Introduction
The epidermal growth factor receptor (EGFR) family
is a transmembrane protein receptor with tyrosine kinase activity
(1,2). EGFR is overexpressed in more than 60%
of triple-negative breast cancers (TNBCs), as well as in non-small
cell lung cancers (NSCLC), colorectal cancer (CRC), and
glioblastoma (3,4). It can activate several downstream
signaling pathways related to cancer when the EGFR extracellular
domain binds to its ligands. Given the important role of EGFR in
multiple cancer processes such as proliferation and metastasis,
various tyrosine kinase inhibitors (TKIs) and monoclonal antibodies
(mAbs) have been developed for targeting EGFR in human cancers.
Although EGFR-targeted therapies have shown success in certain
cases, many patients fail to respond because tumors inevitably
develop acquired resistance (5–7).
Therefore, it is crucial to identify other treatments in the
prevention of cancer.
One of the therapeutic methods that can meet this
need is immunotoxins (ITs). ITs are antibody-cytotoxin chimeric
molecules with specific cell killing ability (8). To date, unprecedented progress in
treating hematological tumors has been achieved. Lumoxiti
(moxetumomab pasudotox-tdfk) is a first-class anti-CD22 recombinant
IT that fuses the binding domain of the anti-CD22 antibody to PE38
(9). It has been approved by the
FDA as an intravenous fluid for the treatment of adult patients
with recurrent or refractory hairy cell leukemia (HCL) (9). However, ITs are not as optimistic as
hematologic malignancies in the treatment of solid tumors (10,11).
The capacity of IT to penetrate solid tumors (12), the generation of antitoxin
antibodies (9,13), and their clearance through kidney
filtration (14) all limit the role
of IT therapy in the treatment of solid tumors.
Nanoantibody, also known as single-domain antibody
(sdAb), is the smallest known antigen-binding fragment available,
preserving the full binding capacity of an intact antibody
(15). Compared with conventional
antibodies, the benefits of nanobodies include ease of expression,
low molecular weights, good tissue penetration, high stability and
solubility, and refolding ability; these benefits have granted
their applications for the medical and biotechnological fields
(15).
A novel typical type I ribosome-inactivating protein
(RIP) known as cucurmosin (CUS) was isolated from the sarcocarp of
Cucurbita moschata by our group with a determined DNA
sequence and spatial structure (16). CUS can inhibit the growth of
different human tumor cells in vitro and in vivo,
including, but not limited to, PANC-1 (17), HepG2 (18), CFPAC-1 (19), and SW-1990 (20), but it presents low toxicity to
normal cells. Compared with other RIPs, such as luffaculin and
trichosanthin, the cytotoxicity of CUS is 4- to 7-fold stronger.
These data indicated that CUS can be used as a toxic component of
ITs targeted at tumor cells.
We previously reported an anti-EGFR nanobody
7D12-based recombinant immunotoxin rE/CUS (21), which can selectively kill
EGFR+ cells in vitro. 9G8 is another anti-EGFR
nanobody with different epitope specificities compared with 7D12
(22). Previous findings showed
that 7D12 and 9G8 did not compete for binding to EGFR (22). To improve the binding ability of
rE/CUS and creation of more potential ITs, the bispecific nanobody
7D12-9G8 was used instead of 7D12, which is capable of binding
EGFR, and conjugated CUS using a flexible linker (G4S)3
by genetic engineering methods. The bispecific recombinant IT known
as Bs/CUS is more sensitive to cancer cell lines with EGFR
expression and has a stronger cytotoxic effect than rE/CUS. The
selectivity and cytotoxicity of Bs/CUS illustrate its potential as
a novel candidate for treating EGFR-overexpressing tumors (22).
The aim of the current study was to examine a novel
targeting EGFR recombinant immunotoxin CUS generated by fusing CUS
to the EGFR-specific nanobody 7D12-9G8. The results indicated that
Bs/CUS is a promising candidate that should be further evaluated as
a cancer therapeutic for the treatment of EGFR-positive tumors.
Materials and methods
Reagents
Cetuximab was obtained from Merck. Goat pAb to human
IgG (FITC) (ab81051; Abcam), Rb pAb to 6X His-tag®
(FITC) (ab1206; Abcam), mouse-IgGK BP-HRP (sc-516102; Santa Cruz
Biotechnology), and mouse anti-6X His Tag® antibody
(ab18184; Abcam) were used in the present study. Goat-anti-mouse
IgG-APC (550826; BD Bioscience), and FITC conjugate goat anti-mouse
IgG (AMS.ASS1105-1000; Boster Biological Technology) were also
used. Mouse anti-CUS antibody, 1G9, was produced by our laboratory.
Escherichia coli strain BL21 (DE3), plasmid pET32a (Sangon
Biotech), and Ni-NTA Sepharose FF (GE Healthcare) were
utilized.
Cell lines
Human hepatoma cell line HepG2, human NSCLC cell
line A549, and human CRC cell lines SW1116 and SW620 were obtained
from the cell bank/stem cell bank of the Chinese Academy of
Sciences. All cells were cultured in RPMI-1640 medium (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 10% fetal bovine
serum (Gibco; Thermo Fisher Scientific, Inc.). All the cell lines
used were authenticated by short tandem repeat-based DNA
profiling.
Production and characterization of
recombinant IT
Anti-EGFR bispecific nanobody 7D12-9G8 was cloned
into pET32a expression plasmid-containing CUS (designed Bs/CUS).
The expression and purification of Bs/CUS were performed following
previously described protocol (21).
Western blot analysis and 12% SDS-PAGE gel were used
to analyze the purified protein. The mouse-anti-His tag antibody
and mouse anti-CUS antibody were used as primary antibodies, and
mouse-IgGK BP-HRP was used as the secondary
antibody.
EGFR expression and Bs/CUS binding
capability
Cell surface EGFR expression of A549, HepG2, SW1116,
and SW620 cells and binding ability of Bs/CUS were assessed by flow
cytometry. To evaluate cell surface EGFR expression, cells were
incubated with cetuximab and detected by anti-human-IgG (FITC). To
verify Bs/CUS binding ability, 1 µg of rE/CUS and Bs/CUS was
incubated with the abovementioned cell lines at 4°C for 30 min,
respectively. The mouse-anti-6X His tag antibody was added,
followed by the detection antibody anti-mouse-IgG (APC), and
analyzed by flow cytometry.
In vitro cytotoxicity assay
Sulforhodamine B (SRB) was applied to assess the
cytotoxic activity of drugs as previously described (21). In brief, cells (3×104/ml
per well) were added into 96-well plates in RPMI-1640 medium and
incubated overnight at 37°C in 5% CO2. CUS, 7D12-9G8,
rE/CUS, Bs/CUS, and CUS+7D12-9G8 at different concentrations were
added for 72 and 120 h, respectively. Absorbance was measured by
using an Epoch Microplate Reader (BioTek instrument, Inc.) at 515
nm. Cytotoxic activity was defined by IC50 values.
Apoptosis assay
The FITC-Annexin V apoptosis kit (KeyGen
Biotechnology Co., Ltd.) was used to evaluate the apoptotic effects
of Bs/CUS on HepG2 cells. Cells at a density of 4×104/ml
per well were seeded in 6-well plates, incubated overnight at 37°C
and treated for 72 h with Bs/CUS or kept untreated (UT) at
concentrations of 1, 4, 16, and 64 nmol/l. Subsequently, the cells
were collected, centrifuged at 500 × g for 5 min at room
temperature, counted, 5 µl Annexin V-FITC and 5 µl PI were added
for 15 min at room temperature in the dark for staining,
resuspended in 500 µl binding buffer, and analyzed by flow
cytometry (BD Biosciences).
Cell cycle analysis
Cell cycle changes induced by Bs/CUS treatment were
studied by flow cytometry. In preparation for the cell cycle assay,
HepG2 cells (1×106 cells/well) were seeded in 6-well
plates and incubated overnight at 37°C. The cells were then treated
with Bs/CUS or kept untreated (UT) at concentrations of 1 and 4
nmol/l and incubated with serum-free medium for 24 and 48 h,
respectively. Cells were collected by EDTA-free trypsinization,
fixed in 70% ethanol, and treated with PI solution (50 µg/ml PI and
100 µg/ml RNase A) (Sigma-Aldrich; Merck KGaA) for 30 min at 37°C.
Flow cytometry was used to analyze the cell cycle status of treated
HepG2 cells.
Confocal microscopy
HepG2 cells were seeded in glass bottom cell culture
dishes at a density of 1×105 cells per well and allowed
to attach overnight. The next day, the cells were incubated with 5
µg/ml Bs/CUS for 3, 6, and 9 h, respectively. The cells were washed
with PBS and fixed with 4% paraformaldehyde for 15 min at room
temperature and 0.5% Triton X-100 permeate for 15 min. The cells
were then washed with cold PBS and incubated in blocking buffer
(PBS containing 5% bovine serum albumin) at room temperature for 30
min, followed by incubation with the primary antibody 1G9 at 4°C
overnight. Cells were washed with cold PBS three times and
incubated with the indicated goat anti-mouse IgG (FITC) secondary
antibody for 1 h at room temperature. DAPI was applied to stain the
cell nuclei at room temperature for 15 min. Images were obtained by
confocal microscopy using an LSM 710 system (Carl Zeiss) with 63×
water C-Apochromat objective.
Statistical analysis
Each experiment was repeated in triplicate to
determine the reproducibility of the results. Experimental data
were presented as mean ± SD. A one-way ANOVA followed by Dunnett's
multiple comparison test and Student's t-test were used to analyze
the statistical comparisons. P<0.05 was considered statistically
significant.
Results
Generation and characterization of
Bs/CUS
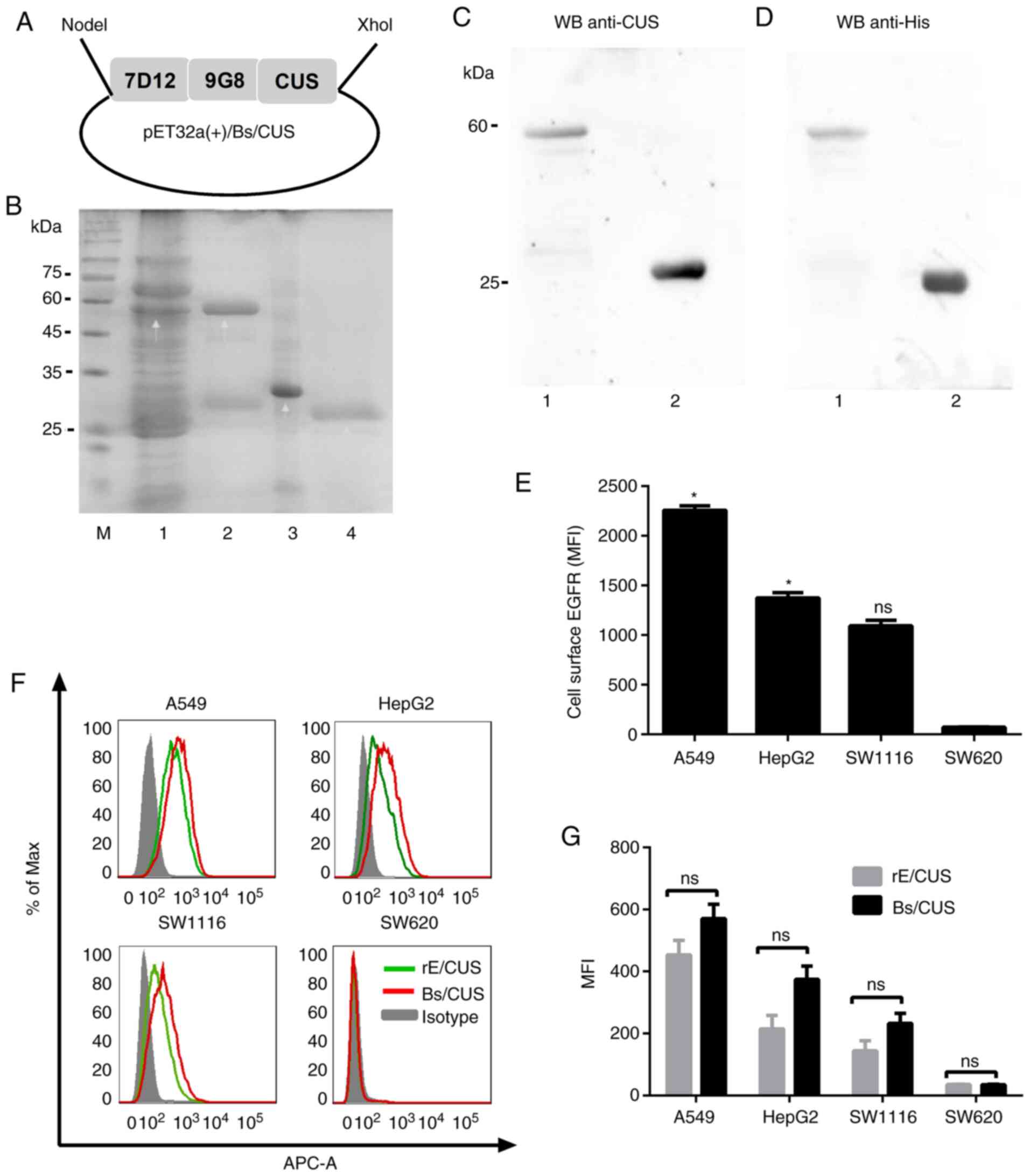
The recombinant plasmid of pET32a(+)/Bs/CUS was
constructed as shown in Fig. 1A.
Bs/CUS and CUS were expressed in E. coli BL21 (DE3) and
analyzed using 12% SDS-PAGE gel (Fig.
1B). Western blot analysis was applied to confirm the
recombinant proteins Bs/CUS and CUS by using 1G9 (Fig. 1C) and mouse-anti-His tag antibody
(Fig. 1D), respectively. The
protein bands were detected at 58 and 28 kDa, which were consistent
with the expected weights of Bs/CUS and CUS, respectively. These
results demonstrated that the bispecific recombinant IT-Bs/CUS was
successfully expressed in the E. coli prokaryotic expression
system and purified.
The EGFR expression at the cell surface level was
analyzed by flow cytometry using cetuximab as a primary antibody.
As shown in Fig. 1E, A549 and HepG2
had the higher EGFR expression level (P<0.05), while SW1116 had
relatively lower expression levels. SW620 cells were considered
EGFR-negative cells.
Flow cytometry was applied to analyze the binding
characteristic of Bs/CUS and compared with that of rE/CUS. Fig. 1F and G show that the fluorescence
intensity of Bs/CUS was higher than that of rE/CUS, indicating that
Bs/CUS had stronger binding activity than rE/CUS. The rank order of
the sensitivities of the cell lines to Bs/CUS in the EGFR
expression level was A549>HepG2>SW1116>SW620, which was
consistent with cetuximab. These results demonstrated that Bs/CUS
retained the binding capacity and specificity with EGFR and was
superior to rE/CUS.
Cytotoxicity in vitro
An SRB-based assay was applied to determine the
specific cytotoxicity of Bs/CUS in vitro. The various tumor
cell lines were incubated with decreasing concentrations of CUS,
7D12-9G8, rE/CUS, Bs/CUS, and CUS + 7D12-9G8 for 72 and 120 h. The
proteins were found to be cytotoxic in a dose-dependent manner on
target cells (Fig. 2A). rE/CUS and
Bs/CUS were significantly more cytotoxic than CUS alone
(P<0.05). For the 72-h experiments, the IC50 of
Bs/CUS to SW1116 was >0.1 nmol/l but decreased to 0.02 nmol/l
for A549 and HepG2 (Table I). At
the 120-h tests, the IC50 values of Bs/CUS to the
abovementioned cell lines were 0.025, 0.008, and 0.008 nmol/l,
respectively, which decreased to 2- to 5-fold lower than that at 72
h (Fig. 3A and Table I), suggesting a time-dependent
cytotoxicity of Bs/CUS. Notably, the ability of Bs/CUS to inhibit
protein synthesis increased by 70-100 times compared with the
rE/CUS previously developed by our group. The highest target index
of Bs/CUS was approximately 22,500, which indicated a 78-fold
increase compared with that of rE/CUS.
 | Table I.IC50 values of cells
treated with drugs for 72 and 120 h. |
Table I.
IC50 values of cells
treated with drugs for 72 and 120 h.
|
| IC50
(Mean ± SD) |
|---|
|
|
|
|---|
| Cell lines | Bs/CUS
(nmol/l) | rE/CUS
(nmol/l) | CUS (nmol/l) | CUS+7D12-9G8
(nmol/l) | IT-1 | IT-2 |
|---|
| A549 (72 h) |
0.018±0.011a |
1.300±0.712b | 215±0.106 | 257±0.097 | 9760 | 102.3 |
| A549 (120 h) |
0.008±0.001a |
0.623±0.172b | 180±0.026 | 170±0.031 | 22500 | 288.9 |
| HepG2 (72 h) |
0.025±0.011a |
2.386±0.624b | 244±0.033 | 337±0.015 | 9760 | 102.3 |
| HepG2 (120 h) |
0.008±0.002a |
0.551±0.163b | 109±0.009 | 187±0.010 | 13625 | 197.8 |
| SW1116 (72 h) |
0.138±0.107a |
7.665±2.602b | 192±0.023 | 261±0.047 | 1391 | 25.05 |
| SW1116 (120 h) |
0.025±0.011a |
1.820±0.884b | 74±0.016 | 72±0.007 | 2960 | 40.66 |
| SW620 (72 h) |
186.8±41.96a |
494.2±73.79b | 550±0.201 | 471±0.089 | 2.944 | 0.001 |
| SW620 (120 h) |
88.89±5.71a |
305±19.83b | 202±0.011 | 169±0.023 | 2.272 | 0.662 |
After CUS and 7D12-9G8 were mixed at a molar ratio
of 1:1, they were incubated with different tumor cells for 72 and
120 h. Results showed that the IC50 of CUS + 7D12-9G8
was consistent with CUS alone. This result suggested that
Bs/CUS-inhibited protein synthesis was not a result of the simple
synergy of the two drugs.
To determine whether the cytotoxic activity of
Bs/CUS is specific, RIT was investigated with the non-target
(EGFR−) cell line SW620. As shown in Fig. 2 and Table I, the cytotoxicity of Bs/CUS did not
increase in comparison with both CUS and CUS + 7D12-9G8.
Annexin V/PI assay was applied to determine whether
the cytotoxicity of Bs/CUS is caused by apoptosis, and the data are
shown in Fig. 2B and C. The results
indicated a significant increase in early and late apoptosis in
Bs/CUS in comparison with the untreated control. The apoptosis rate
increased with the increasing concentration of Bs/CUS. The
percentage of apoptosis was 22.45±0.21, 69.60±0.14, 66.50±0.42 and
74.15±2.33, respectively, in Bs/CUS-treated HepG2 at different
concentrations. When the concentration of Bs/CUS was half that of
rE/CUS (8 nmol/l), the apoptosis rate was higher than that of
rE/CUS. Thus, Bs/CUS was found to be highly efficient in inducing
apoptosis-mediated cell death.
Cell cycle analysis
HepG2 cells were treated with different doses of
Bs/CUS (PBS, 1 and 4 nmol/l) for 24 and 48 h, and flow cytometric
analysis was carried out to detect the cell cycle distribution via
PI staining. For the 24 h test, it was found that Bs/CUS caused
arrest in the G0/G1 phase and the 48 h test compared with the
control group (Fig. 3). The
proportion of cells incubated with 4 nmol/l Bs/CUS significantly
increased in the G0/G1 phase and decreased in the proportion of
cells in the S and G2/M phases (10.58 and 4.81%, respectively;
P<0.0001).
Internalization of Bs/CUS
Indirect immunofluorescence was applied to confirm
the internalization of Bs/CUS in HepG2 cells. As shown in Fig. 4, the fluorescent signal was detected
mainly on the cell surface treated with Bs/CUS for 3 h. For 6 h,
signals started to accumulate in the cytoplasm, and the
fluorescence signal was detected both in the cytoplasm and
membrane. At incubation periods longer than 9 h, the internalized
Bs/CUS protein appeared as evenly distributed dots in the
cytoplasm, while the cell membrane showed no luminescence,
indicating that it had completely entered the cytoplasm.
Discussion
EGFR is overexpressed in various tumors and is
associated with cancer initiation, progression, and poor prognosis,
by mutations, gene amplification, or both through constitutive EGFR
activation (3,4). Cancer therapies targeting EGFR, such
as TKIs and mAbs, have been developed as standard therapies for
several cancers including but not limited to NSCLC and CRC
(23–25). The main limitation of treatments
targeting EGFR is the appearance of acquired resistance.
Immunotoxins function not by suppressing receptor-mediated
signaling but by directly killing the cells. In this study, CUS,
the toxin moiety used is a typical type I RIP. It can hydrolyze the
N-glycosidic bond at A4324 on the 28S rRNA of eukaryotic cells,
irreversibly inactivating the ribosomal 60S subunit, which halts
protein synthesis. Thus, there can be less chance for tumor cells
to upregulate rescue mutations or alternative signaling pathways to
resist immunotoxin therapy (26).
Given the importance of EGFR in solid tumors, we
constructed a recombinant IT by using the anti-EGFR bispecific
nanobody 7D12-9G8 fused to a toxin known as Bs/CUS and demonstrated
the anti-tumor activity of the IT Bs/CUS in vitro. The
conventional recombinant IT molecules are constructed by fusion of
the toxin with scFv or dsFv, which often have the problems of
stability, water solubility, and aggregation (27). Nevertheless, the IT Bs/CUS based on
bispecific nanobody did not have the abovementioned problems,
possibly due to the beneficial outcomes of the nanobody, so it
could be directly expressed in the E. coli system as soluble
proteins.
According to previous findings, when the various
tumor cell lines were incubated with decreasing concentrations of
CUS for 24, 48, 72, 96, and 120 h, the proteins were found to be
cytotoxic in a time-dependent manner on target cells (17–19).
Results of the cytotoxicity assay in vitro showed that the
killing ability of Bs/CUS on different tumor cells was
time-dependent under the conditions of 72 and 120 h incubation.
This result and incubation time are consistent with the research
results of other immunotoxins using CUS as a toxic agent in our
laboratory (28,29).
On the basis of the combination tests of flow
cytometry, SRB cytotoxicity, and apoptosis assay, our results
showed that the IT Bs/CUS had higher EGFR binding capacity and
binding specificity than rE/CUS. These results were consistent with
previous reports showing that the bi-paratopic 9G8-7D12 molecule
had a higher affinity than 7D12 and 9G8 alone, respectively, due to
their different binding sites on the EGFR receptor (22). However, there was no statistical
difference between the affinity of rE/CUS and Bs/CUS. The binding
ability of Bs/CUS was lower than the chemically-linked conjugates,
T-CUS245C, and D-CUS245C, which were
constructed by our group. Therefore, future studies are necessary
to understand the Kd value of Bs/CUS and further improve it. A
comparison with other bispecific ITs also reflected the high
potency against tumors of Bs/CUS, such as dDT2219, an RIT with an
IC50 range of 0.23-1.03 nmol/l against B-cell
malignancies (30).
Nevertheless, we have verified that Bs/CUS exerts
its function of killing tumor cells through the apoptotic pathway
by annexin V/phycoerythrin-based apoptosis assay. To make the
results solid, more verification experiments such as CCK8, DraQ7,
and immunofluorescent staining using apoptosis-related antibody
should be utilized in subsequent investigations. In addition, in
the present study, we preliminarily measured that Bs/CUS induced
HepG2 cell arrest in the G0/G1 phase; however, the molecular
mechanism of cell cycle arrest involved in Bs/CUS remains to be
verified.
Given that the rE/CUS has a molecular weight of
approximately 42 kDa, it could be cleared rapidly in vivo
via the kidneys. Although Bs/CUS is an IT based on the fusion of
bispecific nanobody with a molecular weight of 58 kDa, it may also
face the above problem due to its size under the glomerular
filtration threshold (70 kDa) (31). Accumulating evidence from multiple
trials indicated that binding to albumin is an exceptional option
to prolong the in vivo half-life of small proteins (14,32,33).
Thus, future studies are necessary to verify the effectiveness of
Bs/CUS with the albumin-binding domain in vivo.
Taken together, Bs/CUS demonstrates high
cytotoxicity and selectivity on EGFR-positive cancer cells,
indicating that it could be a promising candidate and it should be
further evaluated as a cancer therapeutic for the treatment of
EGFR-positive tumors.
Acknowledgements
Not applicable.
Funding
This research was funded by the National Science
Foundation of China (no. 30772587), the Natural Science Foundation
of Fujian Province (no. 2016J01769), the Fujian Province Health and
Family Planning Scientific Research Talent Training Project (no.
2018-CX-40), and Fujian Province Student Innovation and
Entrepreneurship Project (no. 201810392045).
Availability of data and materials
The datasets used and analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CZ and YC contributed to study concept and design,
acquisition of data, analysis and interpretation of data, and
drafting of the manuscript; XD performed the experiments; JW
conducted additional experiments; YL contributed to the statistical
analysis; HZ, ML, and JL contributed to the study concept, study
supervision and critical revision of the manuscript; JX contributed
to the study concept and design, study supervision and critical
revision of the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviation
Abbreviations:
|
EGFR
|
epidermal growth factor receptor
|
References
|
1
|
Garrett TPJ, McKern NM, Lou M, Elleman TC,
Adams TE, Lovrecz GO, Zhu HJ, Walker F, Frenkel MJ, Hoyne PA, et
al: Crystal structure of a truncated epidermal growth factor
receptor extracellular domain bound to transforming growth factor
alpha. Cell. 110:763–773. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ogiso H, Ishitani R, Nureki O, Fukai S,
Yamanaka M, Kim JH, Saito K, Sakamoto A, Inoue M, Shirouzu M and
Yokoyama S: Crystal structure of the complex of human epidermal
growth factor and receptor extracellular domains. Cell.
110:775–787. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ciardiello F and Tortora G: EGFR
antagonists in cancer treatment. N Engl J Med. 358:1160–1174. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Furnari FB, Fenton T, Bachoo RM, Mukasa A,
Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, et al:
Malignant astrocytic glioma: Genetics, biology, and paths to
treatment. Genes Dev. 21:2683–2710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Morgillo F, Della Corte CM, Fasano M and
Ciardiello F: Mechanisms of resistance to EGFR-targeted drugs: Lung
cancer. ESMO Open. 1:e0000602016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Misale S, Yaeger R, Hobor S, Scala E,
Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M,
Siravegna G, et al: Emergence of KRAS mutations and acquired
resistance to anti-EGFR therapy in colorectal cancer. Nature.
486:532–536. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lynch TJ, Bell DW, Sordella R,
Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat
SM, Supko JG, Haluska FG, et al: Activating mutations in the
epidermal growth factor receptor underlying responsiveness of
non-small-cell lung cancer to gefitinib. N Engl J Med.
350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Alewine C, Hassan R and Pastan I: Advances
in anticancer immunotoxin therapy. Oncologist. 20:176–185. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kreitman RJ, Tallman MS, Robak T, Coutre
S, Wilson WH, Stetler-Stevenson M, Fitzgerald DJ, Lechleider R and
Pastan I: Phase I trial of anti-CD22 recombinant immunotoxin
moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy
cell leukemia. J Clin Oncol. 30:1822–1828. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hassan R, Bullock S, Premkumar A, Kreitman
RJ, Kindler H, Willingham MC and Pastan I: Phase I study of SS1P, a
recombinant anti-mesothelin immunotoxin given as a bolus I.V.
infusion to patients with mesothelin-expressing mesothelioma,
ovarian, and pancreatic cancers. Clin Cancer Res. 13:5144–5149.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kreitman RJ, Hassan R, Fitzgerald DJ and
Pastan I: Phase I trial of continuous infusion anti-mesothelin
recombinant immunotoxin SS1P. Clin Cancer Res. 15:5274–5279. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mason-Osann E, Hollevoet K, Niederfellner
G and Pastan I: Quantification of recombinant immunotoxin delivery
to solid tumors allows for direct comparison of in vivo and in
vitro results. Sci Rep. 5:108322015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mazor R, Crown D, Addissie S, Jang Y,
Kaplan G and Pastan I: Elimination of murine and human T-cell
epitopes in recombinant immunotoxin eliminates neutralizing and
anti-drug antibodies in vivo. Cell Mol Immunol. 14:432–442. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dennis MS, Zhang M, Meng YG, Kadkhodayan
M, Kirchhofer D, Combs D and Damico LA: Albumin binding as a
general strategy for improving the pharmacokinetics of proteins. J
Biol Chem. 277:35035–35043. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Siontorou CG: Nanobodies as novel agents
for disease diagnosis and therapy. Int J Nanomedicine. 8:4215–4227.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hou X, Meehan EJ, Xie J, Huang M, Chen M
and Chen L: Atomic resolution structure of cucurmosin, a novel type
1 ribosome-inactivating protein from the sarcocarp of Cucurbita
moschata. J Struct Biol. 164:81–87. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang B, Huang H, Xie J, Xu C, Chen M,
Wang C, Yang A and Yin Q: Cucurmosin induces apoptosis of BxPC-3
human pancreatic cancer cells via inactivation of the EGFR
signaling pathway. Oncol Rep. 27:891–897. 2012.PubMed/NCBI
|
|
18
|
Xie J, Que W, Liu H, Liu M, Yang A and
Chen M: Anti-proliferative effects of cucurmosin on human hepatoma
HepG2 cells. Mol Med Rep. 5:196–201. 2012.PubMed/NCBI
|
|
19
|
Xie J, Wang C, Zhang B, Yang A, Yin Q,
Huang H and Chen M: Cucurmosin induces the apoptosis of human
pancreatic cancer CFPAC-1 cells by inactivating the PDGFR-β
signalling pathway. Pharmacol Rep. 65:682–688. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xie J, Wang C, Yang A, Zhang B, Yin Q,
Huang H and Chen M: Cucurmosin kills human pancreatic cancer
SW-1990 cells in vitro and in vivo. Anticancer Agents Med Chem.
13:952–956. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Deng C, Xiong J, Gu X, Chen X, Wu S, Wang
Z, Wang D, Tu J and Xie J: Novel recombinant immunotoxin of EGFR
specific nanobody fused with cucurmosin, construction and antitumor
efficiency in vitro. Oncotarget. 8:38568–38580. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Roovers RC, Vosjan MJ, Laeremans T, el
Khoulati R, de Bruin RC, Ferguson KM, Verkleij AJ, van Dongen GA
and van Bergen en Henegouwen PM: A biparatopic anti-EGFR nanobody
efficiently inhibits solid tumour growth. Int J Cancer.
129:2013–2024. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Greenhalgh J, Dwan K, Boland A, Bates V,
Vecchio F, Dundar Y, Jain P and Green JA: First-line treatment of
advanced epidermal growth factor receptor (EGFR) mutation positive
non-squamous non-small cell lung cancer. Cochrane Database Syst
Rev. CD0103832016.PubMed/NCBI
|
|
24
|
Jean GW and Shah SR: Epidermal growth
factor receptor monoclonal antibodies for the treatment of
metastatic colorectal cancer. Pharmacotherapy. 28:742–754. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee JJ and Chu E: First-line use of
anti-epidermal growth factor receptor monoclonal antibodies in
metastatic colorectal cancer. Clin Colorectal Cancer. 2 (Suppl
6):S42–S46. 2007. View Article : Google Scholar
|
|
26
|
Simon N and FitzGerald D: Immunotoxin
therapies for the treatment of epidermal growth factor
receptor-dependent cancers. Toxins (Basel). 8:1372016. View Article : Google Scholar
|
|
27
|
Cheung LS, Fu J, Kumar P, Kumar A,
Urbanowski ME, Ihms EA, Parveen S, Bullen CK, Patrick GJ, Harrison
R, et al: Second-generation IL-2 receptor-targeted diphtheria
fusion toxin exhibits antitumor activity and synergy with anti-PD-1
in melanoma. Proc Nat Acad Sci U S A. 116:3100–3105. 2019.
View Article : Google Scholar
|
|
28
|
Xiong J, Zhang C, Wu S, Gu X, Cai Y, Xu C,
Chen Z, Sun J, Wu X, You X, et al: Recombinant cucurmosin-based
immunotoxin targeting HER-2 with potent in vitro anti-cancer
cytotoxicity. Biochem Biophys Res Commun. 513:15–21. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang C, Xiong J, Lan Y, Wu J, Wang C,
Huang Z, Lin J and Xie J: Novel cucurmosin-based immunotoxin
targeting programmed cell death 1-ligand 1 with high potency
against human tumor in vitro and in vivo. Cancer Sci.
111:3184–3194. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schmohl JU, Todhunter D, Taras E,
Bachanova V and Vallera DA: Development of a deimmunized bispecific
immunotoxin dDT2219 against B-cell malignancies. Toxins (Basel).
10:322018. View Article : Google Scholar
|
|
31
|
Sanz L, Blanco B and Alvarez-Vallina L:
Antibodies and gene therapy: Teaching old ‘magic bullets’ new
tricks. Trends Immunol. 25:85–91. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Roovers RC, Laeremans T, Huang L, De Taeye
S, Verkleij AJ, Revets H, de Haard HJ and van Bergen en Henegouwen
PMP: Efficient inhibition of EGFR signaling and of tumour growth by
antagonistic anti-EFGR nanobodies. Cancer Immunol Immunother.
56:303–317. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tijink BM, Laeremans T, Budde M,
Stigter-van Walsum M, Dreier T, de Haard HJ, Leemans CR and van
Dongen GAMS: Improved tumor targeting of anti-epidermal growth
factor receptor Nanobodies through albumin binding: Taking
advantage of modular nanobody technology. Mol Cancer Ther.
7:2288–2297. 2008. View Article : Google Scholar : PubMed/NCBI
|