1 and miR-638 expression
levels.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) assay
TRIzol® reagent obtained from Invitrogen;
Thermo Fisher Scientific, Inc. was employed for total RNA
extraction in accordance with the manufacturer's guidelines.
Following the measurement of the concentration and purity of RNA
specimens, RT was performed to synthesize cDNA specimens with U6
and GAPDH as the internal reference. A PCR solution was prepared
with a premixed solution containing forward (F)/reverse (R)
primers, DEPC from Beyotime Institute of Biotechnology, SYBR Green
from Applied Biosystems; Thermo Fisher Scientific, Inc., and
templates. Subsequently, the prepared solution was placed on an
RT-PCR instrument for PCR amplification. miRNAs underwent RT into
cDNAs with the use of a miRNA RT Kit from Tiangen Biotech Co., Ltd.
and then subjected to PCR and quantitative analysis on the basis of
the instructions of the miRNA qPCR kit from the same company. The
primer sequences were as follows: lnc-NEAT1 F, TGTCCCTCGGCTATGTCAGA
and R, GAGGGGACGTGTTTCCTGAG; GAPDH F, TGACCACAGTCCATGCCATCAC and R,
GCCTGCTTCACCACCTTCTTGA; miR-638 F, AAGGGATCGCGGGCGGGT and R,
CAGTGCAGGGTCCGAGGT; and U6 F, CTCGCTTCGGCAGCACATATACTA and R,
ACGAATTTGCGTGTCATCCTTGC.
Western blot analysis
Total proteins were extracted with RIPA lysis buffer
acquired from Beyotime Institute of Biotechnology and then
quantified with a Pierce BCA Protein Assay Kit obtained from Thermo
Fisher Scientific, Inc. SDS-PAGE (10%) was used for the isolation
of 50 µg of proteins, which were then transferred to a PVDF
membrane (EMD Millipore). The membrane was sealed with 5% skim milk
at room temperature for 1 h and incubated with primary antibodies
against proliferating cell nuclear antigen (PCNA product no.
ab92552; dilution 1:1,000), β-actin (product no. ab8226; dilution
1:2,000), proliferation marker protein Ki-67 (Ki-67 product no.
ab92742; dilution 1:2,000), PGK1 (product no. ab38007; dilution
1:1,000), AKT (product no. ab179463; dilution 1:1,000), p-AKT
(product no. ab38449; dilution 1:1,500), mTOR (product no. ab2732;
dilution 1:1,000), p-mTOR (product no. ab109268; dilution 1:1,500),
Bax (product no. ab182733; dilution 1:1,000) and Bcl-2 (product no.
ab196495; dilution 1:1,000) at 4°C overnight. Then, at room
temperature, the membrane was further probed for 1 h using
secondary antibodies (product nos. ab97040 and ab97080; dilution
1:3,000) coupled with HRP. All the antibodies were purchased from
Abcam. Finally, Clarity Max™ Western ECL Substrate obtained from
Bio-Rad Laboratories, Inc. was used to improve particular protein
signals. The antibodies aforementioned were commercially obtained
from Abcam.
CCK-8 assay
Approximately 1×104 HAECs were inoculated
into each well of a 96-well plate overnight. A total of 10 µl of
CCK-8 solution (Dojindo Molecular Technologies, Inc.) was added to
each well following transfection for 0, 24, 48, or 72 h. Next, the
cells were further cultured at 37°C for 3 h. Cell proliferation was
evaluated after the measurement of the absorbance
(A)450.
Colony formation assay
After seeding in a 12-well plate for 15 days of
culture in complete medium, the transfected HAECs were immobilized
with methanol and then dyed with 0.1% crystal violet solution (room
temperature for 20 min) provided by Sigma-Aldrich; Merck KGaA.
Finally, colonies containing at least 50 cells were quantified with
the use of an DMi1 inverted microscope (Leica Microsystems).
Cell apoptosis assays
Cells undergoing apoptosis were examined using
Annexin V FITC/PI apoptosis detection kit procured from Vazyme
Biotech Co., Ltd. in accordance with the manufacturer's
instructions. Briefly, harvested cells were resuspended in 1X
binding buffer (100 µl), and a dyeing solution consisting of PI (5
µl) and Annexin V FITC (5 µl) was added. After mixing, the cells
were incubated for 15 min at room temperature away from light.
Then, prior to the addition of 1X binding buffer (240 µl),
apoptotic cells were counted with the use of a flow cytometric
apparatus supplied by BD Biosciences.
Examination of caspase-3 activity
Caspase-3 activity was assessed using Caspase-3
Assay Kit (Colorimetric) supplied by Abcam in accordance with the
manufacturer's instructions. Briefly, 5×106 transfected
cells were resuspended with 50 µl of cell lysate cooled in advance.
Then, cell supernatant separation and protein concentration
assessment were performed. Subsequently, 4 mM DEVD-p-NA substrate
(5 µl) with a final concentration of 200 µM and 2X reaction buffer
with 10 mM DTT (50 µl) were added to 50 µl of each sample.
Subcellular fractions
In reference to the manufacturer's instructions, RNA
in HAEC cytoplasm or nucleus was isolated and purified with a
cytoplasmic and nuclear RNA purification kit supplied by Norgen
Biotek. Then, NEAT1, U6 and GAPDH expression levels in the
cytoplasmic and nuclear fractions were independently assessed via
qRT-qPCR.
Luciferase activity analysis
The 3′UTR fragments of PGK1 and NEAT1
comprising the binding sites of miR-638 were subjected to PCR
amplification and established with psiCHECK-2 vector supplied by
Promega Corporation to form PGK1-WT and NEAT1-WT
reporters. Additionally, PGK1-MUT and NEAT1-MUT
reporters comprising MUT miR-638 binding sites were produced via a
Quickchange Multi Site-Directed Mutagenesis kit purchased from
Stratagene; Agilent Technologies, Inc. Subsequently, HAECs were
treated with the established luciferase reporters and plasmids or
miRNAs. After 48 h, cell luciferase activity was assessed with the
use of a dual luciferase reporter assay kit supplied by Promega
Corporation in accordance with the manufacturer's instructions.
RNA immunoprecipitation (RIP)
assay
To probe the presence of NEAT1 in the
RNA-induced silencing complex (RISC), an EZ-Magna RIP kit
commercially provided by EMD Millipore was utilized for a RIP assay
with the use of Ago2 antibody (product no. ab32381; dilution 1:150)
from Abcam. Briefly, HAECs were lysed using RIP lysis buffer and
incubated with anti-IgG antibody (cat. no. 12-370; dilution 1:150;
Merck Millipore) supplied by EMD Millipore or anti-Ago2 antibody
obtained from Abcam and protein A/G magnetic beads. RNAs in
complexes linked with magnetic beads were subsequently purified.
Ultimately, the expression levels of miR-638 and NEAT1 were
examined via qRT-PCR using Ago2 or IgG antibody.
Statistical analysis
Each assay was implemented at least thrice, and the
obtained data were presented as the mean ± SEM. Intergroup data
differences were probed using Student's t-test or one-way analysis
of variance (ANOVA) followed by Bonferroni post hoc test. P<0.05
indicated a statistically significant difference.
Results
NEAT1 is increased and miR-638 is
decreased in the serum of patients with AS and ox-LDL-triggered
HAECs
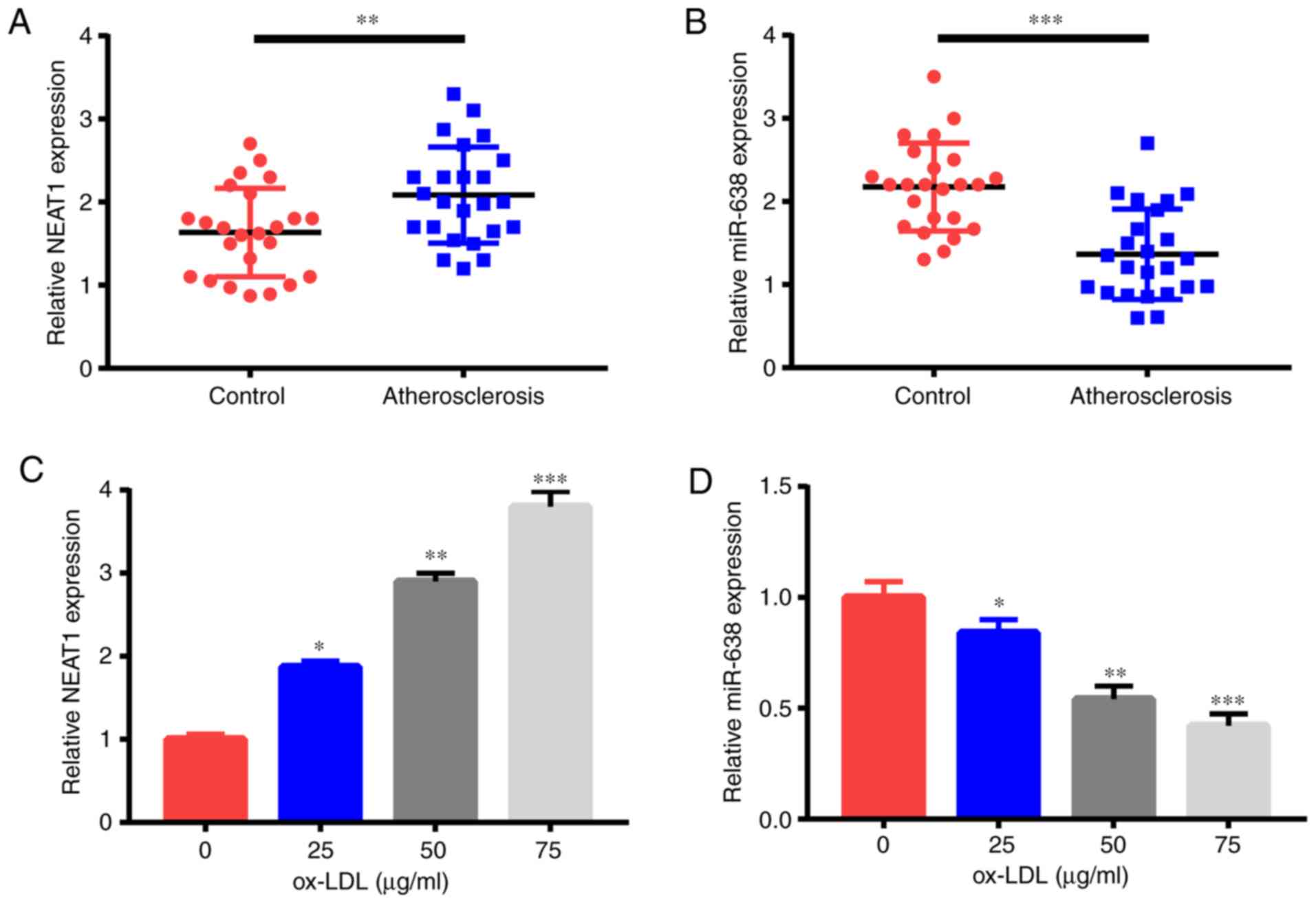
First, NEAT1 and miR-638 expression levels
were detected through qRT-PCR. Compared with the 24 healthy
controls, the 24 patients with AS had significantly increased
NEAT1 expression (Fig. 1A)
but a significantly reduced miR-638 expression (Fig. 1B) in their serum samples. Moreover,
ox-LDL treatment triggered the increase of NEAT1 expression
(Fig. 1C) and the decrease of
miR-638 expression (Fig. 1D) in
HAECs in a dose-dependent manner. Therefore, NEAT1 and
miR-638 were revealed to be possible pivotal controllers in AS
advancement.
NEAT1 downregulation decreases the
proliferation and triggers the apoptosis of ox-LDL-induced
HAECs
The roles of NEAT1 in the apoptosis and
proliferation of ox-LDL-triggered HAECs were investigated by
interfering with NEAT1 expression via siRNA
(si-NEAT1#1 and si-NEAT1#2) treatment. After
transfection with si-NEAT1#1 or si-NEAT1#2, HAECs
were treated with 50 µg/ml ox-LDL for 24 h, and their apoptosis
rate and proliferation capacity were assessed. NEAT1
expression was significantly suppressed by transfection with
si-NEAT1#1 and si-NEAT1#2 (Fig. 2A). As revealed in the CCK-8 assay,
NEAT1 downregulation suppressed HAEC proliferation (Fig. 2B). Furthermore, colony formation
assay ascertained that NEAT1 downregulation impeded the
colony formation ability of HAECs (Fig.
2C). Moreover, the expression levels of proliferation-related
indexes [Ki-67 and proliferating cell nuclear antigen (PCNA)] were
detected. Their expression levels in HAECs were significantly
decreased subsequent to NEAT1 downregulation (Fig. 2D). In addition, the flow cytometric
experiment revealed that apoptosis in HAECs treated with
NEAT1 siRNAs was increased relative to that in the control
(Fig. 2G). Fig. 2E and F revealed that NEAT1
downregulation significantly enhanced caspase-3 activity and Bax
expression and decreased Bcl-2 expression. The aforementioned
findings indicated that NEAT1 downregulation inhibited HAEC
proliferation and stimulated apoptosis.
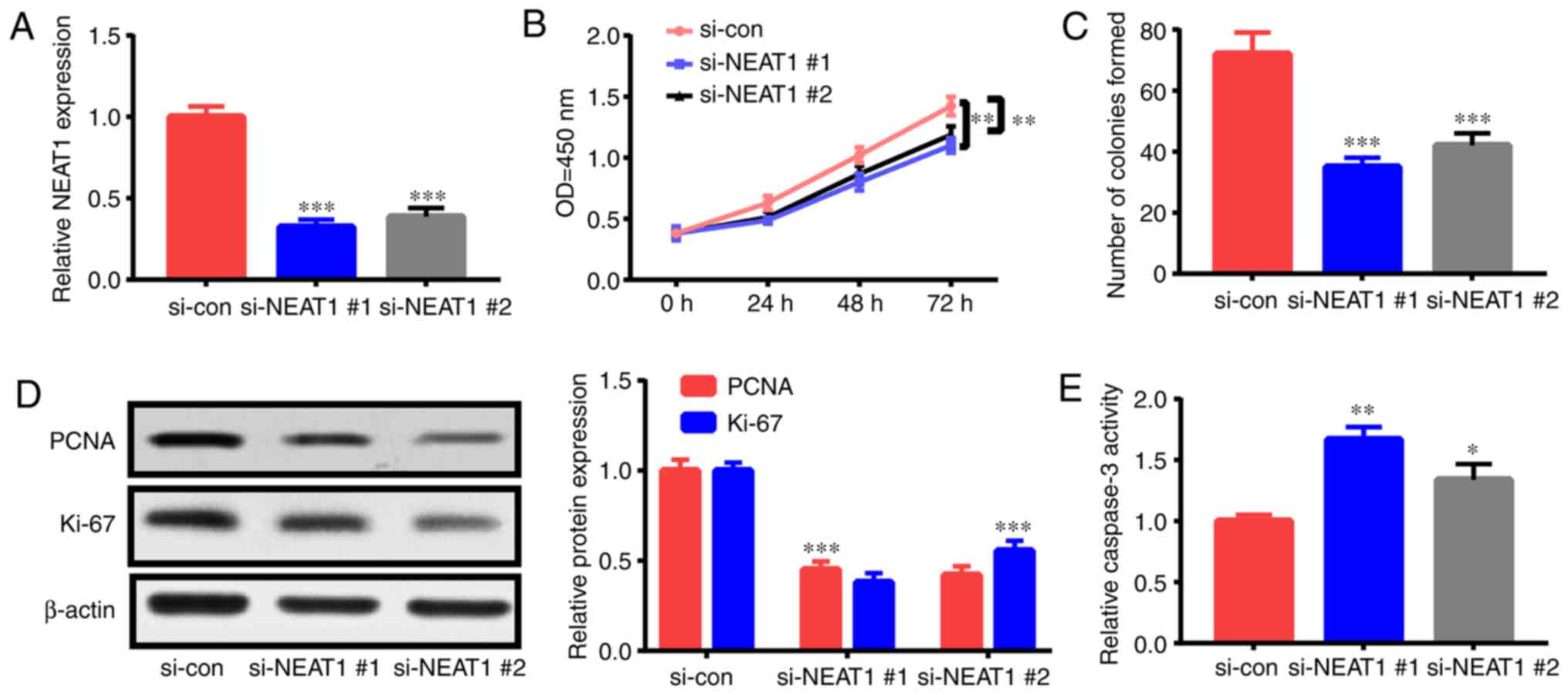 | Figure 2.NEAT1 downregulation decreases
cell proliferation and triggers the apoptosis of ox-LDL-induced
HAECs. HAECs were subjected to si-NEAT1#1,
si-NEAT1#2, or si-con transfection and ox-LDL (50 µg/ml)
treatment for 1 day. Then, (A) NEAT1 expression, (B)
proliferation potential, (C) colony formation ability, (D) PCNA and
Ki-67 expression levels, (E) caspase-3 activity, (F) Bcl-2 and Bax
expression levels and (G) cells undergoing apoptosis were detected.
*P<0.05, **P<0.01 and ***P<0.001. NEAT1, nuclear
paraspeckle assembly transcript 1; ox-LDL, oxidized low-density
lipoprotein; HAECs, human aortic endothelial cells; PCNA,
proliferating cell nuclear antigen. |
NEAT1 reduces miR-638 expression by
direct mutual action
Bioinformatics analysis was applied to verify the
possible miRNA sponged by NEAT1. Some potential binding
sites between miR-638 and NEAT1 were reveealed (Fig. 3A). This speculation was further
demonstrated by the transfection of HAECs with NEAT1-WT or
NEAT1-MUT luciferase reporter and miR-638 or miR-con. The
results of the following luciferase assay revealed that relative to
miR-con treatment, miR-638 treatment decreased by half the
luciferase activity of NEAT1-WT reporter but did not
influence that of the NEAT1-MUT reporter (Fig. 3B). Then, the direct binding
relationship between miR-638 and NEAT1 was elucidated via
RIP assay. Fig. 3C revealed that
miR-638 and NEAT1 were markedly upregulated by Ago2 antibody
relative to the control IgG antibody, revealing the presence of
miR-638 and NEAT1 in the RISC. The fractionation experiment
in subcells revealed that NEAT1 was primarily present in the
cytoplasm of HAECs, indicating that NEAT1 may sponge miR-638
(Fig. 3D). Furthermore, miR-638
expression significantly decreased upon overexpression of
NEAT1 expression; these effects were abrogated by
si-NEAT1 transfection (Fig. 3E
and F). Lastly, an inverse relationship between miR-638
expression and NEAT1 expression was observed in the serum of
patients (Fig. 3G).
 | Figure 3.NEAT1 curbs miR-638 expression
through direct mutual action. (A) Speculated binding sites between
miR-638 and NEAT1 and MUT sites in NEAT1-MUT reporter
are displayed. (B) Luciferase activity was assessed in HAECs
co-transfected with NEAT1-WT or NEAT1-MUT reporter
and miR-638 or miR-con. (C) Efficiency of binding between miR-638
and NEAT1 to Ago2 protein in HAECs was investigated via RIP
experiment. (D) U6, GAPDH, and NEAT1 expression levels in
HAEC cytoplasm and nucleus were assessed. (E and F) HAECs were
treated with pcDNA-NEAT1 overexpression plasmid, pcDNA3.1
empty vector, si-con, si-NEAT1#1, or si-NEAT1#2, and
miR-638 and NEAT1 levels were examined. (G) Correlation
analysis of NEAT1 and miR-638 expression levels. *P<0.05,
**P<0.01, and ***P<0.001. NEAT1, nuclear paraspeckle
assembly transcript 1; HAECs, human aortic endothelial cells; RIP,
RNA immunoprecipitation. |
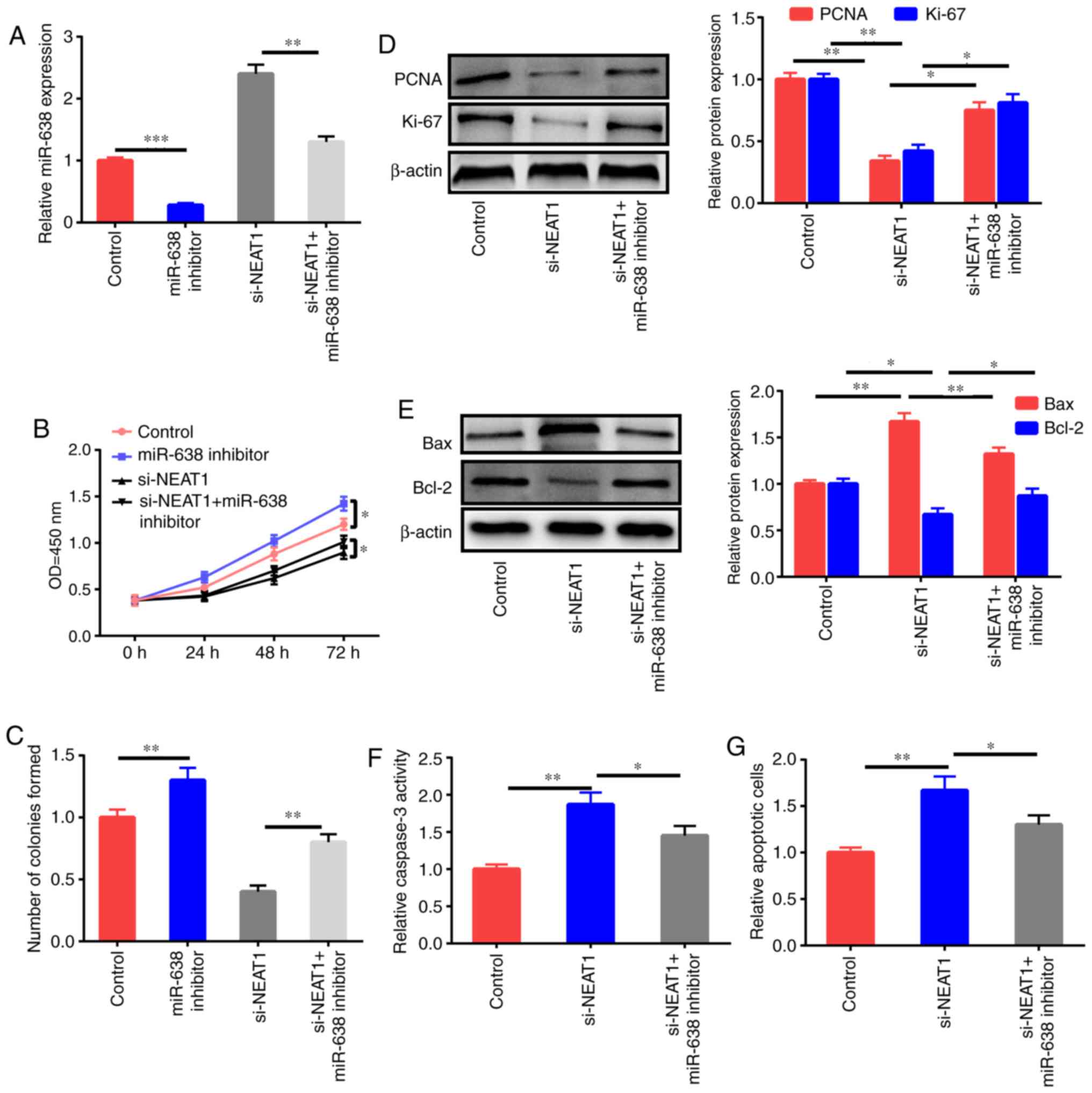
miR-638 inhibitor reverses the effects
of NEAT1 downregulation on the apoptosis and proliferation of
ox-LDL-induced HAECs
Whether the antiproliferation and proapoptosis
effects of NEAT1 downregulation were mediated by miR-638 was
further studied through the treatment of HAECs with the control,
miR-638 inhibitor, si-NEAT1, and si-NEAT1+miR-638
inhibitor after ox-LDL stimulation, and miR-638 expression was
assessed (Fig. 4A). miR-638
inhibition significantly enhanced the proliferation ability and
colony formation ability of HAECs, and NEAT1 downregulation
decreased cell growth; this effect was reversed by the miR-638
inhibitor (Fig. 4B and C). The
downregulation of PCNA and Ki-67 expression after treatment with
si-NEAT1 was partly reversed by inhibition of miR-638
(Fig. 4D). Impeding miR-638
expression partly reversed the increase in Bax expression, the
decrease of Bcl-2, and the upregulation of caspase-3 activity and
cell apoptosis induced by si-NEAT1 (Fig. 4E-G).
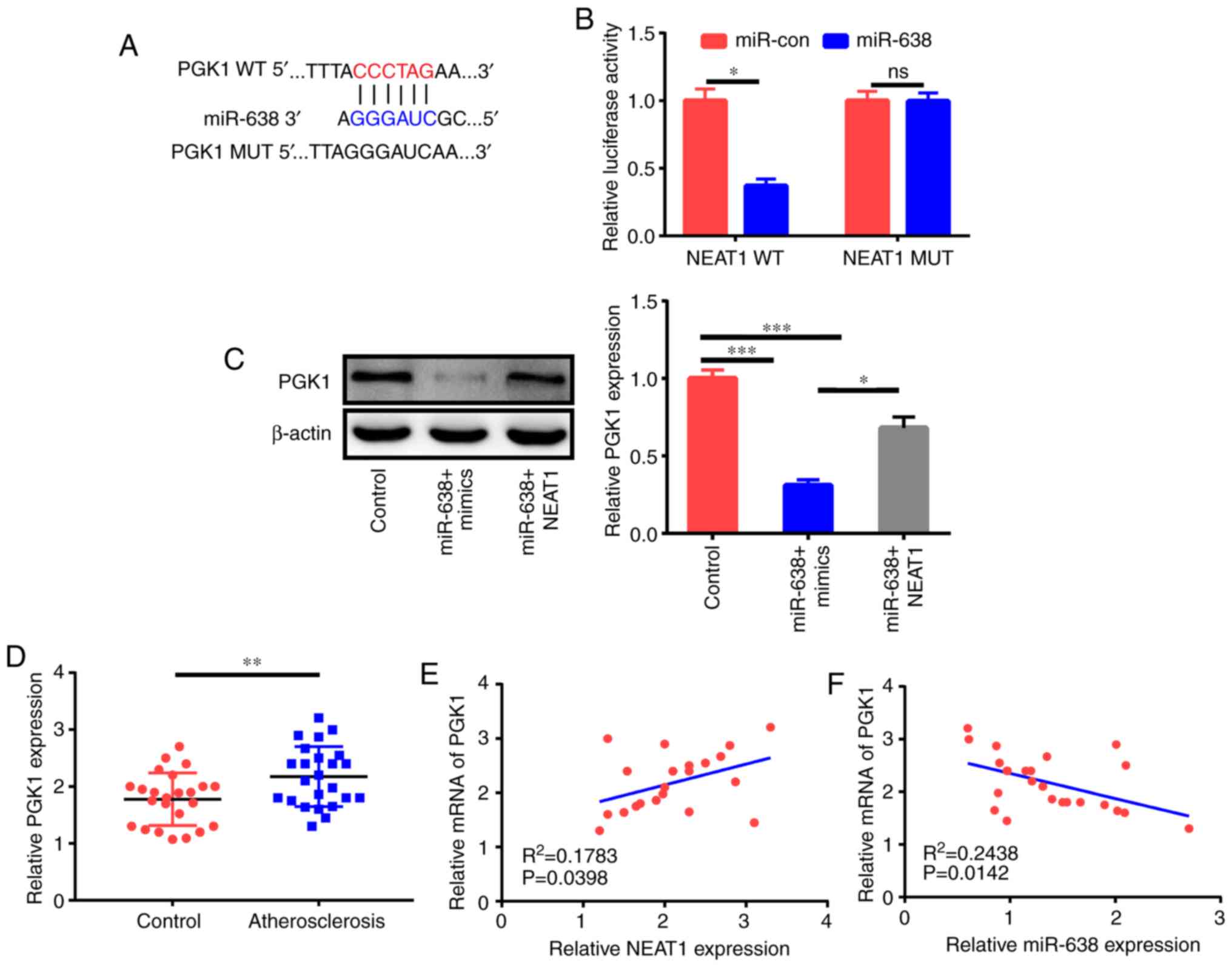
PGK1 is a target of miR-638
LncRNAs function as competing endogenous RNAs
(ceRNAs) of miRNAs to adjust target mRNA expression (17). Thus, bioinformatics analysis was
applied to elucidate the possible targets of miR-638. As revealed
in Fig. 5A, the PGK1 3′UTR
comprised the potential binding sequences of miR-638. The
subsequent luciferase assay demonstrated that miR-638 decreased the
luciferase activity of the PGK1-WT reporter but not that of
the PGK1-MUT reporter (Fig.
5B). The western blot assay further revealed that miR-638
significantly inhibited PGK1 expression, and NEAT1
could partly reverse this effect (Fig.
5C). Clinical data indicated that PGK1 expression in the
serum of patients with AS was higher than that in the serum of
healthy controls (Fig. 5D). In
addition, PGK1 expression had a positive relationship with
NEAT1 expression and an inverse association with miR-638
expression (Fig. 5E and F) in
patients with AS.
miR-638 blocks the proliferation of
ox-LDL-induced HAECs and facilitates their apoptosis by modulating
AKT/mTOR signaling
PGK1 activates the AKT/mTOR pathway (18). In the present study, the effects of
PGK1 and miR-638 on the AKT/mTOR signaling pathway were also
evaluated. Western blot results revealed that the inhibition of
miR-638 increased p-AKT and p-mTOR instead of total AKT and mTOR
(Fig. 6A). miR-638 overexpression
decreased p-AKT and p-mTOR expression but did not affect total AKT
and mTOR expression. In addition, the upregulation of PGK1
could partly reverse the effects of miR-638 on p-AKT and p-mTOR
(Fig. 6A). Then, HAECs were treated
with the control, miR-638 inhibitor, miR-638 mimics, and miR-638
mimics+PGK1 for 24 h. PGK1 upregulation partly reversed the
suppressive effects of miR-638 on proliferation capacity and colony
formation ability (Fig. 6B and C).
The expression levels of PCNA and Ki-67 were also detected
(Fig. 6D). PGK1 partly reversed the
apoptosis of HAECs caused by the upregulation of miR-638 (Fig. 6E-G). To sum up, these findings
indicated that miR-638 exerted its roles via modulation of the
AKT/mTOR signaling by targeting PGK1.
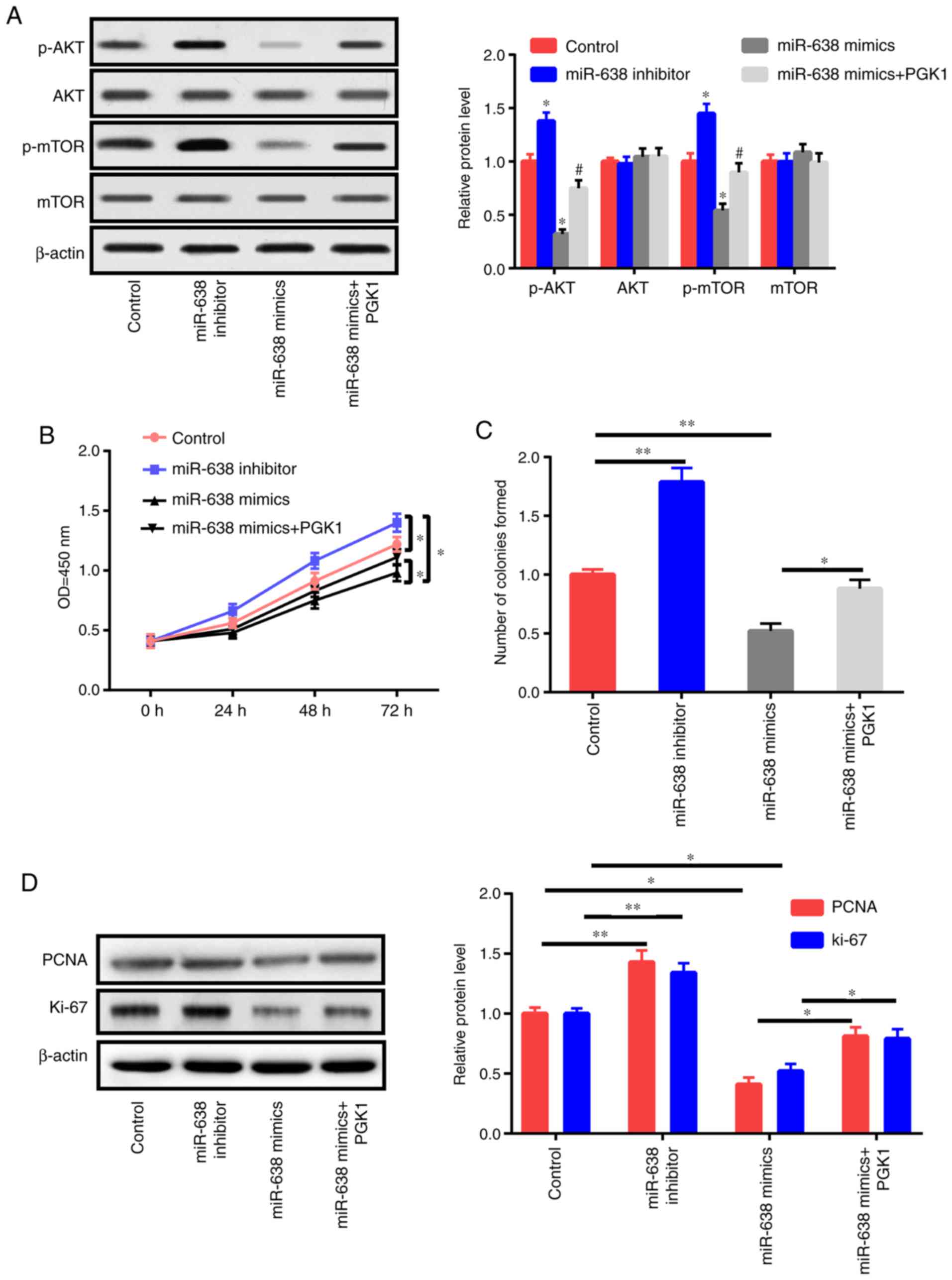 | Figure 6.miR-638 adjusts HAEC proliferation
and apoptosis through Akt/mTOR signaling. (A) HAECs were treated
with the control, miR-638 inhibitor, miR-638 mimics, si-NEAT1, and
si-NEAT1+miR-638 inhibitor mimics+PGK1 after 1 day of ox-LDL (50
µg/ml) stimulation, and mTOR, p-mTOR, AKT, and p-AKT expression
levels were assessed. (B) Cell proliferation abilities were
determined. (C) Colony formation ability. (D) Expression levels of
PCNA and Ki-67. miR-638 adjusts HAEC proliferation and apoptosis
through Akt/mTOR signaling. (A) HAECs were treated with the
control, miR-638 inhibitor, miR-638 mimics, si-NEAT1, and
si-NEAT1+miR-638 inhibitor mimics+PGK1 after 1 day of ox-LDL (50
µg/ml) stimulation, and mTOR, p-mTOR, AKT, and p-AKT expression
levels were assessed. (B) Cell proliferation abilities were
determined. (C) Colony formation ability. (D) Expression levels of
PCNA and Ki-67. (E) Cells undergoing apoptosis. (F) Caspase-3
activity. (G) Bcl-2 and Bax expression levels. *P<0.05,
**P<0.01 vs. control group, #P<0.05 vs. miR-638
mimics group. HAEC, human aortic endothelial cell; NEAT1, nuclear
paraspeckle assembly transcript 1; ox-LDL, oxidized low-density
lipoprotein; PCNA, proliferating cell nuclear antigen. |
Discussion
Despite the advanced treatment and clarified
pathogenesis of AS, AS continues to severely endanger human health
and exhibit high morbidity and mortality rates worldwide (19). A growing body of evidence has
revealed that ncRNAs, including lncRNAs and miRNAs, exert mainstay
effects on AS advancement (20). To
further investigate the molecular mechanism of NEAT1,
bioinformatics analysis was implemented to determine the possible
targets of NEAT1. miR-638 was likely to mutually act with
NEAT1. miR-638 has been revealed to be a tumor suppressor
(21). The downregulation of
miR-638 could promote cell proliferation and inhibit cell apoptosis
(15,22). Ox-LDL could trigger AS by boosting
EC dysfunction and activation, foaming cell formation, adjusting
VSMC behavior, as well as several other mechanisms (23).
Endothelial cells also play a pivotal part in an
early step in AS growth and contribute to the formation and
progression of AS plaques as well as the occurrence of
complications (24). Notably,
endothelial dysfunction is associated with smoking, dyslipidemia,
obesity, diabetes, mental stress, hypertension, and other
cardiovascular risk factors (25).
Endothelial cell apoptosis and death can lead to local lipid
deposition and eventually AS and even thrombosis (26,27).
Thus, the present study aimed to investigate the involvement of
NEAT1 in the modulation of AS advancement by adjusting
miR-638 in ox-LDL-triggered HAECs.
First, the expression of the level of NEAT1
was confirmed to be increased and the expression level of miR-638
to be decreased in serum samples from AS cases. Second, miR-638
expression was decreased but NEAT1 expression was increased
in HAECs after ox-LDL treatment. As revealed by loss-of-function
tests, NEAT1 downregulation significantly suppressed the
proliferation and facilitated the apoptosis of ox-LDL-triggered
HAECs.
Next, qRT-qPCR, fraction assay in subcells,
bioinformatics analysis, luciferase assay, and RIP test further
confirmed that NEAT1 interfered in miR-638 expression by
binding to miR-638. Moreover, an inverse relationship was revealed
between miR-638 expression and NEAT1 expression in the serum
of AS cases. Thus, the mediation of miR-638 in the effects of
NEAT1 on the apoptosis and proliferation of ox-LDL-triggered
HAECs was further probed. Findings revealed that the proapoptosis
and antiproliferation functions mediated by NEAT1
downregulation were markedly undermined subsequent to miR-638
reduction in ox-LDL-triggered HAECs. According to emerging
evidence, miRNAs function by adjusting the stability or translation
of target mRNAs. Thus, bioinformatics was utilized to identify the
possible target mRNAs of miR-638. The findings revealed that
PGK1 was an underlying target of miR-638 as demonstrated by
the subsequent luciferase assay. The present study ascertained that
NEAT1 increased the expression of the target gene
PGK1 in HAECs by functioning as a ceRNA of miR-638. In
addition, the mRNA expression of PGK1 was positively
associated with NEAT1 expression and inversely correlated
with miR-638 expression in the serum of AS cases.
PGK1 plays a key role in glycolytic ATP
production. Several lines of evidence suggest that mitochondrial
ATP production can activate the PI3K/AKT pathway via P2 receptors
in multiple cell types (28,29).
Moreover, an increased level of cytosolic free calcium, the vital
second messenger of the ATP/P2 receptor-signaling pathway, can
activate PI3K through direct linking with its p85 regulatory
subunit, resulting in AKT/mTOR pathway activation (30). PGK1 was revealed to activate
the AKT and ERK pathways in prostate cancer cells, resulting in the
promotion of cell invasion (31).
Thus, the effects of PGK1 and miR-638 on the expression levels of
genes associated with AKT/mTOR signaling (p-AKT, p-mTOR, total AKT,
and total mTOR) were examined in ox-LDL-induced HAECs. miR-638
downregulation activated the AKT/mTOR pathway in ox-LDL-triggered
HAECs. In addition, the function of miR-638 in AKT/mTOR signaling
was undermined subsequent to PGK1 upregulation. These
findings demonstrated that miR-638 suppression increased AKT/mTOR
signaling by adjusting PGK1. Then, whether miR-638
influences the apoptosis and proliferation of ox-LDL-triggered
HAECs was investigated. The results revealed that PGK1 could
partly reverse the effects of miR-638.
To sum up, the present study confirmed that
NEAT1 downregulation restrained ox-LDL-triggered HAECs from
proliferating and induced their apoptosis by inactivating AKT/mTOR
signaling via miR-638. However, the present study has several
limitations. First, the number of samples was small, and the
results would be more convincing if we had collected additional
samples. Second, whether other signaling pathways were affected was
not verified in this study, and other key molecules should be
detected. Third, in vivo experiments were not performed.
In vivo experiments are planned in future studies to
strengthen our conclusion.
The present study determined that the
NEAT1/miR-638/ PGK1/AKT/mTOR regulatory axis is
involved in HAEC proliferation, indicating that NEAT1 has a
possible function in AS prevention. However, the mechanism and
roles of NEAT1 in AS should be determined with the use of
animal models in the future.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
All authors participated in the study and manuscript
preparation. XZ, MXG, ZGW and XHQ performed all experiments, QHJ,
SL and HYZ performed the statistical analysis and drafted the
manuscript. HLC conceived and designed the study and revised the
manuscript. All authors read and approved the manuscript and agree
to be accountable for all aspects of the research in ensuring that
the accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
The study protocol was approved by the Ethics
Committee of Tianjin Chest Hospital. All the patients provided
written informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Laslett LJ, Alagona P Jr, Clark BA III,
Drozda JP Jr, Saldivar F, Wilson SR, Poe C and Hart M: The
worldwide environment of cardiovascular disease: Prevalence,
diagnosis, therapy, and policy issues: A report from the American
College of Cardiology. J Am Coll Cardiol. 60 (25 Suppl):S1–S49.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dharmakidari S, Bhattacharya P and
Chaturvedi S: Carotid artery stenosis: Medical therapy, surgery,
and stenting. Curr Neurol Neurosci Rep. 17:772017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Libby P, Bornfeldt KE and Tall AR:
Atherosclerosis: Successes, surprises, and future challenges. Circ
Res. 118:531–534. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li Y, Yang C, Zhang L and Yang P:
MicroRNA-210 induces endothelial cell apoptosis by directly
targeting PDK1 in the setting of atherosclerosis. Cell Mol Biol
Lett. 22:32017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen M, Ren L, Meng Y, Shi L, Chen L, Yu
B, Wu Q and Qi G: The protease inhibitor E64d improves
ox-LDL-induced endothelial dysfunction in human aortic endothelial
cells. Can J Physiol Pharmacol. 96:120–127. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen X, Yan CC, Zhang X and You ZH: Long
non-coding RNAs and complex diseases: From experimental results to
computational models. Brief Bioinform. 18:558–576. 2017.PubMed/NCBI
|
|
7
|
Liu Y, Zheng L, Wang Q and Hu YW: Emerging
roles and mechanisms of long noncoding RNAs in atherosclerosis. Int
J Cardiol. 228:570–582. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pan JX: LncRNA H19 promotes
atherosclerosis by regulating MAPK and NF-kB signaling pathway. Eur
Rev Med Pharmacol Sci. 21:322–328. 2017.PubMed/NCBI
|
|
9
|
Shan K, Jiang Q, Wang XQ, Wang YN, Yang H,
Yao MD, Liu C, Li XM, Yao J, Liu B, et al: Role of long non-coding
RNA-RNCR3 in atherosclerosis-related vascular dysfunction. Cell
Death Dis. 7:e22482016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yu X, Li Z, Zheng H, Chan MT and Wu WK:
NEAT1: A novel cancer-related long non-coding RNA. Cell Prolif.
50:2017. View Article : Google Scholar
|
|
11
|
Huang-Fu N, Cheng JS, Wang Y, Li ZW and
Wang SH: Neat1 regulates oxidized low-density lipoprotein-induced
inflammation and lipid uptake in macrophages via paraspeckle
formation. Mol Med Rep. 17:3092–3098. 2018.PubMed/NCBI
|
|
12
|
Wang L, Xia JW, Ke ZP and Zhang BH:
Blockade of NEAT1 represses inflammation response and lipid uptake
via modulating miR-342-3p in human macrophages THP-1 cells. J Cell
Physiol. 234:5319–5326. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rupaimoole R and Slack FJ: MicroRNA
therapeutics: Towards a new era for the management of cancer and
other diseases. Nat Rev Drug Discov. 16:203–222. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Aryal B, Singh AK, Rotllan N, Price N and
Fernández-Hernando C: MicroRNAs and lipid metabolism. Curr Opin
Lipidol. 28:273–280. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li M, Wang J and Liu H: Downregulation of
miR-638 promotes progression of breast cancer and is associated
with prognosis of breast cancer patients. Onco Targets Ther.
11:6871–6877. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang Y, Zhang D, Jiang J and Dong L: Loss
of miR-638 promotes invasion and epithelial-mesenchymal transition
by targeting SOX2 in hepatocellular carcinoma. Oncol Rep.
37:323–332. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Paraskevopoulou MD and Hatzigeorgiou AG:
Analyzing MiRNA-LncRNA Interactions. Methods Mol Biol.
1402:271–286. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang J, Ying G, Wang J, Jung Y, Lu J, Zhu
J, Pienta KJ and Taichman RS: Characterization of phosphoglycerate
kinase-1 expression of stromal cells derived from tumor
microenvironment in prostate cancer progression. Cancer Res.
70:471–480. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Weber C and Noels H: Atherosclerosis:
Current pathogenesis and therapeutic options. Nat Med.
17:1410–1422. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou T, Ding JW, Wang XA and Zheng XX:
Long noncoding RNAs and atherosclerosis. Atherosclerosis.
248:51–61. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zheng DH, Wang X, Lu LN, Chen DL, Chen JM,
Lin FM and Xu XB: MiR-638 serves as a tumor suppressor by targeting
HOXA9 in glioma. Eur Rev Med Pharmacol Sci. 22:7798–7806.
2018.PubMed/NCBI
|
|
22
|
Zhao P, Zhang BL, Liu K, Qin B and Li ZH:
Overexpression of miR-638 attenuated the effects of
hypoxia/reoxygenation treatment on cell viability, cell apoptosis
and autophagy by targeting ATG5 in the human cardiomyocytes. Eur
Rev Med Pharmacol Sci. 22:8462–8471. 2018.PubMed/NCBI
|
|
23
|
Nakajima K, Nakano T and Tanaka A: The
oxidative modification hypothesis of atherosclerosis: The
comparison of atherogenic effects on oxidized LDL and remnant
lipoproteins in plasma. Clin Chim Acta. 367:36–47. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dong Y, Fernandes C, Liu Y, Wu Y, Wu H,
Brophy ML, Deng L, Song K, Wen A, Wong S, et al: Role of
endoplasmic reticulum stress signalling in diabetic endothelial
dysfunction and atherosclerosis. Diab Vasc Dis Res. 14:14–23. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gimbrone MA Jr and Garcia-cardena G:
Endothelial cell dysfunction and the pathobiology of
atherosclerosis. Circ Res. 118:620–636. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hainsworth AH, Oommen AT and Bridges LR:
Bridges, Endothelial cells and human cerebral small vessel disease.
Brain Pathol. 25:44–50. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Goveia J, Stapor P and Carmeliet P:
Principles of targeting endothelial cell metabolism to treat
angiogenesis and endothelial cell dysfunction in disease. EMBO Mol
Med. 6:1105–1120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Osorio-Fuentealba C, Contreras-Ferrat AE,
Altamirano F, Espinosa A, Li Q, Niu W, Lavandero S, Klip A and
Jaimovich E: Electrical stimuli release ATP to increase GLUT4
translocation and glucose uptake via PI3Kγ-Akt-AS160 in skeletal
muscle cells. Diabetes. 62:1519–1126. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ishikawa M, Iwamoto T, Nakamura T, Doyle
A, Fukumoto S and Yamada Y: Pannexin 3 functions as an ER Ca(2+)
channel, hemichannel, and gap junction to promote osteoblast
differentiation. J Cell Biol. 193:1257–1274. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cheng A, Wang S, Yang D, Xiao R and
Mattson MP: Calmodulin mediates brain-derived neurotrophic factor
cell survival signaling upstream of Akt kinase in embryonic
neocortical neurons. J Biol Chem. 278:7591–7599. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lay AJ, Jiang XM, Kisker O, Flynn E,
Underwood A, Condron R and Hogg PJ: Phosphoglycerate kinase acts in
tumour angiogenesis as a disulphide reductase. Nature. 408:869–873.
2000. View Article : Google Scholar : PubMed/NCBI
|