Introduction
Hypopharyngeal squamous cell carcinoma (HSCC) is an
aggressive tumor that develops in the hypopharynx (1). It accounts for 5% of all head and neck
squamous cell carcinomas (2).
Although HSCC therapy has markedly advanced over the past few
decades, the 5-year survival rate of patients with HSCC remains
lower than 50% due to diagnosis at late stages and tumor recurrence
or metastasis (3,4). Thus, understanding the oncogenic
mechanisms underlying HSCC and identifying novel biomarkers for the
early diagnosis of HSCC would be critical in improving diagnosis,
therapeutic strategies, and prognosis in HSCC treatment.
Long noncoding RNAs (lncRNAs) account for the
majority of transcripts and functional RNAs with more than 200
nucleotides (5). Mounting evidence
has documented that lncRNAs are critical players in the development
and metastasis of cancers, including HSCC (6,7). Qian
et al reported that lncRNA UCA1 regulates the proliferation,
invasion, and survival of HSCC cells (8). AB209630 and PEG10 have also been
demonstrated to regulate HSCC proliferation, invasion, and
metastasis (9,10). lncRNAs have been reported to
modulate multiple signaling pathways in tumorigenesis and
metastasis, such as VEGFA signaling, Wnt/beta-catenin signaling,
and epithelial-mesenchymal transition (EMT) (11–13).
RP11-156L14.1 is a novel lncRNA that has been identified in
cutaneous anaplastic large cell lymphoma (14). It has also been demonstrated to be
highly expressed in various cancers, including HSCC (15). However, the expression profile and
functional role of RP-11-156L14.1 in HSCC are not clear.
MicroRNAs (miRNAs) are small non-coding RNAs, which
can post-transcriptionally regulate target gene expression in tumor
progression and metastasis (16).
miRNAs including miR-489, miR-451a, and miR-504 have been
demonstrated to serve as tumor suppressors in HSCC (2,17,18).
Notably, lncRNAs can sponge miRNAs and function as competing
endogenous RNAs (ceRNAs) (19).
Multiple studies have described the lncRNA-miRNA-mRNA axis in HSCC
(11,20,21).
Kolenda et al reported that lncRNA ZFAS1 regulated the HSCC
cell phenotype via miR-150-5p (22). Thus, understanding the interplay
between lncRNA and miRNA in HSCC may lead to new therapeutic
strategies for HSCC.
In the present study, a novel lncRNA RP11-156L14.1
was identified that was highly expressed in HSCC tissues and cell
lines. Knockdown of RP11-156L14.1 inhibited HSCC cell
proliferation, migration, and invasion in vitro.
Furthermore, knockdown of RP11-156L14.1 suppressed HSCC tumor
development in vivo. Mechanistically, RP11-156L14.1 directly
interacted with miR-548ao-3p as a ceRNA and regulated the
expression of downstream targets of the signal sequence receptor
subunit 1 (SSR1). In summary, these findings indicated that the
lncRNA RP11-156L14.1/miR-548ao-3p/SSR1 axis may be a new potential
biomarker in the clinical treatment of HSCC.
Materials and methods
Clinical specimens
Thirty pairs of HSCC tissues and the adjacent normal
tissues were obtained from patients (aged, 33–65 years; 15 male and
15 female patients) who underwent surgical treatment at the Second
Affiliated Hospital of Xi'an Jiaotong University. All patients
underwent hypopharyngeal tumor resection plus ipsilateral modified
neck dissection. None of the patients had received
chemoradiotherapy or biotherapy before surgery. Before undergoing
surgery, all patients provided written informed consent for their
tissues to be used in the present study. The Ethics Committee of
the Second Affiliated Hospital of Xi'an Jiaotong University
approved this study. All tissues were snap frozen in liquid
nitrogen before sample procession.
Cell culture
The HSCC cell lines FaDu (derived from a primary
lesion of hypopharyngeal SCC), SAS (derived from a primary lesion
of tongue SCC), and HSC3 (derived from lymph node metastasis of
tongue SCC) were obtained from American Type Culture Collection
(ATCC). The normal epithelial cell line Nthy-ori3-1 was maintained
at our laboratory. All cell lines were cultured in DMEM with 10%
fetal bovine serum (FBS; both from Gibco; Thermo Fisher Scientific,
Inc.) and 1% penicillin-streptomycin in a humidified atmosphere
containing 5% CO2 at 37°C. Mycoplasma contamination was
not detected in any cell line.
Transfection
Briefly, the cells were seeded onto 6- or 96-well
plates. When cell confluency reached ~60–70%, the medium was
changed to DMEM without 10% FBS, and the transfection was conducted
using Lipofectamine™ 3000 (Invitrogen; Thermo Fisher Scientific,
Inc.). The different transfection groups were as follows: i) empty
vector group and pcDNA3.1-RP11-156L14.1 group; ii) negative control
(NC) group and si-RP11-156L14.1–1/2/3 groups; iii) NC+miR-Ctrl
group, pcDNA3.1-RP11-156L14.1+ miR-Ctrl group, NC+miR-548ao-3p
mimic group, and pcDNA3.1-RP11-156L14.1+miR-548ao-3p mimic group;
iv) si-NC+miR-Ctrl group, si-RP11-156L14.1+miR-Ctrl group,
si-NC+miR-548ao-3p group, and si-RP11-156L14.1+miR-548ao-3p mimic
group; v) si-NC+miR-548ao-3p inhibitor NC group,
si-RP11-156L14.1+miR-548ao-3p inhibitor NC group, and
si-RP11-156L14.1+miR-548ao-3p inhibitor group. The cells were
harvested at the indicated time-point (48 h) for further
experimentation. The FaDu cells were transfected with
RP11-156L14.1-knockdown vector or control vector, with or without
miR-548ao-3p inhibitors.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was purified from patient tissues or
cultured cells using TRIzol (Invitrogen; Thermo Fisher Scientific,
Inc.). cDNA was obtained through reverse transcription using a
PrimeScript RT reagent Kit (Takara Bio Inc.). Real-time
quantitative PCR was performed using a SYBR PrimeScript™ PLUS Kit
(Takara Bio, Inc.) to detect the expression levels of
lncRNA-RP11-156L14.1, miR-548ao-3p, and SSR1. GAPDH or U6 was used
as the endogenous control to normalize the expression level through
2−ΔΔCq method (23).
Thermal conditions of PCR reactions were: 95°C for 1 min, and then
12 sec at 95°C and 32 sec at 57°C for 40 cycles. The primers used
were as follows: RP11-156L14.1 forward,
5′-AAAACCACCAGCCCTTTCTCATCC-3′ (forward) and reverse,
5′-TAGCCTTCCTCCTGATCTGCCAAG-3′; miR-548ao-3p,
5′-CGCAAAGACCGTGACTACTTTTGCA-3′ (universal miRNA reverse primer:
cat. no. B532451-0020; Sangon Biotech Co., Ltd.); SSR1 forward,
5′-GGAGCCGCCGAGAGCCTTAG-3′ and reverse,
5′-ACGAGTAAGAGAAGCAGCAGCAAG-3′; GAPDH forward,
5′-GGGAGCCAAAAGGGTCAT-3′ and reverse, 5′-GAGTCCTTCCACGATACCAA-3′.
All assays were performed independently in triplicate.
Cell proliferation assays
Cells were seeded into 96-well plates with a density
of 2×104 cells/well. After transfection, the cells were
cultured for the indicated time-points (0, 24, 48 and 72 h), and
then cell proliferation ability was evaluated by a CCK-8 assay
(Dojindo Molecular Technologies, Inc.) following the manufacturer's
protocol. At each time-point, 10 µl of CCK-8 reagent was added into
each well. Cells were incubated at 37°C subsequently for 2 h.
Against a background control, the sample absorbance was tested at
450 nm via a microplate reader (Bio-Rad Laboratories, Inc.). EdU
staining was performed to evaluate DNA synthesis in proliferating
cells with an EdU assay Kit (Invitrogen; Thermo Fisher Scientific,
Inc.) following the manufacturer's instructions. After
transfection, cells were cultured for 48 h, fixed with 4%
paraformaldehyde and permeabilized by 0.3% Triton X-100. Then cells
were incubated with 10 µM EdU for 2 h, and cell nuclei were stained
with DAPI (5 µg/ml). The number of EdU-positive cells was counted
under a light microscope in five random fields (Olympus
Corporation; magnification, ×10). For colony formation assay, cells
were seeded into 6-well plates at a density of 500 cells per well
and cultured for 10 days. Then the cell colonies were fixed with
methanol for 10 min at room temperature, stained with 0.1% crystal
violet at room temperature for 15 min, and then counted under the
inverted light microscope (Olympus Corporation; magnification,
×10). All assays were performed independently in triplicate.
Cell cycle analysis
Cells (2×103) were fixed with 70% ethanol
at 4°C overnight. The fixed cells were treated with ribonuclease A
(20 µg/ml; Sigma-Aldrich; Merck KGaA) and incubated with 50 µg/ml
propidium iodide (Sigma-Aldrich; Merck KGaA) for 30 min at 37°C.
Then, the populations in G2-M, S, and G0-G1 phases were determined
by flow cytometric analysis (BD Biosciences). All assays were
performed independently in triplicate.
Wound healing assay
After transfection, Ntrhy-ori3-1 or FaDu cells
(5×105) were seeded into 6-well plates and cultured
until 90% confluency. Wounds were stimulated by scratching with a
200-µl sterile pipette tip. The floating cells were washed away
with PBS. Cell migration was monitored by examining the closure of
the wound at 0 and 48 h under an inverted optical microscope
(Olympus Corporation; magnification, ×10). All assays were
performed independently in triplicate.
Transwell assay
Cell invasion was analyzed using a 24-well Transwell
chamber (Costar, Inc.) with Matrigel-coated membranes. Ntrhy-ori3-1
or FaDu cells were seeded into the upper chamber in FBS-free DMEM,
and 500 µl DMEM medium with 10% FBS was added to the bottom
chamber. After 48 h, the invading cells in the bottom chamber were
fixed with formaldehyde (3.7% in PBS) for 5 min at room temperature
and stained with 0.1% crystal violet at room temperature for 15 min
and then counted under the inverted light microscope (Olympus
Corporation; magnification, ×10). All assays were performed
independently in triplicate.
Western blotting
Cells were lysed using a Protein Extraction kit
according to the manufacturer's protocols (Beyotime Institute of
Biotechnology). RIPA buffer (Beijing Solarbio Science and
Technology Co., Ltd.) was used to extract total protein from the
cultured cells. A BCA protein assay kit (Thermo Fisher Scientific,
Inc.) was used to quantify the proteins. Equal amounts of protein
samples (30 µg) were separated with 10% SDS-PAGE and
electro-transferred onto PVDF membranes (EMD Millipore). The
membranes were blocked with 5% non-fat milk/PBS for 1 h at room
temperature and then incubated by primary antibodies:
Anti-E-cadherin (1:2,000; product code ab1416; Abcam),
anti-N-cadherin (1:1,000; product code ab76057; Abcam),
anti-vimentin (1:2,000; product code ab8978; Abcam), anti-SSR1
(1:1,000; product no. 12009; Cell Signaling Technology, Inc.),
anti-Ki67 (1:1,000; product code ab92742; Abcam), and anti-β-actin
(1:5,000; product code ab179467; Abcam) at 4°C overnight. After the
membranes were washed with PBST three times, they were further
incubated with secondary antibodies: Anti-mouse IgG, HRP-linked and
anti-rabbit IgG, HRP-linked (both 1:3,000; cat. nos. 7076 and 7074,
repectively; Cell Signaling Technology, Inc.) for 2 h at room
temperature. The membranes were developed using the ECL system (EMD
Millipore). All assays were performed independently in triplicate
and the desitometric analysis was performed using ImageJ (1.49p;
National Institutes of Health).
Luciferase reporter assay
A luciferase reporter vector containing the 3′-UTR
of RP11-156L14.1 with the predicted miR-548ao-3p binding site was
constructed (pGL3-RP11-156L14.1-wild-type and
pGL3-RP11-156L14.1-Mut). Similarly, luciferase reporter vectors
containing the 3′-UTR of SSR1-wild-type and SSR1-mutated were
constructed. All reporter vectors used the basic pGL3-Luc vector
(Promega) as a backbone vector. Cells were co-transfected with
luciferase reporter plasmids and control Renilla luciferase
vector (Promega, USA) using Lipofectamine™ 3000 (Invitrogen; Thermo
Fisher Scientific, Inc.), and the relative luciferase activities
were analyzed 48 h later using a dual-luciferase reporter assay kit
(Promega Corporation). All assays were performed independently in
triplicate.
Cytoplasmic and nuclear fractions
To determine the subcellular localization of
RP11-156L14.1, cytoplasmic and nuclear fractions of FaDu or SAS
cells were prepared using a PARIS Kit (Life Technologies; Thermo
Fisher Scientific, Inc.) following the manufacturer's protocol. The
fractions were subjected to subsequent RT-qPCR analysis. GAPDH and
U6 were used as internal controls for the cytoplasm and nucleus,
respectively. All assays were performed independently in
triplicate.
RNA immunoprecipitation (RIP)
assay
An RNA immunoprecipitation assay was performed by
Imprint RNA immunoprecipitation kit (Sigma-Aldrich; Merck KGaA)
referring to the recommended protocols of the manufacturer.
Firstly, IgG-induced chondrocytes were collected and resuspended in
RIP lysis bufer (Beijing Solarbio Science and Technology Co.,
Ltd.), subsequently centrifuged at 12,000 × g for 5 min. Ten, cell
lysates were incubated with anti-Argonaute2 (anti-Ago2) or anti-IgG
(negative control) overnight at 4°C, followed by the addition of
Protein A magnetic beads (cat. no. 73778; Cell Signaling
Technology) to obtain the immunoprecipitation complex. Total RNA
was isolated using GenElute™ Total RNA Purifcation Kit
(Sigma-Aldrich; Merck KGaA). Lastly, the relative enrichment of
RP11-156L14.1 and miR-548ao-3p were determined by RT-qPCR analysis.
All assays were performed independently in triplicate.
Tumor xenograft model
The posterior flanks of the BALB/c nude mice (male,
6 weeks old, n=15) were subcutaneously injected with FaDu cells
(2×107) transfected with sh-RP11-156L14.1 or the
negative control. The animals were monitored daily and the
following criteria for humane endpoint was used: Severe tumor
burden (more than 20 mm in diameter), difficulty breathing,
significant body-weight loss, and clinical signs such as
prostration, hypothermia, and significant abdominal distension.
Tumor volumes were examined every four days, the maximum tumor
diameter observed in this study was 1.4 cm. On day 13 after
inoculation, the mice were euthanized by CO2 inhalation
(CO2 flow rate: 10% of cage volume) and the death of
animals were confirmed by cessation of heartbeat. Tumor xenografts
were harvested, photographed, and weighed. The maximum weight loss
of all the mice used in this study was 6.3% of initial body weight.
The maximum tumor weight/body weight ratio observed was 8.6%. The
animal experiments were conducted under the protocol approval of
the Ethics Committee of Animal Welfare of the Second Affiliated
Hospital of Xi'an Jiaotong University.
Target prediction
Potential target miRNAs of RP11-156L14.1 were
predicted by the Lncbase v.2 (http://carolina.imis.athenainnovation.gr/diana_tools/web/index.php?r=lncbasev2/index).
The target genes of miR-548ao-3p were predicted using three
bioinformatics algorithms: TargetScan human 7.2 (http://www.targetscan.org/vert_72/), miRDB
(http://www.mirdb.org) and microT-CDS v5.0
(http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=microT_CDS/index).
Statistical analysis
Statistical analyses were calculated using GraphPad
Prism 7.0 software (GraphPad Software, Inc.), and the results are
presented as the mean ± SD. Statistical comparisons among multiple
groups were analyzed by one-way analysis of variance (ANOVA)
followed by Tukey's post hoc tests, and P<0.05 was considered to
indicate a statistically significant difference.
Results
RP11-156L14.1 is highly expressed in
HSCC tissues and cell lines
To investigate the expression pattern of lncRNA
RP11-156L14.1 in HSCC, RP11-156L14.1 expression was assessed in 30
paired HSCC tumor tissues and adjacent normal tissues. The
RP11-156L14.1 mRNA level was significantly higher in tumor tissues
compared with that in the adjacent normal tissues (Fig. 1A). In addition, the expression
levels of RP11-156L14.1 in the HSCC cell lines FaDu and SAS were
also higher in comparison with those in normal tissues (Fig. 1B). High expression of RP11-156L14.1
was significantly associated with advanced TNM stages and lymph
node metastasis in HSCC (Table
I).
 | Table I.Association of clinicopathological
features with lncRNA RP11-156L14.1 expression. |
Table I.
Association of clinicopathological
features with lncRNA RP11-156L14.1 expression.
|
| RP11-156L14.1
expression |
|
|---|
|
|
|
|
|---|
|
Characteristics | Low (n=15) | High (n=15) | P-value |
|---|
| Age (years) |
|
| 0.4642 |
|
≤60 | 7 | 9 |
|
|
>60 | 8 | 6 |
|
| Sex |
|
| 0.2733 |
|
Male | 9 | 6 |
|
|
Female | 6 | 9 |
|
| TNM stage |
|
| 0.0034 |
|
I+II | 11 | 3 |
|
|
III+IV | 4 | 12 |
|
| Lymph node
metastasis |
|
| 0.0281 |
|
Negative | 10 | 4 |
|
|
Positive | 5 | 11 |
|
Knockdown of RP11-156L14.1 inhibits
cell proliferation and the cell cycle in HSCC cell lines
To explore the function of RP11-156L14.1, RNA
interference studies involving RP11-156L14.1 knockdown in HSCC cell
lines were employed. FaDu cells were transfected with si-Control or
si-RP11-156L14.1–1/2/3, and the knockdown efficiency was evaluated
by qPCR (Fig. 2A). The most
efficient si-RP11-156L14.1–2 was used in the subsequent
experiments. The CCK-8 assay demonstrated that cell proliferation
was significantly suppressed in SAS and FaDu cells transfected with
si-RP11-156L14.1 compared with that in control-transfected cells
(Fig. 2B and C). Furthermore, EDU
incorporation assays and colony formation assays found that DNA
synthesis and colony formation capabilities, respectively, were
decreased after RP11-156L14.1 knockdown in SAS and FaDu cells
(Fig. 2D and E). Cell cycle
analysis was performed on both cell lines using propidium iodide
staining. As revealed in Fig. 2F,
the fraction of cells in the S phase was significantly reduced in
SAS and FaDu cells transfected with si-RP11-156L14.1 compared with
that in cells transfected with the control vector.
Knockdown of RP11-156L14.1 inhibits
the migration, invasion, and EMT in HSCC cells
The function of RP11-156L14.1 on HSCC metastasis was
further evaluated. As demonstrated by the wound healing assay,
knockdown of RP11-156L14.1 decreased the wound healing capabilities
of SAS and FaDu cells (Fig. 3A and
B). It was also determined that knockdown of RP11-156L14.1 in
SAS or FaDu cells inhibited the cell metastasis phenotype since the
migration and invasion abilities were decreased (Fig. 3C and D). Since tumor metastasis has
been revealed to be closely associated to cell-cell adhesion and
EMT (24), the levels of
EMT-related proteins after RP11-156L14.1 knockdown were examined.
Notably, the results revealed that knockdown of RP11-156L14.1
enhanced E-cadherin expression and inhibited N-cadherin and
vimentin expression in SAS and FaDu cells (Fig. 3E). Decreased E-cadherin expression
and enhanced vimentin expression may be associated with the
increased migration and invasion of HSCC cells. Knockdown of
RP11-156L14.1 was consistently revealed to inhibit the protein
expression of the proliferation marker Ki67 in SAS and FaDu cells
(Fig. 3F).
 | Figure 3.Knockdown of lncRNA RP11-156L14.1
inhibits the migration, invasion, and EMT in HSCC cells. SAS or
FaDu cells were transfected with si-Control or si-RP11-156L14.1. (A
and B) Cell migration capability was analyzed by wound healing
assay. (C and D) A Transwell assay was performed to assess cell
migration and invasion in SAS and FaDu cells. (E) EMT-related
proteins E-cadherin, N-cadherin, and vimentin and (F)
proliferation-marker Ki67 were determined by western blotting in
SAS and FaDu cells. β-actin was used as an internal control. The
experiment was repeated three times, and the representative blot
images are presented. *P<0.05, **P<0.01, ***P<0.001.
lncRNA, long non-coding RNA; EMT, epithelial-mesenchymal
transition; HSCC, hypopharyngeal squamous cell carcinoma. |
RP11-156L14.1 directly binds to
miR-548ao-3p in HSCC cell lines
To investigate the potential mechanism of
RP11-156L14.1 in HSCC, subcellular fractionation was performed, and
lncRNA RP11-156L14.1 was mainly located in the cytoplasm,
indicating that RP11-156L14.1 may be involved in the
post-transcriptional regulation of target genes (Fig. 4A). Bioinformatics analyses were
performed using the online tools LncbaseV2 and RegRNA. Multiple
miRNAs were predicted to interact with RP11-156L14.1 (miR-580-3p,
miR-875-3p, miR-548ao-3p, miR-664b-3p, miR-660-3p, and miR-373-5p).
A luciferase reporter assay was performed to verify the interaction
between miRNAs and RP11-156L14.1, whereas miR-548ao-3p mimics
significantly inhibited the luciferase activity in FaDu cells
transfected with a reporter vector containing the 3′-UTR of
RP11-156L14.1 (Fig. 4B).
RP11-156L14.1 exhibited putative binding sequences with
miR-548ao-3p (Fig. 4C). The
luciferase reporter assay demonstrated that miR-548ao-3p mimics
could inhibit the relative luciferase activity in 293 cells with
reporter vectors containing wild-type RP11-156L14.1 sequences but
not in those with mutated RP11-156L14.1 sequences (Fig. 4C). Ago2 immunoprecipitation
experiments revealed that RP11-156L14.1 was identified as a bona
fide miR-548ao-3p target in FaDu cells (Fig. 4D). Moreover, the present study also
demonstrated that knockdown of RP11-156L14.1 enhanced miR-548ao-3p
expression in FaDu cells (Fig. 4E).
Consistently, the expression level of miR-548ao-3p was markedly
lower in HSCC tumor tissues and HSCC cell lines compared with that
in adjacent normal tissues (Fig. 4F and
G).
 | Figure 4.lncRNA RP11-156L14.1 directly binds
to miR-548ao-3p in HSCC cell lines. (A) A subcellular fractionation
assay was performed in FaDu or SAS cells. The relative expression
levels of RP11-156L14.1, GAPDH, and U6 in the cytoplasm or nucleus
were analyzed by qPCR. (B) FaDu cells were transfected with a
luciferase reporter containing the 3′-UTR of RP11-156L14.1, in
combination with different miRNA mimics or a negative control. The
relative luciferase activity was analyzed 48 h later. (C)
Bioinformatics analysis predicted putative binding sequences
between wt RP11-156L14.1 and miR-548ao-3p. The mutated
RP-11-154L14.1 sequences are listed. 293 cells were transfected
with miR-Con or miR-548ao-3p mimics, in combination with a reporter
vector containing RP11-156L14.1-wt or RP11-156L14.1-mut sequences.
Relative luciferase activity was measured 48 h later. (D) Ago2
immunoprecipitation experiments were performed in FaDu cells, and
IgG was used as a control. The relative expression of RP11-156L14.1
or miR-548ao-3p was determined by qPCR. (E) FaDu cells were
transfected with RP11-156L14.1 knockdown vector, along with si-NC
control vectors. The relative expression of RP11-156L14.1 or
miR-548ao-3p was determined by qPCR. (F) The relative expression of
miR-548ao-3p in HSCC tumor tissues and adjacent non-tumor tissues
was determined by qPCR. (G) The relative expression of miR-548ao-3p
in HSCC cell lines and normal tissues was determined by qPCR.
*P<0.05, **P<0.01, ***P<0.001. lncRNA, long non-coding
RNA; HSCC, hypopharyngeal squamous cell carcinoma; wt, wild-type;
qPCR, quantitative PCR. |
RP11-156L14.1 promotes cell
proliferation and the cell cycle via miR-548ao-3p in HSCC
cells
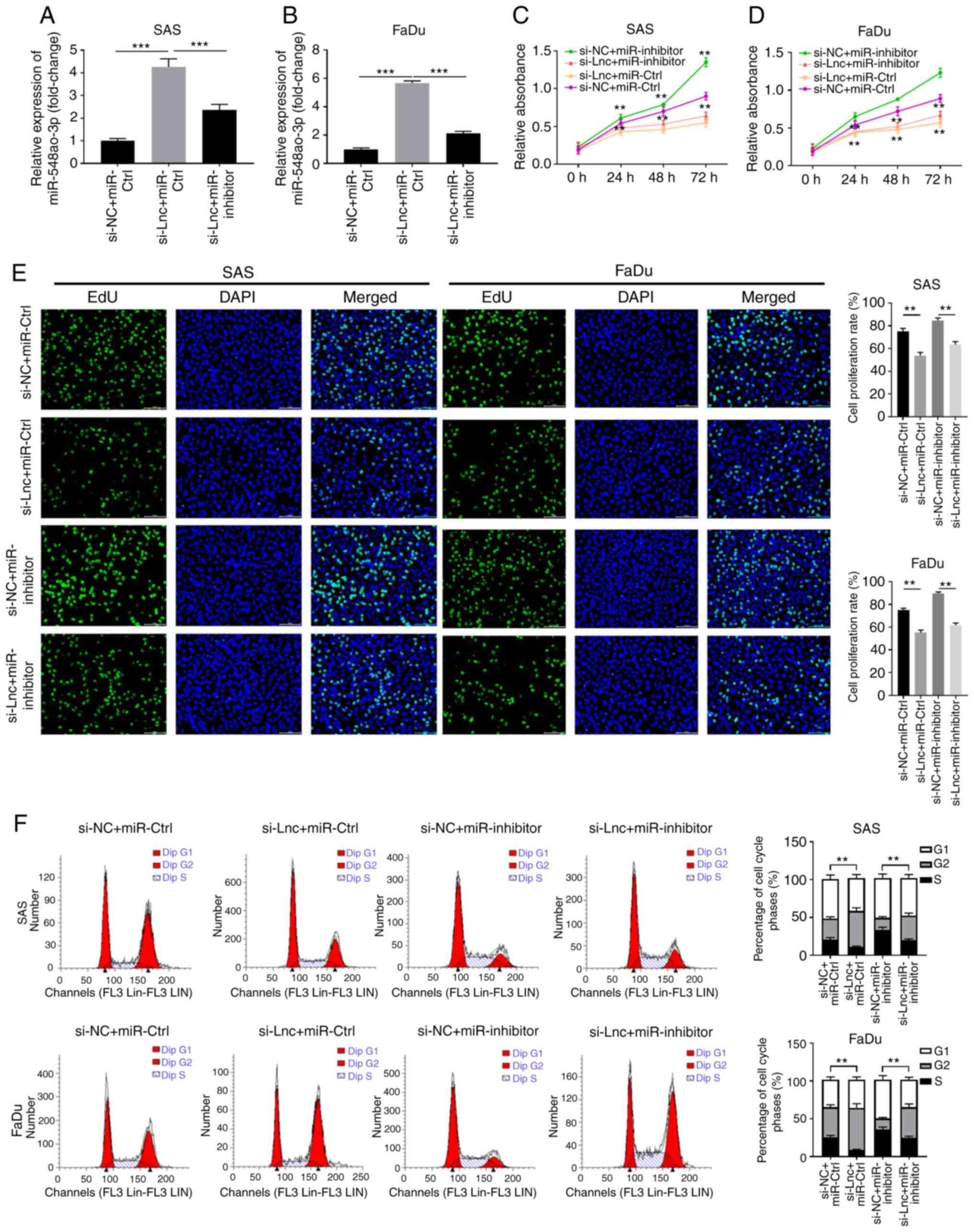
Next, the functional relationship between
RP11-156L14.1 and miR-548ao-3p was further evaluated. While
knockdown of RP11-156L14.1 increased miR-548ao-3p expression,
miR-548ao-3p-inhibitor transfection downregulated the expression
levels of miR-548ao-3p in SAS and FaDu cells (Fig. 5A and B). In SAS and FaDu cells,
knockdown of RP11-156L14.1 inhibited cell proliferation and DNA
synthesis and reduced the number of cells in the S phase, while
transfection of the miR-548ao-3p-inhibitor enhanced cell growth.
However, SAS and FaDu cells transfected with miR-548ao-3p inhibitor
combined with si-RP11-156L14.1 exhibited similar levels of cell
proliferation, DNA synthesis and S phase percentage compared with
the si-NC+miR-Ctrl group (Fig.
5C-F). The combined evidence indicated that lncRNA
RP11-156L14.1 could promote cell proliferation and the cell cycle
by competing with miR-548ao-3p in HSCC cells.
RP11-156L14.1 promotes migration,
invasion, and EMT via miR-548ao-3p in HSCC cells
Further experiments were conducted to assess whether
the function of RP11-156L14.1 in HSCC metastasis also depends on
miR-548ao-3p. In SAS and FaDu cells, knockdown of RP11-156L14.1
inhibited cell migration and invasion, with reduced
N-cadherin/vimentin expression and enhanced E-cadherin expression
(Fig. 6A-E). However, inhibition of
miR-548ao-3p exhibited the opposite biological effects on cell
migration/invasion in SAS and FaDu cells and partially abolished
the effects of RP11-156L14.1 knockdown (Fig. 6A-E).
 | Figure 6.RP11-156L14.1 promotes migration,
invasion, and EMT via miR-548-3p in HSCC cells. SAS or FaDu cells
were transfected with si-NC+miR-Ctrl, si-RP11-156L14.1+miR-Ctrl,
si-NC+miR-548ao-3p inhibitor, or si-RP11-156L14.1+miR-548ao-3p
inhibitor. (A and B) Wound-healing assays were conducted to
evaluate the cell migration of SAS and FaDu cells. (C and D)
Transwell assays were performed to assess cell migration and
invasion in SAS and FaDu cells. (E) EMT-related proteins
E-cadherin, N-cadherin, and vimentin were determined by western
blotting in SAS and FaDu cells. β-actin was used as an internal
control. The experiment was repeated three times, and the
representative blot images are presented. *P<0.05, **P<0.01,
***P<0.001. EMT, epithelial-mesenchymal transition; HSCC,
hypopharyngeal squamous cell carcinoma. |
RP11-156L14.1 functions as a ceRNA in
regulating SSR1 expression by binding to miR-548ao-3p
To investigate the potential target of miR-548ao-ep
in HSCC, the bioinformatics analysis online tool TargetScan was
used, which revealed that miR-548ao-3p targeted the 3′-UTR of SSR1
(Fig. 7A and B). A luciferase
reporter assay was performed, and miR-548ao-3p mimics were found to
inhibit the relative luciferase activity in 293 cells with reporter
vectors transfected with the WT 3′-UTR of SSR1 but not the mutated
3′-UTR of SSR1 (Fig. 7C). To
further confirm the relationship between RP11-156L14.1 and
miR-548ao-3p in regulating SSR1, RP11-156L14.1 combined with
miR-548ao-3p mimics was overexpressed in SAS or FaDu cells. The
results revealed that overexpression of RP11-156L14.1 significantly
upregulated SSR1 expression, while miR-548ao-3p decreased the
expression of SSR1 (Fig. 7D).
Moreover, it was determined that the expression of SSR1 was
significantly higher in HSCC tumor tissues and cell lines, compared
with that in adjacent normal tissues (Fig. 7E-F).
 | Figure 7.RP11-156L14.1 functions as a ceRNA in
regulating SSR1 expression by binding to miR-548ao-3p. (A and B)
Bioinformatics analyses predicted putative binding sequences
between WT SSR1 3′-UTR and miR-548ao-3p. The mutated SSR1 3′-UTR
sequences are listed. (C) 293 cells were transfected with control
mimics or miR-548ao-3p mimics, in combination with reporter vectors
containing Wt 3′-UTR SSR1 or mutated 3′-UTR of SSR1 sequences.
Relative luciferase activity was measured 48 h later. (D) SAS or
FaDu cells were transfected with pcDNA3.1 control,
pcDNA3.1-RP11-156L14.1, or pcDNA3.1-RP11-156L14.1+miR-548ao-3p
mimics, and the protein levels of SSR1 were analyzed. β-actin was
used as an internal control. The experiment was repeated three
times, and the representative blot images are presented. (E)
Relative expression of SSR1 in HSCC tumor tissues and adjacent
non-tumor tissues was determined by qPCR. (F) Relative expression
of SSR1 in HSCC cell lines and normal control cell was determined
by qPCR. *P<0.05, **P<0.01, ***P<0.001. SSR1, signal
sequence receptor subunit 1; WT, wild-type; HSCC, hypopharyngeal
squamous cell carcinoma; qPCR, quantitative PCR. |
RP11-156L14.1 knockdown inhibits tumor
development and metastasis in HSCC in vivo
FaDu cells were stably transfected with the control
vector or RP11-156L14.1 knockdown vector and subcutaneously
implanted into the back flanks of nude mice to set up the xenograft
tumor model. Knockdown of RP11-156L14.1 markedly inhibited tumor
development, and the volumes and weights of xenograft tumors were
significantly lower in the RP11-156L14.1-knockdown group compared
with the sh-NC group (Fig. 8A-D).
Additionally, miR-548ao-3p and SSR1 mRNA expression were analyzed
in tumor tissues. As revealed in Fig.
8E and F, tumor tissues from the sh-RP11-156L14.1 group
exhibited significantly higher expression levels of miR-548ao-3p,
and significantly lower expression levels of SSR1. SSR1 protein
levels were downregulated in the tumor tissues of the
sh-RP11-156L14.1 group (Fig. 8G and
H). Overall, these data indicated that lncRNA RP11-156L14.1
knockdown suppressed HSCC growth in the xenograft model.
Discussion
Mounting studies have revealed that lncRNAs exert
critical functions in HSCC progression and metastasis (6,7).
Notably, the present study delineated the biological function of
RP11-156L14.1 in cell proliferation, migration, and invasion.
Additionally, its interaction network with miR-548ao-3p and SSR1
was identified, indicating that RP11-156L14.1 could be utilized as
a potential biomarker in HSCC.
RP11-156L14.1 is a novel lncRNA that was identified
by genome scale analysis in squamous cell lung cancer (25). Another study revealed that
RP11-156L14.1 was highly expressed in childhood T-cell acute
lymphoblastic leukemia, potentially targeting the genes SMURF2,
MAP3K3, and miRNA-633 (26).
However, the function of RP11-156L14.1 in tumors, especially in
HSCC, was not studied. In the present study, for the first time, to
the best of our knowledge, it was demonstrated that RP11-156L14.1
was up-regulated in HSCC tumor tissues compared with adjacent
non-tumor tissues. The enhanced expression of RP11-156L14.1 was
also confirmed in HSCC cell lines. Due to the limited resources, we
only examined the expression of RP11-156L14.1 in FaDu and SAS cells
in the present study. RNA silencing assays were performed to assess
the functions of RP11-156L14.1 in SAS and FaDu cells. Functional
assays demonstrated that RP-11-156L14.1 acted as an oncogene and
knockdown of RP-11-156L14.1 inhibited HSCC cell proliferation,
migration, and invasion in vitro. The xenograft model
revealed that knockdown of RP11-156L14.1 suppressed HSCC tumor
development in vivo.
In a previous study, miRNA-633 was predicted to be
the potential target of RP11-156L14.1 (26). In the present study, it was
determined that RP11-156L14.1 directly interacted with
miR-548ao-3p. miR-548ao-3p belongs to the large, poorly conserved
primate-specific mir-548 family (27). miR-548-3p has been reported as a
tumor suppressor in breast cancer, while miR-548q was revealed to
be an oncogene and potential prognostic biomarker in gastric cancer
(28). The present study revealed
that miR-548ao-3p functioned as a tumor suppressor and the
inhibition of miR-548ao-3p enhanced HSCC cell proliferation,
migration, and invasion. Moreover, the data revealed that
miR-548ao-3p mimics could antagonize the effects of RP11-156L14.1
overexpression. In contrast to the expression of RP11-156L14.1 in
HSCC tumor tissues, miR-548ao-3p expression was significantly lower
in HSCC tumor tissues compared with that in adjacent normal
tissues.
To elucidate the target genes regulated by
miR-548ao-3p, SSR1 was identified as a direct target gene
controlled by miR-548ao-3p. SSR1 is a glycosylated membrane
receptor related to protein translocation (29). A previous study has demonstrated
that SSR1 may be used as a novel prognostic marker in renal and
liver cancer (30,31). The present study consistently
determined that SSR1 was highly expressed in HSCC and miR-548ao-3p
regulated SSR1 expression by binding to its 3′-UTR. However, the
questions of whether SSR1 is also controlled by other miRNAs and
how SSR1 regulates cell proliferation, migration, and invasion of
HSCC cells require further investigation. Another limitation of the
present study is that the signaling pathways involved in the
regulation of the lncRNA RP11-156L14.1/miR-548ao-ep/SSR1 axis have
not been defined yet.
In summary, the results of the present study
demonstrated that lncRNA RP11-156L14.1 was enhanced in HSCC tissues
and cell lines. Moreover, this study revealed that RP11-156L14.1
promoted HSCC development and metastasis by sponging miR-548ao-3p
to regulate the expression of SSR1. The xenograft model revealed
that knockdown of RP11-156L14.1 inhibited HSCC tumor growth in
vivo. Overall, the lncRNA RP11-156L14.1/miR-548ao-3p/SSR1 axis
provides novel insights into HSCC tumorigenesis and suggests a
potential new biomarker for HSCC patients.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Shaanxi
Province Key R&D Program General Project-Social Development
Field (grant no. 2020SF-024), the Innovation Capability Support
Program of Shaanxi (grant no. 2017KJXX-45), the Natural Science
Foundation Group-style Aided Tibet Medical Project of Tibet
Autonomous Region [grant no. XZ2019ZR-ZY72(Z)] and the Natural
Science Basic Research Program of Shaanxi (grant no.
2018JM7023).
Availability of data and material
The datasets used and/or analyzed in the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
JH and GXZ conceived and designed the experiments.
YY, HNL and XYR performed the experiments. NL, JY and ZHW analyzed
the data. JY and ZHW wrote the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The investigation conformed to the principles
outlined in the Declaration of Helsinki, and written informed
consent was obtained from all participants. The Ethics Committee of
the Second Affiliated Hospital of Xi'an Jiaotong University
approved this study. The animal experiments were conducted under
the protocol approval of the Ethics Committee of Animal Welfare of
the Second Affiliated Hospital of Xi'an Jiaotong University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Katsoulakis E, Riaz N, Hu M, Morris L,
Sherman E, McBride S and Lee N: Hypopharyngeal squamous cell
carcinoma: Three-dimensional or intensity-modulated radiotherapy? A
single institution's experience. Laryngoscope. 126:620–626. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fukumoto I, Kinoshita T, Hanazawa T,
Kikkawa N, Chiyomaru T, Enokida H, Yamamoto N, Goto Y, Nishikawa R,
Nakagawa M, et al: Identification of tumour suppressive
microRNA-451a in hypopharyngeal squamous cell carcinoma based on
microRNA expression signature. Br J Cancer. 111:386–394. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Takes RP, Strojan P, Silver CE, Bradley
PJ, Haigentz M Jr, Wolf GT, Shaha AR, Hartl DM, Olofsson J,
Langendijk JA, et al: Current trends in initial management of
hypopharyngeal cancer: The declining use of open surgery. Head
Neck. 34:270–281. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chan JY and Wei WI: Current management
strategy of hypopharyngeal carcinoma. Auris Nasus Larynx. 40:2–6.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang G, Lu X and Yuan L: LncRNA: A link
between RNA and cancer. Biochim Biophys Acta. 1839:1097–1109. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhou J, Li W, Jin T, Xiang X, Li M, Wang
J, Li G, Pan X and Lei D: Gene microarray analysis of lncRNA and
mRNA expression profiles in patients with hypopharyngeal squamous
cell carcinoma. Int J Clin Exp Med. 8:4862–4882. 2015.PubMed/NCBI
|
|
7
|
Zhou J, Cao S, Li W, Wei D, Wang Z, Li G,
Pan X and Lei D: Time-course differential lncRNA and mRNA
expressions in radioresistant hypopharyngeal cancer cells.
Oncotarget. 8:40994–41010. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Qian Y, Liu D, Cao S, Tao Y, Wei D, Li W,
Li G, Pan X and Lei D: Upregulation of the long noncoding RNA UCA1
affects the proliferation, invasion, and survival of hypopharyngeal
carcinoma. Mol Cancer. 16:682017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhou J, Li M, Yu W, Li W, Wang J, Xiang X,
Li G, Pan X and Lei D: AB209630, a long non-coding RNA decreased
expression in hypopharyngeal squamous cell carcinoma, influences
proliferation, invasion, metastasis, and survival. Oncotarget.
7:14628–14638. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao M, Sun D, Li X, Xu Y, Zhang H, Qin Y
and Xia M: Overexpression of long noncoding RNA PEG10 promotes
proliferation, invasion and metastasis of hypopharyngeal squamous
cell carcinoma. Oncol Lett. 14:2919–2925. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang CZ: Long intergenic non-coding RNA
668 regulates VEGFA signaling through inhibition of miR-297 in oral
squamous cell carcinoma. Biochem Biophys Res Commun. 489:404–412.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee SH, Koo BS, Kim JM, Huang S, Rho YS,
Bae WJ, Kang HJ, Kim YS, Moon JH and Lim YC: Wnt/β-catenin
signalling maintains self-renewal and tumourigenicity of head and
neck squamous cell carcinoma stem-like cells by activating Oct4. J
Pathol. 234:99–107. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Huang W, Cui X, Chen J, Feng Y, Song E, Li
J and Liu Y: Long non-coding RNA NKILA inhibits migration and
invasion of tongue squamous cell carcinoma cells via suppressing
epithelial-mesenchymal transition. Oncotarget. 7:62520–62532. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
van Kester MS, Tensen CP, Vermeer MH,
Dijkman R, Mulder AA, Szuhai K, Willemze R and van Doorn R:
Cutaneous anaplastic large cell lymphoma and peripheral T-cell
lymphoma NOS show distinct chromosomal alterations and differential
expression of chemokine receptors and apoptosis regulators. J
Invest Dermatol. 130:563–575. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ali MM, Akhade VS, Kosalai ST, Subhash S,
Statello L, Meryet-Figuiere M, Abrahamsson J, Mondal T and Kanduri
C: PAN-cancer analysis of S-phase enriched lncRNAs identifies
oncogenic drivers and biomarkers. Nat Commun. 9:8832018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nair VS, Maeda LS and Ioannidis JP:
Clinical outcome prediction by microRNAs in human cancer: A
systematic review. J Natl Cancer Inst. 104:528–540. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kikkawa N, Hanazawa T, Fujimura L, Nohata
N, Suzuki H, Chazono H, Sakurai D, Horiguchi S, Okamoto Y and Seki
N: miR-489 is a tumour-suppressive miRNA target PTPN11 in
hypopharyngeal squamous cell carcinoma (HSCC). Br J Cancer.
103:877–884. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kikkawa N, Kinoshita T, Nohata N, Hanazawa
T, Yamamoto N, Fukumoto I, Chiyomaru T, Enokida H, Nakagawa M,
Okamoto Y and Seki N: microRNA-504 inhibits cancer cell
proliferation via targeting CDK6 in hypopharyngeal squamous cell
carcinoma. Int J Oncol. 44:2085–2092. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang C, Wang C, Jia Z, Tong W, Liu D, He
C, Huang X and Xu W: Differentially expressed mRNAs, lncRNAs, and
miRNAs with associated co-expression and ceRNA networks in
ankylosing spondylitis. Oncotarget. 8:113543–113557. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guan GF, Zhang DJ, Wen LJ, Xin D, Liu Y,
Yu DJ, Su K, Zhu L, Guo YY and Wang K: Overexpression of lncRNA
H19/miR-675 promotes tumorigenesis in head and neck squamous cell
carcinoma. Int J Med Sci. 13:914–922. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhuang K, Wu Q, Jiang S, Yuan H, Huang S
and Li H: CCAT1 promotes laryngeal squamous cell carcinoma cell
proliferation and invasion. Am J Transl Res. 8:4338–4345.
2016.PubMed/NCBI
|
|
22
|
Kolenda T, Guglas K, Kopczyńska M,
Teresiak A, Bliźniak R, Mackiewicz A, Lamperska K and Mackiewicz J:
Oncogenic role of ZFAS1 lncRNA in head and neck squamous cell
carcinomas. Cells. 8:3662019. View Article : Google Scholar
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nijkamp MM, Span PN, Hoogsteen IJ, van der
Kogel AJ, Kaanders JH and Bussink J: Expression of E-cadherin and
vimentin correlates with metastasis formation in head and neck
squamous cell carcinoma patients. Radiother Oncol. 99:344–348.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang Y, Qian CY, Li XP, Zhang Y, He H,
Wang J, Chen J, Cui JJ, Liu R, Zhou H, et al: Genome-scale long
noncoding RNA expression pattern in squamous cell lung cancer. Sci
Rep. 5:116712015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Remke M, Pfister S, Kox C, Toedt G, Becker
N, Benner A, Werft W, Breit S, Liu S, Engel F, et al:
High-resolution genomic profiling of childhood T-ALL reveals
frequent copy-number alterations affecting the TGF-beta and
PI3K-AKT pathways and deletions at 6q15-16.1 as a genomic marker
for unfavorable early treatment response. Blood. 114:1053–1062.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liang T, Guo L and Liu C: Genome-wide
analysis of mir-548 gene family reveals evolutionary and functional
implications. J Biomed Biotechnol. 2012:6795632012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shi Y, Qiu M, Wu Y and Hai L: MiR-548-3p
functions as an anti-oncogenic regulator in breast cancer. Biomed
Pharmacother. 75:111–116. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vogel F, Hartmann E, Görlich D and
Rapoport TA: Segregation of the signal sequence receptor protein in
the rough endoplasmic reticulum membrane. Eur J Cell Biol.
53:197–202. 1990.PubMed/NCBI
|
|
30
|
Poplawski P, Wiśniewski JR, Rijntjes E,
Richards K, Rybicka B, Köhrle J and Piekiełko-Witkowska A:
Restoration of type 1 iodothyronine deiodinase expression in renal
cancer cells downregulates oncoproteins and affects key metabolic
pathways as well as anti-oxidative system. PLoS One.
12:e01901792017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rao CV, Asch AS and Yamada HY: Frequently
mutated genes/pathways and genomic instability as prevention
targets in liver cancer. Carcinogenesis. 38:2–11. 2017. View Article : Google Scholar : PubMed/NCBI
|