Introduction
Ovarian carcinoma, which is one of the most common
gynecological cancers, is the seventh most lethal malignancy
worldwide (1). The efficacy of
platinum-based therapy such as cisplatin (CDDP) in ovarian
carcinoma is hindered by the occurrence of drug resistance, a
phenomenon often associated with an increased metastatic potential
(2,3). Shedding light onto the mechanisms of
chemoresistance may render insights into novel therapeutic targets
and improved treatment strategies (2–5).
A number of aberrantly expressed gene products
mediate tumor chemoresistance, including the upregulation of
ATP-binding cassette transporters, such as P-glycoprotein (P-gp)
(6). Additional mechanisms,
including apoptosis evasion and tumor cell survival, may also
contribute to CDDP resistance, among other cancer hallmarks,
including tumor cell heterogeneity, redundancy of growth-promoting
pathways, increased mutation rate and/or epigenetic alterations
(4). Similarly, a process termed
the epithelial-mesenchymal transition (EMT) has been demonstrated
to contribute to drug resistance (7–9). A
recent study has also reported that interleukins produced by the
tumor microenvironment promote the EMT through altered expression
of several inducers of this process, including Snail and STAT
transcription factors (5).
Osteopontin (OPN) emerges in the interface between
chemoresistance and EMT; it is a matrix glycophosphoprotein that
serves important roles in tumor cell proliferation and chemotherapy
resistance (10–15). The OPN primary transcript undergoes
alternative splicing, generating at least three main splicing
isoforms (OPN-SIs), termed OPNa, OPNb and OPNc (16). In addition, OPN is subjected to
post-translational modifications, generating several additional
isoforms (17,18). The sum of all these isoforms
comprises total OPN (tOPN). Although tOPN has been implicated in
cancer cell resistance to a wide range of anticancer agents
(10), the underlying molecular
mechanisms through which the full-length OPN and each OPN-SI
specifically perform their roles in chemoresistance remain
unclear.
Nakamura et al (19) have demonstrated that prostate cancer
cells overexpressing OPNb and OPNc are more resistant to docetaxel
compared with cells transfected with an empty vector and exhibit a
typical mesenchymal phenotype. Our recent study demonstrated that
OPNc was upregulated in distinct B-acute lymphoblastic leukemia
(B-ALL) cell lines (20). Our other
previous study revealed that OPNc expression levels in B-ALL cells
were significantly increased in response to treatment with
chemotherapeutic agents that have been used in several backbone
treatment strategies for B-ALL, namely vincristine or etoposide
(21). Based on these findings, the
present study aimed to investigate whether different OPN-SIs may
differentially modulate chemoresistance in an ovarian carcinoma
cell line model as well as their potential functional roles in the
chemoresistant phenotype.
Materials and methods
Study design
The present study used ACRP, an ovarian cancer cell
line resistant to CDDP, as well as its corresponding parental
control cell line A2780 as models. Some data obtained using the
ACRP cell line have been validated by also testing
OVCar-8/DoxR, an ovarian cancer cell line resistant to
doxorubicin (Dox), which originated from OVCar-8 cells. Both
ovarian cancer cell lines were used to assess the roles of OPNc in
chemoresistance. The expression of OPN-SIs and P-gp was assessed
using reverse transcription-quantitative PCR (RT-qPCR). After
evaluating OPNc expression in the CDDP and Dox resistance models,
the OPNc isoform was silenced in order to evaluate its roles in the
resistant phenotype by transfecting ACRP and
OVCar-8/DoxR cells with a specific anti-OPNc DNA
oligomer modified with phosphorothiotates. In these cell lines,
functional assays were performed using
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT),
trypan blue and clonogenic assays. The biological effects were
validated by analyzing the mRNA expression levels of EMT markers
and cytokines. To validate the cytotoxicity results observed using
the knockdown approach, experimental assays were performed in the
A2780 parental cell line ectopically overexpressing OPNc
(OPNc+). OPNc and P-gp expression levels were determined
in the A2780 OPNc+ cell line, and additional functional
assays were performed, including MTT, trypan blue exclusion and
clonogenic assays in the absence or presence of CDDP.
Cell lines and culture conditions
The epithelial ovarian cancer cell line A2780 and
the corresponding CDDP-resistant cell line ACRP were generously
provided by Dr Pat J. Morin (National Institutes of Health,
Bethesda, MD, USA). ACRP cells were selected for progressive
resistance to CDDP as previously described (22). The cells were maintained in
RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc.) at 37°C in a humidified atmosphere of 5%
CO2. The human ovarian cell line OVCar-8 was acquired
from the American Type Culture Collection. The OVCar-8 cell line
resistant to Dox, termed OVCar-8/DoxR resistant cell
line, was originated by progressively culturing OVCar-8 cells with
increasing concentrations of Dox for 6 months. The doses were
incrementally increased upon selection of Dox-resistant clones up
to 17 µM Dox, which was used to maintain the
OVCar-8/DoxR cells.
Isolation of total RNA and
RT-qPCR
Total cellular RNA was isolated from the cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. The RNA was
reverse-transcribed using SuperScript™ II Reverse Transcriptase kit
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. mRNA expression analysis was performed by
qPCR using the Eco Real-Time PCR System (Illumina, Inc.) and Power
SYBR® Green PCR Master mix (Applied Biosystems; Thermo
Fisher Scientific, Inc.). The thermocycling conditions were as
follows: Initial incubation at 50°C for 2 min and 95°C for 10 min,
followed by additional incubation at 94°C for 5 min; 40 cycles of
94°C for 30 sec, 60°C for 30 sec and 72°C for 45 sec; and final
melting curve analysis at 72°C for 15 sec, 90°C for 5 sec, 55°C for
5 sec and 90°C for 5 sec. Fold-changes in the expression levels of
each mRNA was calculated using the 2−∆∆Cq method
(23). GAPDH or β-actin were used
as the normalization controls. The PCR primers sequences are listed
in Table I.
 | Table I.Forward and reverse oligonucleotide
sequences. |
Table I.
Forward and reverse oligonucleotide
sequences.
| Gene | Sequence
(5′→3′) |
|---|
| P-gp | F:
CCCATCATTGCAATAGCAGG |
|
| R:
GTTCAAACTTCTGCTCCTGA |
| OPNa | F:
ATCTCCTAGCCCCACAGAAT |
|
| R:
CATCAGACTGGTGAGAATCATC |
| OPNb | F:
CTCCTAGCCCCACAGACCCT |
|
| R:
TATCACCTCGGCCATCATATG |
| OPNc | F:
CTGAGGAAAAGCAGAATG |
|
| R:
AATGGAGTCCTGGCTGT |
| GAPDH | F:
TCCCATCACCATCTTTCAGGAGCA |
|
| R:
TTCTACATGGTGGTGAAGACGCCA |
| β-actin | F:
GGCGGCACCACCATGTACCCT |
|
| R:
AGGGGCCGGACTCGTCATACT |
| E-cadherin | F:
GAATGACAACAAGCCCGAAT |
|
| R: GAC
CTCCATCACAGAGGTTCC |
| Cytokeratin-18 | F:
GCGAGAAGGAGACCATGCA |
|
| R:
GGTGTTCCCGGATTTTGATCT |
| Vimentin | F:
GACAATGCGTCTCTGGCACGTCTT |
|
| R: TCC
TCCGCCTCCTGCAGGTTCTT |
| N-cadherin | F:
GGTGGAGGAGAAGAAGACCAG |
|
| R: GCA
TCAGGCTCCACAGT |
| Claudin-3 | F:
CTGCTCTGCTGCTCGTGTCC |
|
| R:
TTAGACGTAGTCCTTGCGGTCGTAG |
| Slug | F:
TTCGGACCCACACATTACCT |
|
| R:
GCAGTGAGGGCAAGAAAAAG |
| Snail | F:
TTCCAGCAGCCCTACGACCAG |
|
| R:
CTTTCCCACTGTCCTCATC |
| Twist | F:
CCCAACTCCCAGACACCTC |
|
| R:
CAAAAAGAAAGCGCCCACC |
| IL-6 | F:
CATTTGTGGTTGGGTCAGG |
|
| R:
AGTGAGGAACAAGCCAGAGC |
| IL-8 | F:
CTTGGCAGCCTTCCTGATTT |
|
| R:
GGGTGGAAAGGTTTGGAGTATG |
| IL1-α | F:
CATCCTCCACAATAGCAGACAG |
|
| R:
GAGTTTCCTGGCTATGGGATAAG |
| IL1-β | F:
CAAAGGCGGCCAGGATATAA |
|
| R:
CTAGGGATTGAGTCCACATTCAG |
| GP130 | F:
TGCCTCCAGAAAAACCTAAAAA |
|
| R:
TTTGTCTCCAAGTGTGTTTCC |
OPNc knockdown using anti-OPNc
oligomers
After reaching 90-95% confluency, ACRP or OVCar-8
DoxR cells were transfected with 100 nM anti-OPNc
phosphorothiotate-modified antisense DNA oligomer (ASO anti-OPNc;
5′-A*C*A*AC*GCATTCTGCTTT*T*C*C-3′) or with a non-specific/scrambled
control sequence (ASO SCR; 5′-C*C*T*T*TTCGTCTTACGAC*A*C*A-3′),
using Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions. The
phosphorothiotate-modified bases are marked by an asterisk in the
ASO sequences. Transfections were performed with 24 µg oligomers
and 24 µl Lipofectamine® 2000 diluted in RPMI-1640
medium and Opti-MEM in 10-cm plates in a 5% CO2
incubator at 37°C for 24 h. The transfected cells were collected at
24 h post-transfection for subsequent experiments. The
downregulation of the OPNc mRNA expression levels was confirmed by
RT-qPCR.
OPNc overexpression
To generate a cell line stably overexpressing OPNc
(A2780 OPNc+), 4×105 A2780 cells were plated
in 6-well plates and maintained for 24 h in a 5% CO2
incubator at 37°C. Following adhesion, the cells were transfected
with 2 µg pCR3.1 expression plasmid containing the complete cDNA
encoding OPNc or with the empty vector plus 4 µl
Lipofectamine® 2000, which were diluted in RPMI-1640
medium and Opti-MEM. The cells were then maintained in a 5%
CO2 incubator at 37°C for 24 h. Subsequently, 300 µg/ml
geneticin was added to the culture medium to select the transfected
clones. The culture medium containing 300 µg/ml geneticin was
changed every 2 days until clones stably expressing the OPNc
isoform were selected, which lasted 15 days. Subsequent experiments
were performed following the selection or A2780 OPNc+
cell line in selection culture media. Following selection, A2780
OPNc+ and A2780 pCR3.1 cells were routinely maintained
with 300 µg/ml geneticin to ensure OPNc overexpression.
MTT assay
The MTT assay (Uniscience Corp.) was used to measure
the cytotoxic effects of CDDP (Libbs Farmaceutica, Ltda.) on
ovarian cancer cell lines. Briefly, ACRP, OVCar-8 DoxR,
A2780 OPNc+ cells or A2780 cells transfected with the
pCR3.1 empty vector were seeded in 96-well plates (1×104
cells/well). After adhesion, the cells were exposed to 10, 20, 40,
60 and 100 µM CDDP or drug-free complete medium for 24 or 96 h.
Drug-free medium was added to the control wells. A total of 20 µl
MTT (5 mg/ml) was added to each well 4 h prior to the end of the
time intervals. MTT solution was then removed at 24 or 96 h, and
the formazan crystals were dissolved in 150 µl DMSO (Sigma-Aldrich;
Merck KGaA). The optical density at 570 nm was determined using a
SpectraMax 190 microplate reader. All experiments were performed in
triplicate.
Clonogenic assay
To determine clonogenicity, a total of
2×103 ACRP, OVCar-8 DoxR, A2780
OPNc+ cells or A2780 cells transfected with the pCR3.1
empty vector were seeded in 6-well plates and maintained in culture
for ~14 days, until colonies could be observed. Colonies were fixed
in absolute ethanol for 10 min and stained with 0.5% crystal violet
(Sigma-Aldrich; Merck KGaA) for 1 h at room temperature. The
crystals were dissolved in 33% glacial acetic acid solution, and
the optical density was measured at 595 nm using a SpectraMax 190
microplate reader.
Statistical analysis
Data are presented as the mean ± SD of three
independent experiments. Differences in various parameters between
two groups were analyzed by two-tailed Student's t-test. GraphPad
Prism version 5.02 (GraphPad Software, Inc.) was used to plot the
graphs. Statistical analysis was performed using Microsoft Excel
2010 (Microsoft Corporation). P<0.05 was considered to indicate
a statistically significant difference.
Results
OPNc is upregulated in ACRP
chemoresistant cells
As a first approach to determine whether the three
tested OPN-SIs differently modulated CDDP resistance in ovarian
cancer cell lines, the transcriptional levels of each OPN-SI were
analyzed in ACRP cells and compared with those in the corresponding
parental cells. Among the three tested OPN-SIs, only OPNc
expression levels were upregulated in ACRP cells compared with
those in the parental A2780 cell line, although the results were
not statistically significant (Fig.
1A).
Since tOPN can modulate P-gp expression (6), P-gp expression was subsequently tested
in ACRP cells. The results demonstrated that the mRNA expression
levels of P-gp were also upregulated in ACRP cells compared with
those in the parental control cells, although the results did not
reach statistical significance (Fig.
1B).
OPNc knockdown sensitizes ACRP cells
to CDDP treatment
To further characterize the involvement of OPNc in
CDDP-resistant ACRP cells, the OPNc isoform was knocked down using
the ASO anti-OPNc. OPNc mRNA expression levels were reduced
compared with those in cells transfected with the ASO-SCR (Fig. 2A). By contrast, OPNc knockdown in
ACRP cells resulted in the upregulation of OPNa and OPNb mRNA
expression levels compared with those in the ASO-SCR group,
although this result did not achieve statistical significance
(Fig. S1). Notably, following OPNc
silencing, P-gp expression levels were also downregulated compared
with those in cells transfected with the ASO-SCR (Fig. 2B), suggesting that OPNc may modulate
P-gp expression.
The present study then tested whether OPNc knockdown
may modulate ACRP cell viability in response to CDDP exposure.
Following OPNc knockdown, ACRP cells were more sensitive to 10 and
40 mM CDDP treatment compared with the ASO SCR-transfected cells 24
h after drug treatment (Fig. 2C and
D).
To further determine whether the roles of OPNc in
drug resistance were dependent on the cell line model and the class
of chemotherapeutic drug, the same experiments were performed in
the OVCar-8 DoxR cell line. The results demonstrated a
strong but non-significant downregulation of the P-gp expression
levels following OPNc knockdown in these cells compared with those
in cells transfected with the ASO SCR (Fig. S2A and B), which was similarly
associated with Dox sensitization following 24-h (0.05 and 0.1 µM)
and 96-h (0.1, 0.2, 0.5 and 1 µM) drug exposure (Fig. S2C and D). These results suggested
that OPNc may modulate drug resistance and cell viability in
distinct models of drug-resistant ovarian cancer cells.
OPNc modulates cell viability and
colony formation in ACRP cells
The effects of OPNc knockdown on the chemoresistant
ovarian cancer cell line viability were next assessed. Notably, the
number of ACRP cells was decreased in response to OPNc silencing
compared with that of the ASO SCR-transfected cells (Fig. 2E). The colony formation capacity of
ACRP cells transfected with the ASO anti-OPNc was analyzed, and the
results demonstrated that OPNc knockdown reduced the colony
formation capacity in ACRP cells compared with that of the control
group, but the difference was not statistically significant
(Fig. 2F). Similarly, the cell
number and clonogenicity were impaired in OVCar-8 DoxR
cells transfected with the ASO anti-OPNc compared with those in the
control cells (Fig. S3A and B).
These results suggested that OPNc may not only modulates drug
sensitivity, but also promote cell viability.
OPNc affects the expression of EMT
markers in ACRP cells
The present study further investigated whether OPNc
may modulate the EMT, and the RT-qPCR results demonstrated that
ACRP cells exhibited an intermediate or partial EMT phenotype
compared with that of the parental A2780 cell line. In ACRP cells,
the expression levels of the epithelial markers E-cadherin,
claudin-3 and cytokeratin-18 were upregulated compared with those
in A2780 cells (Fig. 3A). In
addition, vimentin and N-cadherin levels were also upregulated,
whereas Slug, Snail and Twist exhibited similar or lower expression
levels compared with those in A2780 cells (Fig. 3B). However, none of these
differences were statistically significant. In response to OPNc
silencing, changes in the expression patterns of the epithelial and
mesenchymal markers were observed. E-cadherin expression levels
were non-significantly upregulated in ACRP cells following OPNc
silencing compared with those in the control cells. By contrast,
the levels of claudin-3 and cytokeratin-18 were non-significantly
downregulated in the same experimental conditions (Fig. 3C). Among the mesenchymal markers,
the vimentin and N-cadherin transcriptional levels remained largely
unchanged following OPNc silencing in ACRP cells, whereas the
levels of Slug, Snail and Twist transcripts were downregulated
compared with those in the ASO SCR-transfected cells (Fig. 3D). These results demonstrated
alterations in the EMT marker transcriptional patterns in response
to OPNc downregulation in ACRP resistant cell line, suggesting that
OPNc may modulate CDDP resistance-associated EMT in these
cells.
 | Figure 3.OPNc expression levels modulate the
epithelial-mesenchymal transition marker and associated interleukin
expression patterns in cisplatin-resistant ACRP ovarian cancer
cells. (A, B and E) A2780 and ACRP cells were harvested, and the
expression levels of (A) epithelial and (B) mesenchymal markers, as
well as (E) IL-6, IL-8, IL-1α, IL-1β and the GP130 receptor were
analyzed by RT-qPCR. (C, D and F) ACRP cells were transfected with
ASO SCR or ASO anti-OPNc, and RT-qPCR was used to determine the
mRNA expression levels of (C) epithelial and (D) mesenchymal
markers, as well as (F) IL6, IL8, IL-1α, IL-1β and the GP130
receptor. The mRNA expression levels of E-cad, Cl3, ck18, Vim,
N-cad, Slug, Snail and Twist were normalized to those of GAPDH, and
the expression levels of IL6, IL8, IL-1α, IL-1β and the GP130
receptor were normalized to those of β-actin. Relative expression
levels were calculated using the 2−∆∆Cq method. *P≤0.05
and **P≤0.01; ns, non-significant. OPNc, osteopontin-c isoform;
RT-qPCR, reverse transcription-quantitative PCR; ASO, antisense
oligonucleotide; SCR, scramble; E-cad, E-cadherin; Cl3, claudin-3;
ck18, cytokeratin-18; Vim, vimentin; N-cad, N-cadherin. |
OPNc affects the expression of
EMT-related cytokines
The present study next tested the expression levels
of the EMT-related cytokines IL-6, IL-8, IL-1α, IL-1β and the GP130
receptor. Notably, a general non-significantly upregulated
expression pattern was observed for the EMT-related cytokines in
ACRP cells compared with that in the parental cell line A2870
(Fig. 3E). Consistently, the
expression levels of IL-6, IL-8 and IL-1α were significantly
downregulated, and the levels of IL-1β and the GP130 receptor were
non-significantly downregulated in the OPNc-silenced ACRP cells
compared with those in the cells transfected with the ASO SCR
(Fig. 3F). In addition, decreased
levels of EMT-associated cytokines were observed in OVCar-8
DoxR cells following OPNc knockdown compared with those
in ASO SCR-transfected control cells (Fig. S4), further suggesting that OPNc may
be associated with drug resistance as well as with the EMT
phenotype.
OPNc+ cells exhibit
enhanced proliferative capacity and sensitivity to CDDP
To further determine the cellular effects of OPNc,
this splice variant was stably overexpressed in the A2780 parental
cells (Fig. 4A). The results
demonstrated that the OPNc+ cells also displayed high
P-gp expression levels (Fig. 4B).
Notably, OPNc overexpression improved the cell proliferative
capacity, as denoted by a higher number of cells (Fig. 4C), cell viability at 96 h (Fig. 4D) and clonogenic potential (Fig. 4E) in the OPNc+ group
compared with those in the empty vector-transfected control
group.
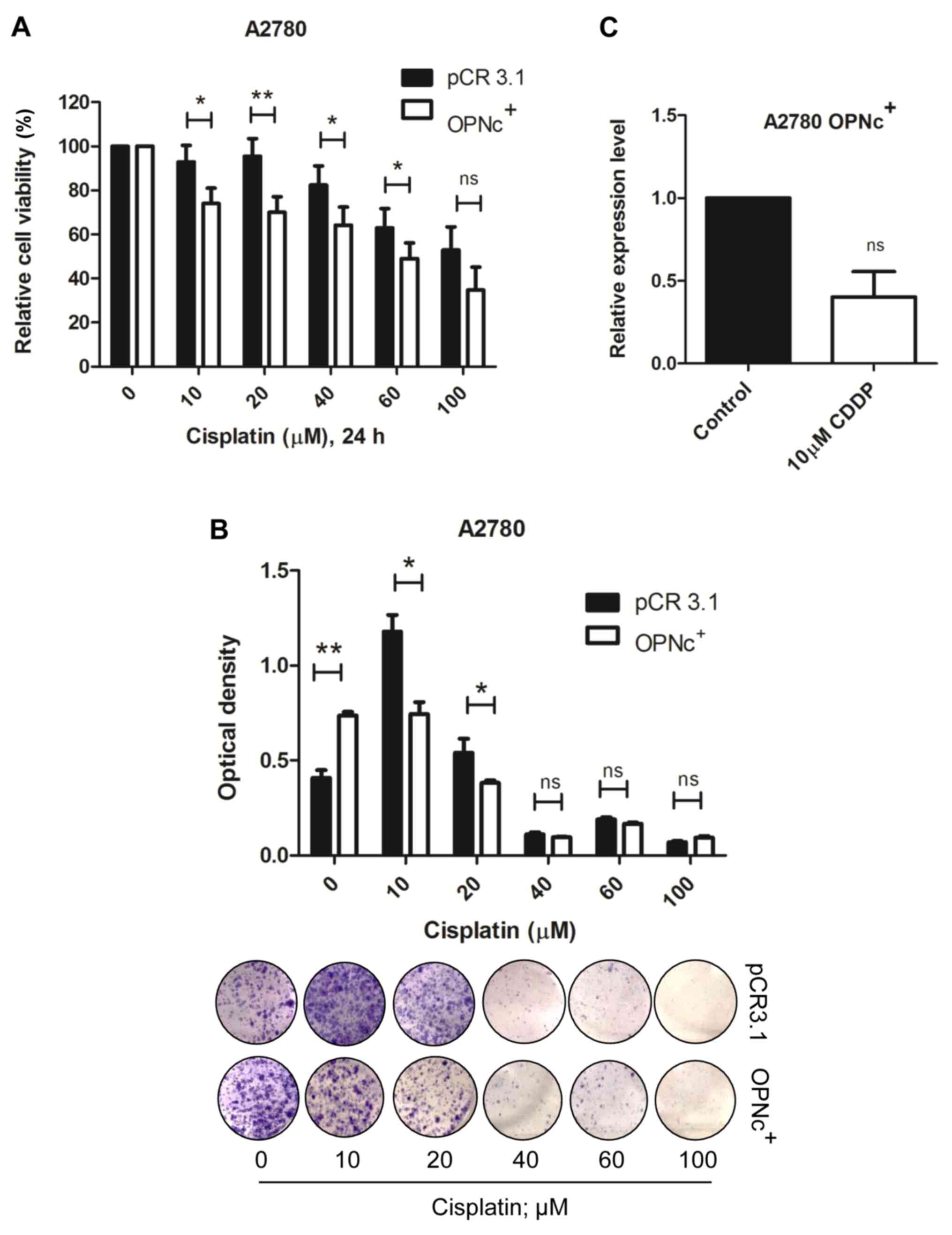
In order to address whether OPNc overexpression
modulated the response to CDDP, OPNc+ cells were exposed
to increasing CDDP concentrations for 24 and 96 h. Notably, OPNc+
cells became more susceptible to CDDP treatment compared with cells
transfected with the empty PCR3.1 expression vector after 24-h drug
treatment (Fig. 5A). These results
were confirmed in a long-term assessment of cell viability, which
demonstrated that OPNc+ cells formed fewer colonies in response to
CDDP treatment (Fig. 5B). We then
hypothesized that CDDP may negatively modulate the expression of
OPNc, further sensitizing OPNc+ cells to this drug. The
results of RT-qPCR analysis demonstrated that CDDP downregulated
the OPNc mRNA levels in OPNc+ cells (Fig. 5C) following 24-h CDDP treatment.
However, these effects were less pronounced following 96-h CDDP
exposure (Fig. S5A and B). These
results demonstrated that CDDP inhibited OPNc expression in OPNc+
cells, increasing cell sensitivity and further decreasing cell
viability.
Discussion
The present study aimed to evaluate whether OPN-SIs
were differentially expressed in CDDP-resistant ovarian cancer
cells compared with their parental cells. Based on the differential
expression patterns, the contribution of OPNc to the modulation of
chemoresistance was selected for further investigation in this cell
line model. The results demonstrated that the expression levels of
the OPNc splice variant were upregulated in ACRP cells compared
with those in the parental cell line. Using an OPNc-specific
knockdown approach in the ACRP cell line, the present study
revealed that OPNc modulated various aspects of drug resistance,
such as cell viability, sensitivity to CDDP, and colony formation,
as well as the EMT phenotype and the expression of the associated
cytokines. A number of these features were also validated by stably
overexpressing OPNc in the A2780 parental cell line.
The results of the present study demonstrated that
ACRP cells presented with higher expression levels of OPNc compared
with those in A2780 cells. Similarly to these results, previous
studies have demonstrated that tOPN expression was induced in human
hepatocellular carcinoma cells resistant to CDDP compared with
non-resistant cells (11), in
colorectal cancer cells resistant to oxaliplatin (13) and in glioma cells resistant to
temozolomide (TMZ) and CDDP (15)
and alkylphosphocholines (24). In
a number of experimental models, tOPN expression levels were
upregulated in response to drug treatment (11,13,15).
In addition, tOPN knockdown sensitized cells to drug exposure
(15). It has also been
demonstrated that oncogenic signaling pathways, such as PI3K,
NF-κB/Bcl-2 and Raf/MEK/ERK, are activated in response to tOPN
overexpression, favoring drug resistance (15). Similarly to tOPN and OPNc,
overexpression of other gene products have also been associated
with resistance to distinct therapeutic drugs, including GATA
binding protein 1 (25), secretory
clusterin (26), Her-2 (27) and drug efflux pumps, including P-gp
(28,29). Although studies have reported an
association between tOPN expression and chemoresistance (10,12–15),
the functional roles of specific OPN-SIs in chemoresistant cells
have not been previously determined. The results of the present
study suggested a potential role for OPNc in chemoresistance in the
ACRP and OVCar-8/DoxR cell line models. In accordance
with this hypothesis, our recent study demonstrated that ectopic
overexpression of OPNb and OPNc in PC3 prostate cancer cells
induced resistance to docetaxel (19), suggesting that overexpression of
specific OPN-SIs may differentially contribute to tumor drug
resistance. The expression levels of OPNa and OPNb were also
upregulated in the ACRP cell line compared with those in A2780
cells in the present study, although at lower levels than OPNc.
Future work should evaluate their putative contribution to the
resistant phenotype. Notably, in the present study, when OPNc was
knocked down, OPNa and OPNb were upregulated; the indirect effects
of OPNc knockdown on the expression of the two other splice
variants may be explained by cellular compensation due to low OPNc
levels. However, the specific mechanisms by which OPNc may favor
chemoresistance are currently unknown. Based on available data
regarding the roles of tOPN in several tumor types, we hypothesize
that in the experimental model used in the present study, OPNc may
activate signaling pathways that contribute to the survival of
ovarian tumor cells, favoring increased viability and
colony-forming capacity. Similarly, our previous study demonstrated
that ectopic OPNc overexpression in the ovarian cancer line OVCar-3
contributed to increased migration, proliferation, cell invasion,
independent anchorage and tumor formation in vivo compared
with those in OVCar-3 cells transfected with an empty vector
(30). It was also observed that
certain OPNc-dependent protumorigenic functions were mediated by
the PI3K/AKT signaling pathway, demonstrating the role of OPNc in
ovarian cancer tumor progression through the activation of these
signaling pathways (30). The
protumorigenic functions of the PI3K/AKT pathway also involve the
regulation of survival and apoptosis inhibition, which are
associated with drug resistance in ovarian cancer cells (31,32).
Therefore, we hypothesize that the inhibition of the PI3K/AKT
signaling pathway may be one of the mechanisms underlying the
biological effects observed in the OPNc-knockdown cells.
tOPN has been reported to be a key regulator of the
EMT (33). The specific mechanism
involves the modulation of the expression of transcription factors
that contribute to EMT initiation, such as Twist, Snail and Slug
(33). Therefore, tOPN favors EMT
initiation, which in turn contributes to tumor progression and
chemoresistance. Although these findings refer only to tOPN, OPNc
may similarly modulate cellular plasticity in the ACRP and OVCar-8
DoxR cell lines, further contributing to tumor
progression.
tOPN modulated the expression of ILs through binding
to their receptors in the tumor microenvironment, contributing to
the chemoresistance and tumor progression (10). tOPN has also been demonstrated to
bind to the C-C motif chemokine receptor 1 (CCR1), leading to the
activation of the PI3K/AKT/hypoxia-inducible factor 1α signaling
pathway in hepatocellular carcinoma cell lines (34). The same study also reported that
blocking the OPN-CCR1 interaction with a CCR1 antagonist restricted
the effects of tOPN on tumor progression and metastatic capacity in
this tumor model (34). Therefore,
OPNc upregulation may contribute to the increased expression of the
cytokines analyzed in the present study through the activation of
their corresponding cellular receptors. Indeed, the mRNA expression
levels of the GP130 receptor were downregulated in the
OPNc-knockdown cells in the present study. Thus, OPNc may act
directly and indirectly by promoting the activation of signaling
pathways associated with cell survival, inhibition of apoptosis,
tumor progression and chemoresistance in ovarian cancer.
In addition to the upregulation of OPNc expression
in ACRP cells, the results of the present study demonstrated that
P-gp expression levels were upregulated in these cells compared
with those in the parental A2780 cells, which has been previously
demonstrated in chemoresistant cells in other tumor models
(35). Hsieh et al (6) have reported that tOPN modulates the
expression of P-gp in a concentration- and time-dependent manner
through binding to αvβ3 integrin (6). However, the contribution of specific
OPN-SIs to the regulation of P-gp expression has not been
previously addressed. The results of the present study also
demonstrated that the upregulation of P-gp in ACRP cells was
reversed when OPNc was silenced. Similarly, P-gp expression was
induced following OPNc overexpression. These results were in
agreement with a previous study, in which knockdown of endogenous
tOPN potentiated various P-gp drug substrate-induced apoptosis of
prostate cancer cells (6). These
results suggest that OPNc may be a modulator of P-gp expression and
a potential target to modulate drug efflux by P-gp in cancer cells,
facilitating tumor resistance. However, the molecular mechanisms by
which tOPN, and specifically OPNc, regulates the expression of P-gp
remain unknown and should be determined in future studies. High
levels of P-gp have been demonstrated to induce TMZ resistance in a
glioblastoma (GBM) model (36).
Increasing concentrations of TMZ competed with calcein for P-gp,
resulting in reduced efflux in the Adriamycin-resistant DC3F cells;
however, various inhibitors of P-gp reversed TMZ resistance in two
GBM cell lines by increasing the levels of active caspase-3,
suggesting that P-gp may be a key regulator of TMZ resistance in
GBM (36). High expression of P-gp
has been reported to confer resistance to a wide range of
structurally and functionally unrelated chemotherapeutic agents
(37). In the present study, OPNc
was demonstrated to regulate P-gp gene expression, suggesting that
targeting the OPNc/P-gp signaling axis may be a potential approach
for ovarian cancer cell sensitization to Dox and CDDP.
The results of the present study also demonstrated
that OPNc expression levels modulate the survival of drug-resistant
ovarian carcinoma cells. In response to OPNc knockdown, the
viability of drug-resistant ovarian cancer cells decreased,
resulting in higher sensitivity to CDDP and Dox exposure compared
with that of the control cells. These results were in accordance to
a role of OPNc as a putative novel gene product able to activate
the key steps towards resistance to these chemotherapeutic drugs.
Similar results have been observed for other proteins that mediated
resistance to chemotherapy by stimulating cell survival and
inhibiting apoptosis, as well as promoting the cell cycle,
including Bcl-2, inhibitor of apoptosis proteins and the heat shock
protein family (5). In addition,
tOPN has also been described as a modulator of cell survival in
response to chemotherapeutic drugs (10,38).
In light of these results, among the three OPN-SIs tested in the
present study, OPNc may be a key contributor to the cellular
processes mediating chemoresistance, such as cell survival and
viability, at least in chemoresistant ovarian cancer cells. Future
studies should further characterize the targets and pathways by
which OPNc may activate cell survival in response to
chemotherapeutic drugs.
Following silencing OPNc expression in OVCar-8
DoxR cells, increased sensitivity to Dox treatment
compared with that in the control cells was observed in the present
study. Similarly, the results demonstrated decreased viability and
colony formation capacity in these cells, along with a modulation
in the expression of EMT markers and EMT-related cytokines.
Considering similar experimental findings in CDDP- and
Dox-resistant cell lines following OPNc silencing, we hypothesize
that targeting OPNc may counteract resistance to various
chemotherapeutic drugs, similarly to Dox and CDDP. However, further
studies should investigate whether these findings can be extended
to other drugs.
The results of the present study demonstrated that
OPNc regulated the EMT in the ACRP cell line. OPNc knockdown
altered the pattern of the intermediate EMT phenotype observed in
ACRP cells. These results were in accordance with a putative role
of OPNc as a modulator of EMT, which was previously assigned to
tOPN in several experimental models, such as in breast cancer,
hepatocellular carcinoma, ovarian, gastric and colorectal cancer,
as well as melanoma and brain tumors (33). In the lung cancer cell line A549,
Huang et al (39) have
demonstrated that OPNc overexpression induces increased cell
migration and invasion, and alters the expression of EMT markers
(decreases E-cadherin and increases vimentin expression) in
response to TGF-β treatment, indicating that OPNc stimulates
cellular plasticity of A549 tumor cells. The roles of OPNc in
non-small cell lung cancer cells were mediated by Runt-related
transcription factor 2 via a histone deacetylase-dependent pathway,
further suggesting that OPNc may be a marker of the EMT and tumor
progression in lung cancer. Our previous study in prostate cancer
cells resistant to docetaxel supported these results, as cells
overexpressing OPNb or OPNc presented with an EMT-related phenotype
typical of chemoresistant cells, as observed in other tumor models
(19). The intermediate EMT
phenotype observed in CDDP-resistant ACRP ovarian carcinoma cells
in the present study was in agreement with a previous study
demonstrating that ovarian carcinoma cells present various degrees
of EMT (40). An intermediate EMT
phenotype has been demonstrated to occur in breast, colon and
ovarian carcinoma (41). In
addition, the results of the present study demonstrated that OPNc
silencing modulate the intermediate EMT phenotype. Notably, ovarian
carcinoma has unique biological characteristics, and whether
ovarian cancer cells in the primary tumor undergo a complete
transition to a mesenchymal state is still under discussion
(40,42). In the present study, the EMT does
not fully occur in ovarian carcinoma, and is even reversed in tumor
cells present in malignant peritoneal and pleural effusions
(40). Additionally, Carduner et
al (43) have reported that
ascites in patients with ovarian cancer undergo a shift toward an
unstable intermediate state of the epithelial-mesenchymal spectrum,
conferring aggressive cell behavior, depending on the initial
epithelial-mesenchymal background (43). Future studies are required to fully
characterize the intermediate EMT phenotype in ACRP cells and its
association with OPNc expression by further evaluating the protein
expression and subcellular localization of EMT markers.
In the present study, OPNc knockdown reverted the
expression pattern of EMT-related cytokines in ACRP cells. It has
been demonstrated that a number of interleukins or cytokines induce
the EMT, including IL-6, IL-8 and TGF-β (44,45).
The role of cytokines in cancer has been widely studied,
particularly in ovarian cancer (46). In this context, certain cytokines
have been demonstrated to induce phenotypes consistent with the EMT
in transformed epithelial as well as tumor cell lines (47,48).
The results of the present study demonstrated that the expression
levels of OPNc appeared to modulate the expression of EMT-related
cytokines in the drug-resistant cell lines, which in turn induced
changes in the expression of EMT markers, as has been previously
observed (44,45). A previous study has demonstrated
that tOPN modulates cytokine expression, contributing to tumor
progression; the interaction between tOPN-IL-6-STAT3 contributes to
the invasive and migratory features of osteosarcoma tumor cells,
enhancing their metastatic potential (49). Since these results refer to tOPN,
the present results are the first to report the role of OPN-SIs,
mainly OPNc, in the modulation of the expression of EMT-related
cytokines. Further studies are needed to determine the regulatory
mechanisms between OPNc and the investigated cytokines in the in
vitro chemoresistance models. We hypothesize that upregulation
of OPNc may contribute to the increased expression of the cytokines
analyzed in the present study through the activation of their
corresponding cellular receptors. This was observed to occur for
the GP130 receptor, the mRNA levels of which were downregulated in
the OPNc-knockdown cells in the present study. Thus, OPNc may act
directly and indirectly by promoting the activation of signaling
pathways associated with cell survival, inhibition of apoptosis,
tumor progression and chemoresistance in ovarian cancer.
To validate the data from OPNc silencing
experiments, the present study overexpressed OPNc in the A2780 cell
line. Cells overexpressing OPNc displayed increased cell viability
and colony formation capacity compared with those in cells
transfected with the empty expression vector. These data reinforced
the pro-survival roles of OPNc in these ovarian cancer cells.
Similar to ACRP cells in which OPNc was silenced, A2780
OPNc+ cells were also more sensitive to CDDP treatment
compared with the control cells. These results suggested that cells
overexpressing OPNc may be preferentially targeted for CDDP
sensitization. Further corroborating our hypothesis, the results of
the present study demonstrated that CDDP downregulated the
expression of OPNc in OPNc+ cells. As samples from
patients with ovarian cancer exhibit high levels of OPNc (30), and the results of the present study
demonstrated that CDDP-resistant cells present upregulated levels
of OPNc, we hypothesized that targeting OPNc in ovarian cancer
cells with high levels of OPNc may be a promising new therapeutic
approach to overcome resistance to CPPD. In addition, combining
OPNc silencing with CDDP treatment may possibly enhance
cytotoxicity in these resistant cells. One explanation for this
effect may be that CDDP, in addition to inhibiting OPNc expression,
may also somehow prevent OPNc binding to classical OPN receptors,
such as CD44 and integrin heterodimers, which is worth
investigating in the future.
The cytotoxicity assays performed in the present
study revealed that ACRP cells became more sensitive to CDDP
compared with A2780 cells when CDDP concentrations were
progressively increased (data not shown). Accordingly, drug
adaptation and thus, resistance, are considered to be a
dose-dependent condition for certain drug classes and in
vitro models. In addition, the present study demonstrated that
OPNc silencing in ACRP cells counteracted cell viability and
resistance to CDDP. Notably, OPNc+ cells exhibited
increased sensitivity to CDDP compared with that in the control
cells, which may be associated with CDDP-induced OPNc
downregulation. In the experimental settings of the present study
involving the ACRP cell line which exhibited upregulation of
endogenous OPNc, as well as A2780 OPNc+ cells, targeting
OPNc sensitized cells to CDDP. Based on these results, we
hypothesized that OPNc+ cells may be suitable candidates
for novel approaches to CDDP treatment. Translating these findings
into the clinical practice, high OPNc expression in patients with
ovarian cancer at diagnosis may be a potential predictor for a
favorable response to CDDP treatment. This idea follows the
rationale reported for other drug targets, such as Her-2 (50–52).
In addition, approaches aiming at inducing cell toxicity based on
oncogene expression are already used in other tumor models, such as
in Her2-positive breast, gastric and esophageal cancer, in which
distinct strategies have been proposed to block Her2-induced tumor
growth (53–55).
In conclusion, the results of the present study
demonstrated that OPNc was upregulated in CDDP-resistant ACRP cells
and may serve important roles in modulating various biological
processes associated with drug resistance, including cell
viability, the EMT phenotype and the expression of EMT-related
cytokines. OPNc+ cells were more sensitive to CDDP
cytotoxicity compared with the negative control cells, suggesting
that OPNc overexpression may be a potential strategy for targeted
therapy to improve CDDP sensitivity in ovarian cancer cells.
Therefore, we propose a model in which OPNc is a key modulator of
resistance to CDDP in ovarian cancer cells (Fig. 6). These early insights on the role
of OPNc in cancer drug resistance provide a rationale for using
this variant as a novel potential molecular target to sensitize
ovarian cancer cells to Dox and CDDP treatment.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was funded by Fundação de Amparo à
Pesquisa do Estado do Rio de Janeiro (grant nos. E-26/210.394/2014,
E-26/010.002007/2014 and E-26/203.204/2015), Conselho Nacional de
Desenvolvimento Científico e Tecnológico (grant no. 310591/2014-7),
Ministério da Sáude, Universidade Federal Fluminense/Pró-Reitoria
de Pesquisa e Inovação, the L'Oréal-UNESCO-ABC prize for Women in
Science and Fundação do Câncer (Programa de Oncobiologia).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MCMB participated in the design of the study,
performed the majority of the experiments and analyzed the data.
LBF participated in the data analysis, reviewed and edited the
manuscript. IDSG participated in in the design of the study and
data analysis, as well as reviewed and edited the manuscript. LBAR
participated in the design of the study and provided support to
this work. RCM provided support to this work, reviewed and edited
the manuscript. GNDM and ERPG are co-senior authors and equally
participated in the design of the study, analyzed and summarized
the data, provided financial support, wrote, reviewed and edited of
the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
International Agency for Research on
Cancer (IARC). Global Cancer Observatory. IARC; Lyon: 2020,
simplehttps://gco.iarc.fr/
|
|
2
|
Oronsky B, Ray CM, Spira AI, Trepel JB,
Carter CA and Cottrill HM: A brief review of the management of
platinum-resistant-platinum-refractory ovarian cancer. Med Oncol.
34:1032017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Freimund AE, Beach JA, Christie EL and
Bowtell DDL: Mechanisms of drug resistance in high-grade serous
ovarian cancer. Hematol Oncol Clin North Am. 32:983–996. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gottesman MM, Lavi O, Hall MD and Gillet
JP: Toward a better understanding of the complexity of cancer drug
resistance. Annu Rev Pharmacol Toxicol. 56:85–102. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pandey MK, Prasad S, Tyagi AK, Deb L,
Huang J, Karelia DN, Amin SG and Aggarwal BB: Targeting cell
survival proteins for cancer cell death. Pharmaceuticals (Basel).
9:112016. View Article : Google Scholar
|
|
6
|
Hsieh IS, Huang WH, Liou HC, Chuang WJ,
Yang RS and Fu WM: Upregulation of drug transporter expression by
osteopontin in prostate cancer cells. Mol Pharmacol. 83:968–977.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Du B and Shim JS: Targeting
epithelial-mesenchymal transition (EMT) to overcome drug resistance
in cancer. Molecules. 21:9652016. View Article : Google Scholar
|
|
8
|
Elaskalani O, Razak NBA, Falasca M and
Metharom P: Epithelial-mesenchymal transition as a therapeutic
target for overcoming chemoresistance in pancreatic cancer. World J
Gastrointest Oncol. 9:37–41. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
He Y, Xie H, Yu P, Jiang S and Wei L:
FOXC2 promotes epithelial-mesenchymal transition and cisplatin
resistance of non-small cell lung cancer cells. Cancer Chemother
Pharmacol. 82:1049–1059. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gimba ERP, Brum MCM and Nestal De Moraes
G: Full-length osteopontin and its splice variants as modulators of
chemoresistance and radioresistance (Review). Int J Oncol.
54:420–430. 2019.PubMed/NCBI
|
|
11
|
Ding K, Fan L, Chen S, Wang Y, Yu H, Sun
Y, Yu J, Wang L, Liu X and Liu Y: Overexpression of osteopontin
promotes resistance to cisplatin treatment in HCC. Oncol Rep.
34:3297–3303. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Grbčić P, Tomljanović I, Klobučar M,
Kraljević Pavelić S, Lučin K and Sedić M: Dual sphingosine kinase
inhibitor SKI–II enhances sensitivity to 5-fluorouracil in
hepatocellular carcinoma cells via suppression of osteopontin and
FAK/IGF-1R signalling. Biochem Biophys Res Commun. 487:782–788.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ng L, Wan T, Chow A, Iyer D, Man J, Chen
G, Yau TC, Lo O, Foo CC, Poon JT, et al: Osteopontin overexpression
induced tumor progression and chemoresistance to oxaliplatin
through induction of stem-like properties in human colorectal
cancer. Stem Cells Int. 2015:2478922015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pang H, Cai L, Yang Y, Chen X, Sui G and
Zhao C: Knockdown of osteopontin chemosensitizes MDA-MB-231 cells
to cyclophosphamide by enhancing apoptosis through activating p38
MAPK pathway. Cancer Biother Radiopharm. 26:165–173. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Qian C, Li P, Yan W, Shi L, Zhang J, Wang
Y, Liu H and You Y: Downregulation of osteopontin enhances the
sensitivity of glioma U251 cells to temozolomide and cisplatin by
targeting the NF-κB/Bcl-2 pathway. Mol Med Rep. 11:1951–1955. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gimba ER and Tilli TM: Human osteopontin
splicing isoforms: Known roles, potential clinical applications and
activated signaling pathways. Cancer Lett. 331:11–17. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hui T, Sørensen ES and Rittling SR:
Osteopontin binding to the alpha 4 integrin requires highest
affinity integrin conformation, but is independent of
post-translational modifications of osteopontin. Matrix Biol.
41:19–25. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kazanecki CC, Uzwiak DJ and Denhardt DT:
Control of osteopontin signaling and function by post-translational
phosphorylation and protein folding. J Cell Biochem. 102:912–924.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nakamura KDM, Tilli TM, Wanderley JL,
Palumbo A Jr, Mattos RM, Ferreira AC, Klumb CE, Nasciutti LE and
Gimba ER: Osteopontin splice variants expression is involved on
docetaxel resistance in PC3 prostate cancer cells. Tumour Biol.
37:2655–2663. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bastos ACSF, Blunck CB, Emerenciano M and
Gimba ERP: Osteopontin and their roles in hematological
malignancies: Splice variants on the new avenues. Cancer Lett.
408:138–143. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Santoro JC, Bastos ACSF, Gimba ERP and
Emerenciano M: Reinforcing osteopontin as a marker of central
nervous system relapse in paediatric B-cell acute lymphoblastic
leukaemia: SPP1 splice variant 3 in the spotlight. Br J Haematol.
186:e88–e91. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sherman-Baust CA, Weeraratna AT, Rangel
LBA, Pizer ES, Cho KR, Schwartz DR, Shock T and Morin PJ:
Remodeling of the extracellular matrix through overexpression of
collagen VI contributes to cisplatin resistance in ovarian cancer
cells. Cancer Cell. 3:377–386. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yosifov DY, Reufsteck C, Konstantinov SM
and Berger MR: Interleukin-6, osteopontin and Raf/MEK/ERK signaling
modulate the sensitivity of human myeloma cells to
alkylphosphocholines. Leuk Res. 36:764–772. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Caldwell JT, Edwards H, Dombkowski AA,
Buck SA, Matherly LH, Ge Y and Taub JW: Overexpression of GATA1
confers resistance to chemotherapy in acute megakaryocytic
Leukemia. PLoS One. 8:e686012013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Niu ZH, Wang Y, Chun B, Li CX and Wu L:
Secretory clusterin (sCLU) overexpression is associated with
resistance to preoperative neoadjuvant chemotherapy in primary
breast cancer. Eur Rev Med Pharmacol Sci. 17:1337–1344.
2013.PubMed/NCBI
|
|
27
|
Wilks ST: Potential of overcoming
resistance to HER2-targeted therapies through the PI3K/Akt/mTOR
pathway. Breast. 24:548–555. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Montazami N, Aghapour M, Farajnia S and
Baradaran B: New insights into the mechanisms of multidrug
resistance in cancers. Cell Mol Biol (Noisy-le-grand). 61:70–80.
2015.PubMed/NCBI
|
|
29
|
Yang X, Yi C, Luo N and Gong C:
Nanomedicine to overcome cancer multidrug resistance. Curr Drug
Metab. 15:632–649. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tilli TM, Franco VF, Robbs BK, Wanderley
JL, da Silva FR, de Mello KD, Viola JP, Weber GF and Gimba ER:
Osteopontin-c splicing isoform contributes to ovarian cancer
progression. Mol Cancer Res. 9:280–293. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee S, Choi EJ, Jin C and Kim DH:
Activation of PI3K/Akt pathway by PTEN reduction and PIK3CA mRNA
amplification contributes to cisplatin resistance in an ovarian
cancer cell line. Gynecol Oncol. 97:26–34. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhou B, Sun C, Li N, Shan W, Lu H, Guo L,
Guo E, Xia M, Weng D, Meng L, et al: Cisplatin-induced CCL5
secretion from CAFs promotes cisplatin-resistance in ovarian cancer
via regulation of the STAT3 and PI3K/Akt signaling pathways. Int J
Oncol. 48:2087–2097. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kothari AN, Arffa ML, Chang V, Blackwell
RH, Syn WK, Zhang J, Mi Z and Kuo PC: Osteopontin-A Master
Regulator of Epithelial-Mesenchymal Transition. J Clin Med.
5:52016. View Article : Google Scholar
|
|
34
|
Zhu Y, Gao XM, Yang J, Xu D, Zhang Y, Lu
M, Zhang Z, Sheng YY, Li JH, Yu XX, et al: C-C chemokine receptor
type 1 mediates osteopontin-promoted metastasis in hepatocellular
carcinoma. Cancer Sci. 109:710–723. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Breier A, Gibalova L, Seres M, Barancik M
and Sulova Z: New insight into p-glycoprotein as a drug target.
Anticancer Agents Med Chem. 13:159–170. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Munoz JL, Walker ND, Scotto KW and
Rameshwar P: Temozolomide competes for P-glycoprotein and
contributes to chemoresistance in glioblastoma cells. Cancer Lett.
367:69–75. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Robey RW, Pluchino KM, Hall MD, Fojo AT,
Bates SE and Gottesman MM: Revisiting the role of ABC transporters
in multidrug-resistant cancer. Nat Rev Cancer. 18:452–464. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Luo SD, Chen YJ, Liu CT, Rau KM, Chen YC,
Tsai HT, Chen CH and Chiu TJ: Osteopontin involves cisplatin
resistance and poor prognosis in oral squamous cell carcinoma.
BioMed Res Int. 2015:5085872015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Huang J, Chang S, Lu Y, Wang J, Si Y,
Zhang L, Cheng S and Jiang WG: Enhanced osteopontin splicing
regulated by RUNX2 is HDAC-dependent and induces invasive
phenotypes in NSCLC cells. Cancer Cell Int. 19:3062019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Davidson B, Tropé CG and Reich R:
Epithelial-mesenchymal transition in ovarian carcinoma. Front
Oncol. 2:332012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Huang RYJ, Wong MK, Tan TZ, Kuay KT, Ng
AH, Chung VY, Chu YS, Matsumura N, Lai HC, Lee YF, et al: An EMT
spectrum defines an anoikis-resistant and spheroidogenic
intermediate mesenchymal state that is sensitive to e-cadherin
restoration by a src-kinase inhibitor, saracatinib (AZD0530). Cell
Death Dis. 4:e9152013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chebouti I, Kasimir-Bauer S, Buderath P,
Wimberger P, Hauch S, Kimmig R and Kuhlmann JD: EMT-like
circulating tumor cells in ovarian cancer patients are enriched by
platinum-based chemotherapy. Oncotarget. 8:48820–48831. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Carduner L, Leroy-Dudal J, Picot CR,
Gallet O, Carreiras F and Kellouche S: Ascites-induced shift along
epithelial-mesenchymal spectrum in ovarian cancer cells:
Enhancement of their invasive behavior partly dependant on αv
integrins. Clin Exp Metastasis. 31:675–688. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Techasen A, Loilome W, Namwat N, Dokduang
H, Jongthawin J and Yongvanit P: Cytokines released from activated
human macrophages induce epithelial mesenchymal transition markers
of cholangiocarcinoma cells. Asian Pac J Cancer Prev. 13
(Suppl):115–118. 2012.PubMed/NCBI
|
|
45
|
Sistigu A, Di Modugno F, Manic G and
Nisticò P: Deciphering the loop of epithelial-mesenchymal
transition, inflammatory cytokines and cancer immunoediting.
Cytokine Growth Factor Rev. 36:67–77. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rezaeifard S, Razmkhah M, Robati M,
Momtahan M and Ghaderi A: Cytokines, chemokines, and chemokine
receptors quantitative expressions in patients with ovarian cancer.
Iran J Med Sci. 40:225–232. 2015.PubMed/NCBI
|
|
47
|
Sullivan NJ, Sasser AK, Axel AE, Vesuna F,
Raman V, Ramirez N, Oberyszyn TM and Hall BM: Interleukin-6 induces
an epithelial-mesenchymal transition phenotype in human breast
cancer cells. Oncogene. 28:2940–2947. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fernando RI, Hamilton DH, Huang B and
Palena C: Interleukin-8 drives epithelial-mesenchymal transition of
human carcinomas. J Immunother Cancer. 1 (S1):1872013. View Article : Google Scholar
|
|
49
|
Zhang C, Ma K and Li WY: IL-6 promotes
cancer stemness and oncogenicity in U2OS and MG-63 osteosarcoma
cells by upregulating the OPN-STAT3 pathway. J Cancer.
10:6511–6525. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Learn PA, Yeh IT, McNutt M, Chisholm GB,
Pollock BH, Rousseau DL Jr, Sharkey FE, Cruz AB and Kahlenberg MS:
HER-2/neu expression as a predictor of response to neoadjuvant
docetaxel in patients with operable breast carcinoma. Cancer.
103:2252–2260. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Omarini C, Bettelli S, Caprera C,
Manfredini S, Caggia F, Guaitoli G, Moscetti L, Toss A, Cortesi L,
Kaleci S, et al: Clinical and molecular predictors of long-term
response in HER2 positive metastatic breast cancer patients. Cancer
Biol Ther. 19:879–886. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kwon MJ, Soh JS, Lim SW, Kang HS and Lim
H: HER2 as a limited predictor of the therapeutic response to
neoadjuvant therapy in locally advanced rectal cancer. Pathol Res
Pract. 215:910–917. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Gerson JN, Skariah S, Denlinger CS and
Astsaturov I: Perspectives of HER2-targeting in gastric and
esophageal cancer. Expert Opin Investig Drugs. 26:531–540. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
El Dika I and Ilson DH: Current and future
therapies for targeting HER2 mutations in gastrointestinal cancer.
Expert Rev Anticancer Ther. 18:1085–1092. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Dong Y, Li W, Gu Z, Xing R, Ma Y, Zhang Q
and Liu Z: Inhibition of HER2-positive breast cancer growth by
blocking the HER2 signaling pathway with HER2-glycan-imprinted
nanoparticles. Angew Chem Int Ed Engl. 58:10621–10625. 2019.
View Article : Google Scholar : PubMed/NCBI
|