Introduction
Colorectal cancer (CRC) is the third most frequently
diagnosed type of cancer (38.7 per 100,000 individuals between 2012
and 2016 in the USA), causing more than half a million deaths per
year (1). Stage II CRC is an
early-stage in which the tumor has not yet spread to lymph nodes or
distant sites, and that has a low risk of recurrence (2). Stage II CRC is a heterogeneous disease
both clinically and biologically and, in view of this
heterogeneity, the benefits of adjuvant chemotherapy after complete
surgical resection vary widely depending on histopathological and
molecular tumor features (3).
Despite multiple clinical trials and meta-analyses (4), adjuvant chemotherapy in stage II CRC
remains an area of great controversy. To date, identifying patients
who may benefit from adjuvant therapy is difficult. The decision is
mainly based on the presence or absence of high-risk features, such
as poorly differentiated histology, the presence of lymphovascular
and/or perineural invasion, resection of <12 lymph nodes, bowel
obstruction, local perforation or positive margins (5). Consensus Molecular Subtypes of CRC
have been proposed based on genome-scale analyses (6), but CRC cases carrying common driver
events can vary markedly in their biology (7). Nevertheless, at the biological level,
stage II CRC represents a valuable in vivo model for
studying the effect of chemotherapy on comparable cohorts of
patients treated or not treated with chemotherapy.
Homeodomain-interacting protein kinase 2 (HIPK2) is
a tyrosine-regulated serine/threonine kinase that modulates
different cellular processes, including p53-dependent and
-independent apoptosis, differentiation and development (8–11).
HIPK2 has long been considered as an oncosuppressor that serves a
critical role in cell-fate determination during development and in
the response to different genotoxic stresses, such as UV, ionizing
radiation and treatment with chemotherapeutic drugs (12–15).
Consistent with this hypothesis, low HIPK2 expression has been
observed in several types of tumor, including thyroid, bladder,
breast, ovarian and esophageal cancer (16–19).
As a proof of principle, HIPK2-knockdown impairs p53-dependent and
-independent response to therapies, and induces chemoresistance,
angiogenesis and tumorigenicity in hepatocellular carcinoma
(20), whereas HIPK2 overexpression
promotes cell cycle arrest and/or apoptosis, counteracts hypoxia,
inhibits angiogenesis and induces chemosensitivity in esophageal
squamous cell carcinoma and CRC (10,19,21–23).
Conversely, other studies have revealed increased HIPK2 expression
and its association with tumor progression in tumor samples from
patients with pilocytic astrocytoma, aggressive meningioma,
prostate, cervical and colorectal cancer (21,24–27).
The aforementioned data suggest that HIPK2 may behave differently
depending on the cell context or the tumor histotype; however, the
molecular mechanisms need to be defined. Additionally, the HIPK2
protein can undergo post-translational modifications according to
its redox state, changing its activity (28). The recently identified crosstalk
between HIPK2 and nuclear factor erythroid 2-related factor 2
(NRF2), a master regulator of the antioxidant response considered a
‘double-faced’ molecule, revealed for the first time HIPK2
regulation at the mRNA level (29),
and suggested unexpected pro-survival activity by the
NRF2/HIPK2/p53 interplay (30–32).
NRF2 regulates the transcription of drug metabolizing enzymes,
antioxidant enzymes and drug transporters that allow cell
adaptation and survival in oxidant stress conditions (33). When transiently activated in early
tumorigenesis steps, NRF2 works as a cytoprotective factor and is
associated with chemoprevention (34). By contrast, sustained NRF2
activation, as observed in NRF2-overxpressing cancers including
lung cancer and CRC (35–37), is associated with an unfavorable
prognosis (37,38), promotes metabolic activities that
support cell proliferation and tumor growth, and serves a crucial
role in determining drug resistance (34,39).
Therefore, the NRF2 inhibitor brusatol, a natural quassinoid
isolated from a traditional Chinese herbal medicine, Brucea
javanica, and originally employed to treat amebiasis and
malaria (40,41), has been proposed for cancer therapy
in combination with current chemotherapies (42). In particular, it has been
demonstrated that brusatol decreases the levels of NRF2 by
ubiquitin-mediated degradation, impairs the cytoprotective response
and sensitizes a broad spectrum of xenografts and cancer cells,
such as lung adenocarcinoma A549 cells, cervix epithelioid
carcinoma HeLa cells and triple-negative breast cancer MDA-MB-231
cells, to different chemotherapeutic drugs (43,44).
In the present study, tissue microarray (TMA) from
patients with stage II CRC treated or not treated with adjuvant
chemotherapy, and CRISPR/Cas9-mediated gene editing in CRC cells
were used to address the contribution of HIPK2 in drug-response and
its possible use as a predictive marker.
Materials and methods
Patients
The study group consisted of a retrospective series
of stage II CRC cases (n=84) belonging to a cohort of 270 patients
with CRC who underwent curative-intent surgical resection at the
IRCCS Regina Elena National Cancer Institute (Rome, Italy) between
January 2000 and December 2013. The 84 patients with stage II CRC
included in the present study had a median follow-up of 58.46
months (range, 8–144 months). The follow-up estimated using the
Kaplan-Meier reverse method was 61 months (95% CI, 48–75 months),
representing the 5-year period after which the patients that did
not show disease relapse were no longer included in the follow-up.
Tumors were staged according to the tumor-node-metastasis (TNM)
system criteria (45), and all
patients were diagnosed with stage II CRC (T3/T4, N0, M0). Clinical
data were obtained from hospital medical records. As shown in
Table I, 55 patients were males and
29 were females. The median age of the patients at the time of
surgery was 67 years (range, 35–83 years), with 29 patients <65
and 55 patients >65 years old. Of the 84 patients, 24 were
diagnosed with rectal cancer and 60 with colon cancer (23 patients
had right-side colon cancer, 34 had left-side colon cancer and 3
had transverse colon cancer). Most of the patients were stage T3
(83%) and grade G2. According to the presence or absence of ≥1
high-risk features (T4 tumors, obstruction or perforation, <13
examined lymph nodes, positive margins, high-grade tumor,
lympho-vascular and/or perineural invasion), 54 patients underwent
surgery only and 30 underwent surgery plus adjuvant chemotherapy
(Table II). The study was approved
by the Central Ethics Committee of IRCCS Lazio (approval no.
1058/18). All patients signed an informed consent form for their
tissues and clinical information to be used for research purposes
based on previous approved study for tissue banking by the IFO
Ethics Committee (July 7th 2003 and subsequent amendments and
additions).
 | Table I.Clinicopathological characteristics
of patients with stage II colorectal cancer (n=84). |
Table I.
Clinicopathological characteristics
of patients with stage II colorectal cancer (n=84).
| Characteristic | N | % |
|---|
| Age, years |
|
≤65 | 29 | 35 |
|
>65 | 55 | 65 |
| Sex |
|
|
|
Male | 55 | 65 |
|
Female | 29 | 35 |
| Tumor site |
|
|
| Right
colon | 23 | 27 |
| Left
colon | 34 | 40 |
|
Transverse colon | 3 | 4 |
|
Rectum | 24 | 29 |
| T stage |
|
|
| T3 | 71 | 84 |
| T4 | 13 | 16 |
| Grade |
|
|
| G1 | 4 | 5 |
| G2 | 69 | 82 |
| G3 | 11 | 13 |
| Adjuvant
therapy |
|
|
| No | 54 | 64 |
|
Yes | 30 | 36 |
| Table II.Type of adjuvant therapy administered
to patients (n=30). |
Table II.
Type of adjuvant therapy administered
to patients (n=30).
| Type of adjuvant
therapy | Number of treated
patients |
|---|
| FOLFOX (folic acid
+ 5-FU + oxaliplatin) | 9 |
| FOLFOX +
Vectibix | 1 |
| FOLFOX + Vectibix +
radiotherapy | 1 |
| De Gramont (folic
acid + 5-FU) | 9 |
| De Gramont +
radiotherapy | 1 |
| Xeloda
(capecitabine) | 4 |
| Xeloda +
radiotherapy | 2 |
| Not
specifieda | 3 |
TMA construction
For this retrospective cross-sectional study, the
CRC samples were histopathologically re-evaluated on
hematoxylin/eosin stained slides and representative areas were
marked prior to TMA construction. In cases where informative
results on TMA were absent due to missing tissue, no tumor tissue
or unsuccessful staining, the correspondent routine tissue section
was re-analyzed. Two core cylinders (1-mm diameter) were taken from
the CRC samples using a specific arraying device (Alphelys;
Euroclone S.p.A.) and placed into two separate recipient paraffin
blocks. In addition to tumor tissues, recipient blocks received
normal colon tissues from the aforementioned patients (5 cm from
tumor tissues) as negative controls. Sections (2-µm-thick) of the
resulting microarray blocks were made, transferred to SuperFrost
Plus slides (Menzel-Gläser; VWR International, LLC) and used for
immunohistochemistry (IHC).
IHC
IHC staining on TMA was manually performed using an
anti-HIPK2 rat monoclonal antibody (moAb) 5C6 (46) [kindly provided by Professor M.
Lienhard Schmitz (Justus-Liebig-University, Giessen, Germany)]
diluted 1:50 and incubated at room temperature for 30 min, and
detected using an anti-polyvalent diaminobenzidine staining system
containing both blocking reagent and secondary antibody (ULTRATEK
HRP; ScyTek Laboratories, Inc.) according to the manufacturer's
protocol. Images were obtained at a magnification of ×20 using a
light microscope (DM2000 LED; Leica Microsystems GmbH) equipped
with a software able to capture images (Leica Application Suite V4;
version 4.8.0; Leica Microsystems GmbH). The percentages of
HIPK2+ cells were evaluated by manually counting ≥200
cells per sample at high magnification (×40). Evaluation of the IHC
results was performed independently by two blinded
investigators.
Next generation sequencing (NGS)
Paraffin-embedded tumor specimens, fixed for at
least 24 h in 10% buffered formalin at room temperature, were
reviewed for histological verification, as well as to ensure a
minimum tumor cell content of 20%. Genomic DNA was extracted on the
QIAcube® platform using the QIAamp DNA FFPE tissue kit
(cat. no. 56404; Qiagen GmbH) according to the manufacturer's
protocol. Library preparation was performed using an Ion Chef
System (Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. Briefly, barcoded libraries were
generated from 10 ng of DNA per sample using the Ion Ampliseq
Library kit 2.0 and the Ion Ampliseq Cancer Hotspot Panel v2 (both
from Thermo Fisher Scientific, Inc.). Amplified libraries were
quantified using the Qubit 2.0 Fluorometer and the high sensitivity
Qubit Assay kit (both from Thermo Fisher Scientific, Inc.) and
combined to a final concentration of 100 pM. TP53 sequencing
was performed on an Ion S5 Sequencer using an Ion 530 Chip and an
Ion 530 kit-Chef (all from Thermo Fisher Scientific, Inc.). In
particular, to prepare DNA/RNA samples for sequencing, the
following reagents and conditions were employed: Ion Ampliseq™
Library kit plus (cat. no. A35907; Thermo Fisher Scientific, Inc.),
Ion Ampliseq™ Cancer hotspot panel v2 (cat. no. 4475346; Thermo
Fisher Scientific, Inc.), Ion Xpress barcode Adapter 1–16 kit (cat.
no. A4471250; Thermo Fisher Scientific, Inc.), AMPure XP 60 ml
Agencourt (cat. no. A63881; Beckman Coulter, Inc.) and the Ion
library TaqMan Quantification kit (cat. no. 4468802; Thermo Fisher
Scientific, Inc.), which was also used to verify the
quality/integrity of the processed samples. To prepare the
sequencing library, the following reagents and kits were used (all
from Thermo Fisher Scientific, Inc.): Ion S5™ Chef Solutions (cat.
no. A27754), Ion S5™ Chef Supplies (cat. no. A27755), Ion 510™ 520™
530™ Chef Reagents (cat. no. A34018), Ion 530™ Chip kit (cat. no.
A27763), Ion S5™ Sequencing solutions (cat. no. 27767), Ion S5™
Sequencing reagents (cat. no. 27768). The loading concentration of
the final library for DNA sequencing was 33 pM. The nucleotide
length of sequencing was 200 bp and the direction of sequencing was
paired end. Analysis was carried out using Ion Torrent Suite™
Software v5.4 and Ion Reporter™ v5.4 (both Thermo Fisher
Scientific, Inc.). The Torrent Suite™ Software was used to perform
initial quality control, including chip loading density, median
read length and number of mapped reads. The Coverage Analysis
plugin was applied to all data and used to assess amplicon coverage
for regions of interest. Variants were identified by Ion Reporter
filter with a detection threshold of 5% variants. A cut-off of 200X
coverage was applied to all analyses. Only single nucleotide
variants resulting in a non-synonymous amino acid change or a
premature stop codon, and all short indels resulting in either a
frameshift or insertion/deletion of amino acids were selected. The
NGS data have been uploaded at https://gbox.garr.it/garrbox/index.php/s/GVmwVDVYy3FdtZn.
Cells, reagents and transfection
Human HCT116 cells (kindly provided by Professor
Bert Vogelstein (John Hopkins University School of Medicine,
Baltimore, Maryland, USA), HeLa and HeLa HIPK2-null cells (47) (kindly provided by Professor M.
Lienhard Schmitz) and RKO cells stably transfected with
p-Super-control (Ctrl-i) or p-Super-HIPK2-short hairpin (sh)RNA
(HIPK2-i) (21) [kindly provided by
Professor Gabriella D'Orazi (University of Chieti, Chieti, Italy)]
were cultured in DMEM-GlutaMAX (HCT116 and HeLa cells) or
RPMI-GlutaMAX (RKO cells) supplemented with 10% FBS, 100 U/ml
penicillin and 100 µg/ml streptomycin (all from Thermo Fisher
Scientific, Inc.) and maintained in a humid incubator at 37°C in a
5% CO2 environment. Stabilized patient-derived
colorectal tumor-initiating cells, also known as cancer stem cells
(CSCs), kindly provided by Professor Giorgio Stassi (University of
Palermo, Palermo, Italy) and Professor Ruggero De Maria (Università
Cattolica del Sacro Cuore, Rome, Italy), were cultured as
previously described (48). Cells
were X-irradiated with a dose of 20 Gy using an IBL437C irradiator
(CIS Bio International) following the manufacturer's instructions.
Cell death was measured using the Trypan blue exclusion assay (cat.
no. 15250-061; Thermo Fisher Scientific, Inc.) following the
manufacturer's protocol. The following drugs were used in the
present study: 5-fluorouracil (5-FU) and oxaliplatin (OXA)
(supplied by the Regina Elena Pharmacy), and brusatol (cat. no.
SML1868; Sigma-Aldrich; Merck KGaA).
RNA interference was performed using a commercially
available pool of three NRF2-specific small interfering (si)RNAs
(cat. no. sc-37030) and a negative control siRNA (cat. no.
sc-37007) (both from Santa Cruz Biotechnology, Inc.). HCT116 cells
were transfected for 24 h at 37°C with 20 nM siRNA using RNAi-MAX
Lipofectamine® (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions. After 24 h of
transfection, the cells were collected for subsequent analyses.
Cytotoxicity assay
Cell sensitivity to drugs was quantified using the
Cell Proliferation kit II (Roche Diagnostics) according to the
manufacturer's protocol. The method is based on the ability of
viable cells to cleave the tetrazolium ring of
2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxyanilide
(XTT) inner salt yielding orange formazan crystals, which are
soluble in aqueous solutions. Absorbance of formazan was measured
at 450 nm and cell viability was expressed as a percentage of
absorbance measured in the treated wells compared with that in the
untreated control wells. HCT116 cells were treated at 37°C for 48 h
with 2.5, 5, 12.5, 25, 50, 100 and 200 µM 5-FU and OXA. HeLa cells
were treated at 37°C for 48 h with 5, 10, 25, 50 and 100 µM 5-FU
and OXA. RKO cells were treated at 37°C for 48 h with 6.25, 12.5,
25, 50, 100, 200 and 400 µM 5-FU, and 6.25, 12.5, 25 and 50 µM OXA.
Briefly, 8,000 cells/well (HCT116, HeLa and RKO) were seeded in
96-well plates and allowed to recover for 24 h before drug
treatment. The drug concentrations and incubation times are also
indicated in figures and legends. In combination experiments,
brusatol (15 nM for Ctrl-Cas9 cells and 15 and 40 nM for HIPK2-Cas9
cells) was added 4 h before other chemotherapeutics. Subsequently,
HCT116 Ctrl-Cas9 cells were treated with 1, 2.5, 5 and 10 µM 5-FU
and OXA. HCT116 HIPK2-Cas9 cells were treated with 2.5, 5, 10, 20,
40 and 80 µM 5-FU, and 1, 2.5, 5, 10, 20 and 40 µM OXA. RKO-Ctrli
and RKO-HIPK2i cells were treated with 3, 6, 12.5 and 25 µM 5-FU
and OXA.
CRISPR-Cas9 genome editing and
isolation of knock-out clonal cells
HCT116 cells were cultured in 6-well dishes to reach
60–70% confluence and transfected using Lipofectamine®
LTX reagent (Invitrogen; Thermo Fisher Scientific, Inc.) with
plasmids pX459 pSpCas9(BB)-2A-Puro (cat. no. 62988; Addgene, Inc.)
or pX459 Cas9-HIPK2 single guide (sg)RNA
(5′-GTTCCAACTGGGACATGACTG-3′) (kindly provided by Professor M.
Lienhard Schmitz), that targets the second exon of the HIPK2 gene
affecting the kinase domain (47).
Transfected cells were selected the next day using puromycin (ICN
Biomedicals, Inc.) for 3 days. After selection, cells were cloned
into a 96-well plate, expanded and screened for HIPK2 protein
expression via western blotting using the aforementioned anti-HIPK2
rat 5C6 moAb. Genomic DNA was isolated from each clone using the
Quick-gDNA MiniPrep kit (Zymo Research Corp.) according to the
manufacturer's instructions. PCR was performed using the GoTaq DNA
polymerase (Promega Corporation) using primers flanking the edited
region of the HIPK2 gene: HIPK2 exon 2 (294–753) forward,
5′-TGGCCTCACATGTGCAAGTT-3′ and reverse, 5′-GCCCCGCTTGCATTATTCTG-3′.
The following thermocycling conditions were used: one cycle at 94°C
for 3 min, followed by 40 cycles at 94°C for 3 min, 60°C for 30 sec
and 72°C for 30 sec, and a final cycle at 72°C for 7 min. Sanger
sequencing was performed on the genomic DNA PCR products by
Eurofins Genomics.
Characterization of allele-specific
mutations of HIPK2-Cas9 cells by TOPO TA cloning
To identify the allele-specific mutations of HIPK2
present in the HIPK2-Cas9 cells, the TOPO TA Cloning kit
(Invitrogen; Thermo Fisher Scientific, Inc.) was used to directly
insert Taq polymerase-amplified PCR products amplified from the
edited region into the pcDNA 3.1/V5-His-TOPO vector according to
the manufacturer's protocol. The TOPO cloning reactions were then
transformed into TOP10 E. coli competent cells (Invitrogen;
Thermo Fisher Scientific, Inc.). Single colonies were picked, and
the plasmids were isolated by miniprep (Qiagen GmbH). Restriction
analysis using BamHI 10 U/µl and XhoI 10 U/µl (New
England Biolabs, Inc.) was performed to determine the correct
insertion of the PCR product. Correctly inserted clones were
subsequently sequenced and analyzed for the presence of indel
mutations. Nucleotide Blast alignment tool (49) was used to identify the indel
mutations in the HIPK2-Cas9 cells. Bacterial agar plates were
supplemented with 50 µg/ml ampicillin (Sigma-Aldrich; Merck KGaA)
and bacterial colonies were grown in LB broth (Invitrogen; Thermo
Fisher Scientific, Inc.) with ampicillin (Sigma-Aldrich; Merck
KGaA).
Reverse transcription-quantitative
PCR
Total RNA was isolated using the RNeasy mini kit
(cat. no. 74106; Qiagen GmbH) according to the manufacturer's
protocol. RNA was reverse transcribed into cDNA using a M-MLTV
RTase (cat. no. 28025-0.13; Invitrogen; Thermo Fisher Scientific,
Inc.), 5X First Strand buffer (cat. no. Y02321; Invitrogen; Thermo
Fisher Scientific, Inc.), 0.1 M DTT (cat. no. Y00147; Invitrogen;
Thermo Fisher Scientific, Inc.), Primer random P(dN)6
(cat. no. 1034731; Roche Diagnostics), RNase OUT 5,000 units (cat.
no. 100000840; Invitrogen; Thermo Fisher Scientific, Inc.), 100 mM
dCTP solution (cat. no. 55083), 100 mM dTTP solution (cat. no.
55085), 100 mM dATP solution (cat. no. 55082) and 100 mM dGTP
solution (cat. no. 55084) (all Invitrogen; Thermo Fisher
Scientific, Inc.) for 90 min at 37°C. For quantitative PCR
analysis, mRNA expression levels were evaluated using Power
SYBR-Green PCR master mix (cat. no. 4367659; Applied Biosystems;
Thermo Fisher Scientific, Inc.) with ABI Prism 7500HT Fast
Real-Time PCR System Detector (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The thermocycling conditions were: one cycle at
50°C for 2 min; one cycle at 95°C for 10 min; 40 cycles at 95°C for
15 sec and 60°C for 1 min. Gene expression was quantified using the
2−∆∆Cq method (50).
GAPDH was used as the endogenous reference gene. Primer sequences
were as follows: HIPK2 forward, 5′-AGGAAGAGTAAGCAGCACCAG-3′ and
reverse, 5′-TGCTGATGGTGATGACACTGA-3′; GAPDH forward,
5′-TCCCTGAGCTGAACGGGAAG-3′ and reverse,
5′-GGAGGAGTGGGTGTCGCTGT-3′.
Western blotting
Whole-cell lysates were prepared using RIPA lysis
buffer [50 mM Tris-HCl (pH 8), 150 mM NaCl, 0.5% sodium
deoxycholate, 0.1% SDS, 1% NP-40 and 1 mM EDTA] supplemented with
protease-inhibitor mix (Roche Diagnostics) and Halt Phosphatase
Inhibitor Cocktail (cat. no. 1861277; Thermo Fisher Scientific,
Inc.). The extracted proteins were quantified using the Bio-Rad
Protein assay dye (cat. no. 5000006; Bio-Rad Laboratories, Inc.)
and protein samples (20 µg/lane) were separated via SDS-PAGE onto
4–12% gels (cat. no. NW04122B0X; Invitrogen; Thermo Fisher
Scientific, Inc.) and then transferred onto nitrocellulose
membranes (cat. no. 1620112; Bio-Rad Laboratories, Inc.). After
blocking with 5% skimmed dry milk for 1 h at room temperature (cat.
no. 170-6404; Bio-Rad Laboratories, Inc.), membranes were incubated
overnight at 4°C with the aforementioned anti-HIPK2 rat moAb 5C6
(1:200), anti-NRF2 rabbit moAb (1:1,000; cat. no. ab62352; Abcam),
anti-heat shock protein 70 (51)
mouse moAb (1:1,000; cat. no. SAB4200714; Sigma-Aldrich; Merck
KGaA), anti-GADPH mouse moAb (1:1,000; cat. no. sc-32233; Santa
Cruz Biotechnology, Inc.), anti-poly (ADP-ribose) polymerase (PARP)
rabbit polyclonal Ab (1:1,000; cat. no. 9542; Cell Signaling
Technology, Inc.), anti-catalase mouse moAb (H-9; 1:500; cat. no.
sc-271803; Santa Cruz Biotechnology, Inc.) and anti-αtubulin mouse
moAb (1:1,000; cat. no. MAB-10285; Immunological Sciences).
Following incubation with anti-mouse (cat. no. 7076), anti-rabbit
(cat. no. 7074) (both Cell Signaling Technology, Inc.) or anti-rat
IgG (cat. no. 31470; Thermo Fisher Scientific, Inc.) HRP-linked
secondary antibodies (all 1:10,000) for 1 h at room temperature,
immunoreactions were detected using an ECL WB Detection System
(cat. no. RPN2209; GE Healthcare). Western blot bands were
quantified using ImageJ v1.47 (National Institutes of Health).
Statistical analysis
SPSS version 21.0 (IBM Corp.) was used for
statistical evaluations. Data are presented as the mean ± standard
error (SE). Cell viability data were statistically compared using
the unpaired Student's t-test. The association between variables
was tested using Pearson's χ2 test or Fisher's exact
test, as appropriate. The maximally selected Log-Rank statistics
analysis was applied to the HIPK2 (using 5C6 Ab staining)
continuous variable in order to estimate the most appropriate
cut-off values in which the amount of HIPK2 expression is able to
divide patients into groups with different DFS probabilities
(52). Survival curves were
calculated using the Kaplan-Meier method and the log-rank test was
used to assess differences between subgroups. P≤0.05 was considered
to indicate a statistically significant difference.
Results
High HIPK2 expression is associated
with an improved prognosis in adjuvant-treated patients with stage
II CRC
To evaluate the contribution of HIPK2 in
chemotherapy response in humans, HIPK2 expression was analyzed by
IHC on cancer samples from patients with stage II CRC. After
curative-intent surgical resection of the primary tumors, 30/84
patients were treated with adjuvant therapy according to the
presence of ≥1 high-risk features (T4 tumors, obstruction or
perforation, <13 examined lymph nodes, positive margins,
high-grade tumor, lympho-vascular and perineural invasion). The
remaining 54 patients were only routinely checked for tumor
recurrence. TMA sections of the 84 stage II CRC samples were
labelled with anti-HIPK2 moAb and scored based on the number of
HIPK2+ cells. Consistent with previous observations
(53), TMAs from normal colon
tissues exhibited a low level (≤5%) of HIPK2+ cells
(Fig. 1A). By contrast, TMAs from
tumor samples exhibited a broad range of HIPK2 positivity with up
to 50% positive cells (Fig. 1A).
The cut-off value >10% of HIPK2+ positive cells was
statistically determined (52) and
employed to divide the patients into two groups (HIPK2 ≤10 and
>10). In the total cohort of stage II CRC, 32% (27/84) of the
cases exhibited HIPK2 >10, while analyzing the adjuvant-treated
(Chemo) and untreated (No Chemo) subgroups separately, 47% (14/30)
of the cases exhibited HIPK2 >10 in the adjuvant-treated group
and 24% (13/54) in the untreated one (Fig. 1B).
Next, the patients with HIPK2 ≤10 and >10 were
retrospectively evaluated for their 5-year disease-free survival
(DFS) rate. When the total cohort of unselected stage II CRC cases
was analyzed using Kaplan-Meier curves, a complete overlap was
observed between the HIPK2 ≤10 and >10 groups (Fig. 1C). Notably, when the patients were
divided in the two subgroups of adjuvant-treated and untreated
cases, the following was observed: In the adjuvant treated group,
an improved prognosis was observed in the HIPK2 >10 group
compared with in the ≤10 group (Fig.
1D), while in the untreated group, an improved prognosis was
observed with HIPK2 ≤10 compared with >10 (Fig. 1E). Although a statistically
significant difference was not reached due to the small number of
patients in the subgroups, the divergent curves suggest an
association between a high percentage of HIPK2+ cells
and an improved response to therapy.
HIPK2-associated response to adjuvant
therapy is independent of the TP53 gene status
A major target of HIPK2 in cell response to
different types of stress is the tumor suppressor p53 (8–10).
Mutations of the TP53 gene induce resistance to therapy in
in vitro and in vivo models, as well as in patients
with cancer (54,55). Thus, the present study investigated
whether the aforementioned results may be associated with an
enrichment in wild-type TP53 gene status in patients with a
more favorable prognosis. The TP53 gene was sequenced by NGS
in all 84 tumor samples. In the total cohort of stage II CRC,
TP53 mutations were detected in 38% (32/84) of cases
(Fig. 2A). Similar percentages were
maintained among adjuvant-treated and untreated patients, with
mutations detected in 33.3% (10/30) and 40.7% (22/54) of cases,
respectively (Fig. 2A). Next, the
present study analyzed whether there was an association between
HIPK2 expression and the TP53 gene status. The percentage of
cases with TP53 mutations was 35% (20/57) in the HIPK2 ≤10
subgroup and 44% (12/27) in the HIPK2 >10 group (Fig. 2B). This trend was maintained when
the adjuvant-treated and untreated groups were subdivided in the
HIPK2 ≤10 and >10 subgroups (Fig.
2C), indicating that the association between a high percentage
of HIPK2+ cells and improved response to therapy may be
independent of the TP53 gene status.
Generation of HIPK2-null HCT116 CRC
cells
To investigate the contribution of HIPK2 in
chemotherapy response in CRC cells, the expression levels of
endogenous HIPK2 were first evaluated in different CRC cells,
including patient-derived CSCs, via western blotting. As shown in
Fig. 3A, HIPK2 protein expression
was detected in all cell lines and slightly increased after
irradiation (used as a genotoxic stress to detect the activation of
HIPK2) (14) in three out of seven
cell lines (HCT116, CSC1 and RKO). Next, the CRISPR/Cas9 technology
was used to generate HIPK2-null HCT116 CRC cells. HCT116 cells were
transfected with the Cas9-control plasmid
(HCT116Ctrl-Cas9) or the Cas9-HIPK2 sgRNA plasmid
carrying a sgRNA targeting the second exon of the HIPK2 gene
(HCT116HIPK2-Cas9) (Fig.
3B). After puromycin selection, single-cell clones were
expanded and screened for HIPK2 protein expression via western
blotting. Compared with the HCT116Ctrl-Cas9 cells, the
HIPK2 specific band of 130 kDa was not detectable in the
HCT116HIPK2-Cas9 cells (Fig.
3C). As a further control, the genomic DNA from each clone was
isolated and amplified using PCR with primers flanking the edited
region in the HIPK2 gene, within exon 2. The PCR products
were verified by Sanger sequencing to determine the precise editing
that occurred in each clone. From the sequencing analysis, it was
confirmed that a frameshift mutation within exon 2, leading to a
premature stop codon, was present in both HIPK2 alleles of
HCT116HIPK2-Cas9 cells (Fig. S1).
HIPK2-knockout induces resistance of
CRC cells to 5-FU and OXA
5-FU and OXA are frequently used in the treatment of
CRC, including stage II cases (56). To investigate the contribution of
HIPK2 in the sensitivity of CRC cells to these drugs,
HCT116Ctrl-Cas9 and HCT116HIPK2-Cas9 cells
(herein Ctrl-Cas9 and HIPK2-Cas9 cells) were treated with
increasing concentrations of 5-FU or OXA and cell viability was
assessed using XTT 48 h post-treatment. The results revealed that,
compared with Ctrl-Cas9 cells, HIPK2-knockout significantly
decreased cell sensitivity to both drugs (Fig. 4A). Similar results were obtained
with Ctrl-Cas9 and HIPK2-Cas9 HeLa cells (Fig. S2A) and CRC RKO cells stably
transfected with an HIPK2-specific shRNA (21) (Fig.
S2B), indicating that the drug resistance induced by HIPK2
depletion is not cell-specific and independent of the
gene-depletion method.
 | Figure 4.HIPK2 expression and drug response.
(A) Ctrl-Cas9 and HIPK2-Cas9 HCT116 cells (8×103/well)
were exposed to increasing doses of 5-FU (left panel) and OXA
(right panel) for 48 h, and cell viability was measured by XTT
assay. For each point, the percentage compared with the untreated
sample was calculated. Each point represents the mean ± SE of cell
viability at each dose of 5-FU and OXA. (B) Ctrl-Cas9 and
HIPK2-Cas9 HCT116 cells were treated with increasing doses of 5-FU
(left panel) and OXA (right panel) for 48 h, and counted by Trypan
blue-exclusion test. The percentage of cell death for each sample
is reported as the mean ± SE of three independent experiments.
*P<0.05; **P<0.01; ***P<0.001. (C) PARP cleavage was
detected by western blotting in Ctrl-Cas9 and HIPK2-Cas9 HCT116
cells treated with the indicated doses of 5-FU and OXA for 48 h
from one representative experiment. GADPH was used as a loading
control. Densitometric values reported below the PARP blot were
first normalized according to protein amount in the loading
control, then calculated taking the relative NT control as the
reference value. ImageJ software was employed. SE, standard error;
PARP, poly (ADP-ribose) polymerase; 5-FU, 5-fluorouracil; OXA,
oxaliplatin; HIPK2, homeodomain-interacting protein kinase 2; Ctrl,
control; NT, non-tumor; XTT,
2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide. |
Next, the number of dead cells after 48 h of
treatment was evaluated. As expected, a dose-dependent increment in
cell death was observed in the Ctrl-Cas9 cells; by contrast, no
increase in cell death was observed in the HIPK2-Cas9 cells
(Fig. 4B). These results were
confirmed by PARP cleavage analysis. As shown in Fig. 4C, PARP cleavage was clearly
detectable in Ctrl-Cas9 cells in the presence of either drug,
whereas it was scarcely detectable in HIPK2-Cas9 cells, even though
they had been treated with higher doses of OXA and 5-FU than
control cells due to their drug-resistance. Consistently with
previous data (10), the present
results revealed that HIPK2-knockout induced resistance to 5-FU and
OXA in HCT116 CRC cells.
Chemoresistance induced by
HIPK2-knockout is not overcome using the NRF2 inhibitor
brusatol
Sustained activation of NRF2 contributes to
chemoresistance in different types of human cancer, including CRC
(57,58), and NRF2 inactivation by brusatol
sensitizes several cancer cells to different chemotherapeutic
drugs, such as CRC cells to Adriamycin or lung cancer cells to
cisplatin (30,43,44).
Therefore, brusatol was used in combination with 5-FU and OXA to
assess the effect in the current cellular system. It was revealed
that brusatol decreased the protein expression levels of NRF2 in
HCT116 cells in a dose-dependent manner (Fig. 5A). Subsequently, the effect of
brusatol alone was analyzed to identify a dose that did not affect
cell survival on its own. Ctrl-Cas9 and HIPK2-Cas9 HCT116 cells
were treated with increasing concentrations of brusatol (15–100
nM), and cell viability was assessed using XTT. The results
revealed that viability in Ctrl-Cas9 cells was significantly
decreased in response to doses of brusatol >20 nM, while
HIPK2-Cas9 cells were insensitive to brusatol even at the highest
tested concentration (100 nM; Fig.
5B). Thus, a brusatol dose of 15 nM that did not affect cell
viability in Ctrl-Cas9 cells was employed for combination
experiments. Due to the observed brusatol resistance, HIPK2-Cas9
cells were also treated with a higher dose (40 nM). After 4 h of
brusatol treatment, Ctrl-Cas9 and HIPK2-Cas9 cells were treated
with increasing concentrations of 5-FU and OXA and analyzed using
XTT. It was observed that brusatol significantly increased cell
sensitivity to both drugs in HIPK2-proficient cells (Fig. 5C). However, no significant effect
was induced by brusatol treatment, even at the 40 nM dose, in
HIPK2-Cas9 cells (Fig. 5D),
indicating that brusatol did not overcome the resistance to 5-FU
and OXA in HIPK2-knockout cells. Similar results were obtained
using the Ctrl-i and HIPK2-i RKO cells (Fig. S3), excluding the possibility of
cell-specificity and depletion strategy-specificity. Next, to
explore the potential mechanism of this divergent response to
brusatol, the present study analyzed whether the recently described
transcriptional regulation of HIPK2 by NRF2 (26) was also detectable in HCT116 cells.
It was found that NRF2 depletion by RNA interference resulted in
repression of HIPK2 expression both at the protein (Fig. 5E) and mRNA (Fig. 5F) levels, confirming the
NRF2-mediated transcriptional regulation of HIPK2. Additionally, a
constitutive decrease in the expression levels of NRF2 and its
transcriptional target, catalase, was observed in HIPK2-Cas9 cells
(Fig. 5G). This highlighted a
crosstalk between NRF2 and HIPK2, suggesting that brusatol may be
ineffective in HIPK2-Cas9 cells due to the already decreased
expression levels of NRF2. Overall, the present data suggested that
HIPK2 may be required for the chemotherapy sensitization induced by
brusatol.
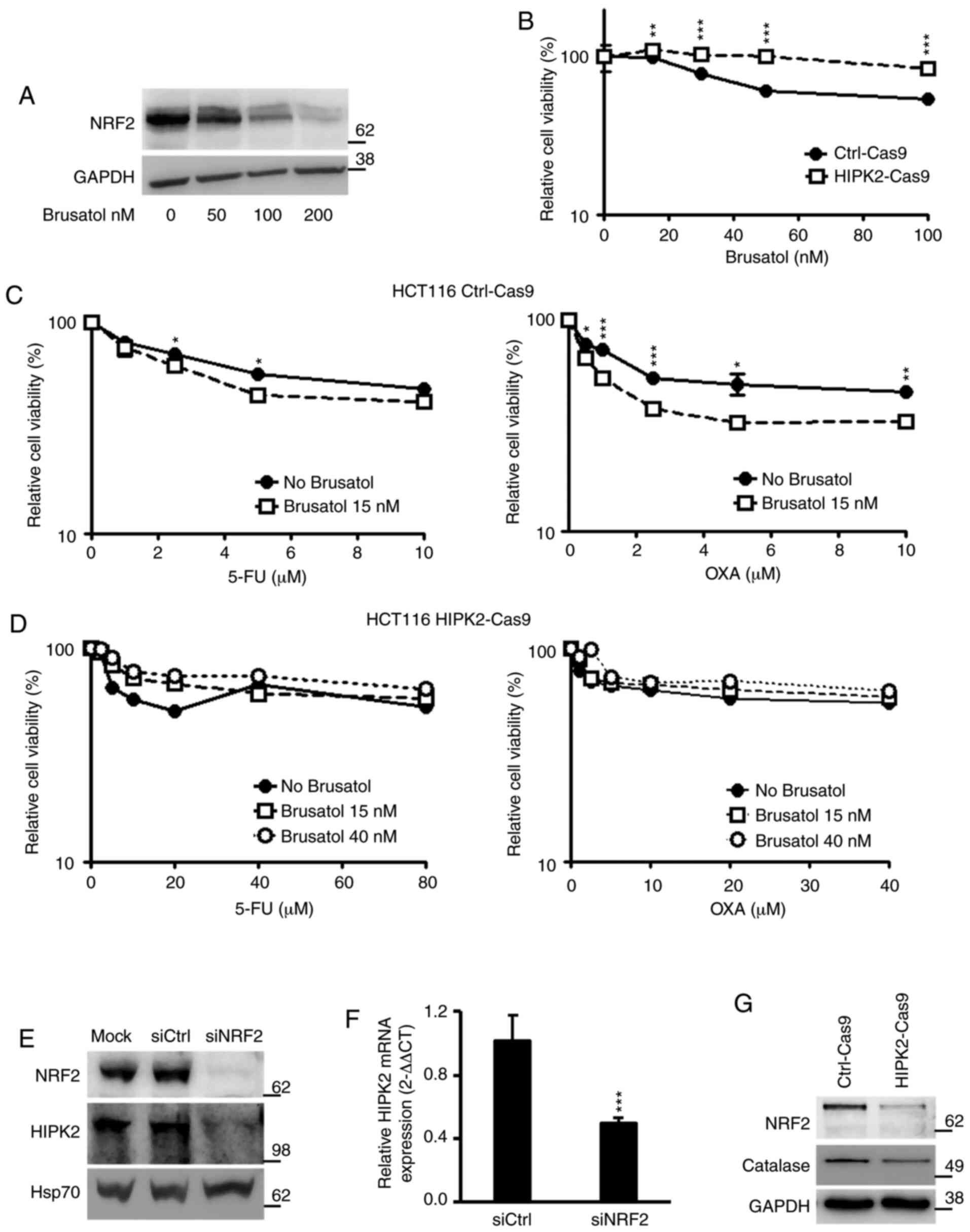 | Figure 5.HIPK2 expression and drug response in
combination with NRF2 inhibition. (A) Parental, HIPK2-proficient
HCT116 cells were treated with increasing doses of brusatol for 4
h, and NRF2 expression was analyzed by WB. GADPH was used as the
protein loading control. (B) Ctrl-Cas9 and HIPK2-Cas9 HCT116 cells
were treated with increasing doses of brusatol, and cell viability
was assessed by XTT after 48 h. Each point represents the
percentage (mean ± SE) of cell viability compared with the
untreated sample. (C) Ctrl-Cas9 cells were treated with increasing
doses of 5-FU (left panel) and OXA (right panel) for 48 h in the
presence or absence of brusatol (15 nM) added 4 h before the other
drugs. The percentage of cell viability compared with the untreated
sample (mean ± SE) was assessed by XTT. (D) HIPK2-Cas9 cells were
treated with 5-FU (left panel) and OXA (right panel) for 48 h in
the presence or absence of brusatol. Two doses of brusatol (15 and
40 nM) were employed on these cells due to their resistance. (E)
siNRF2 and siCtrl were transfected to interfere with NRF2
expression in parental, HIPK2-proficient HCT116 cells. NRF2 and
HIPK2 expression levels were assessed by WB. HSP70 was used as the
protein loading control. (F) HCT116 cells transfected with siNRF2
or siCtrl RNAs were analyzed by reverse transcription-quantitative
PCR to quantify HIPK2 mRNA expression. (G) Protein expression
levels of NRF2 and catalase were detected by WB in Ctrl-Cas9 and
HIPK2-Cas9 HCT116 cells. GADPH was used as the protein loading
control. *P<0.05; **P<0.01; ***P<0.001. XTT,
2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide;
WB, western blotting; si, small interfering; Ctrl, control; SE,
standard error; 5-FU, 5-fluorouracil; OXA, oxaliplatin; HIPK2,
homeodomain-interacting protein kinase 2; NRF2, nuclear factor
erythroid 2-related factor 2; HSP70, heat shock protein 70. |
Discussion
Stage II CRC is an early-stage CRC with a low risk
of recurrence. While therapeutic strategies for the more advanced
CRC stages III and IV are standardized, those for stage II remain
questionable (5). Intense efforts
have focused on developing molecular biomarkers useful as
prognostic factors for identifying stage II patients at increased
risk of disease recurrence, but not for predicting whether stage II
patients would truly benefit from adjuvant chemotherapy (3).
Since HIPK2 is a key player in the cellular response
to different DNA damage-inducing stimuli, including genotoxic
drugs, it is consequently involved in the response of tumor cells
to chemotherapy (10,59). The present study analyzed the role
of HIPK2 in response to chemotherapy and assessed its predictive
role on DFS of patients with stage II CRC. Despite limitations due
to the small sample size that did not allow to obtain statistically
significant differences, the 5-year DFS curves displayed two
opposite trends in adjuvant-treated and untreated subgroups. In the
untreated subgroup, a high percentage of HIPK2+ cells in
the tumor samples (HIPK2 >10) was associated with a lower DFS
time than in those with HIPK2 ≤10. By contrast, HIPK2 >10 cases
in the adjuvant-treated subgroup exhibited a higher DFS time than
the HIPK2 ≤10 cases. The current data are consistent with the
pro-apoptotic and chemosensitivity functions of HIPK2 reported in
several types of cancer cells in vitro and in xenografts,
such as esophageal squamous cell carcinoma cells (19). These data suggest that HIPK2 may
work in a similar manner in experimental systems and at least in
early stage CRC. Furthermore, the present study generated
HIPK2-null HCT116 CRC cells using the CRISPR/Cas9 technology and
assessed their sensitivity to 5-FU and OXA.
Conflicting roles of HIPK2 have been described in
relation to cancer, implicating a tumor-specific and
context-dependent activity of HIPK2, as well as suggesting that it
may work as a versatile protein, which in turn acts as a fine-tuner
of different signaling pathways (60). For instance, a redox-regulated HIPK2
acetylation may restrict its pro-apoptotic activity (28). HIPK2 may regulate the same target
differently, including the oncosuppressor p53, depending on the
cellular context and type, as well as on the stimulus intensity
(28). In addition, mutations in
the TP53 gene contribute to both cancer development
(61) and resistance to cancer
therapy (62). Thus, the present
study investigated whether the TP53 gene status could
explain the different outcomes between adjuvant-treated and
untreated subgroups. No enrichment of the wild-type TP53
gene was observed in the best chemo-responding subgroup. By
contrast, a mild, although not statistically significant,
accumulation of TP53 mutations was observed in
adjuvant-treated patients with HIPK2 >10 and a more favorable
prognosis, suggesting that the drug sensitivity in cells expressing
high HIPK2 expression may be independent of the TP53 gene
status.
Since the TP53 status did not explain the
difference in drug sensitivity, Ctrl-Cas9 and HIPK2-Cas9 HCT116
cells were used to evaluate the possible contribution of the
cytoprotective factor NRF2. This hypothesis stems from different
considerations, including the constitutive activation of NRF2 in
CRC, the role of NRF2 in tumor progression and drug resistance
(58), and the recently identified
pro-survival crosstalk between NRF2 and HIPK2 (29). NRF2 is considered a potential
therapeutic target for improving chemotherapy sensitivity of cancer
cells (63), and the NRF2 inhibitor
brusatol has been demonstrated to possess biological activity in
various types of cancer, including CRC (64), and to improve their chemotherapeutic
response (30). The previously
reported brusatol-mediated repression of NRF2 expression (43) and the NRF2-mediated transcriptional
regulation of HIPK2 (29) were
confirmed in the present study. Notably, when brusatol was used in
combination with 5-FU and OXA, increased chemosensitivity was only
observed in the HIPK2-proficient cells, while no effect was
detected in the HIPK2-null cells, which exhibited decreased NRF2
expression, in accordance with the previous results of the
crosstalk between HIPK2 and NRF2 (26,29).
The current results are in line with the hypothesis that targeting
NRF2 may restore HIPK2 apoptotic activity by increasing the
generation of reactive oxygen species (32), which strongly indicates that NRF2
and HIPK2 expression may be considered as useful markers for
selecting therapeutic options.
In conclusion, the present data revealed that HIPK2
expression was associated with chemo-response in early-stage CRC,
which represents an initial step toward defining a novel predictive
marker for patients with stage II CRC who may benefit from adjuvant
therapy. In addition, the current data demonstrated that the
inhibition of NRF2 induced an increase in chemotherapy response
only when HIPK2 was expressed, indicating that HIPK2 may be a
crucial factor to be considered for combination therapy with NRF2
modulators.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Professor M.
Lienhard Schmitz (Justus-Liebig-University, Giessen, Germany),
Professor Bert Vogelstein (John Hopkins University School of
Medicine, Baltimore, Maryland, USA), Professor Gabriella D'Orazi
(University of Chieti, Chieti, Italy), Professor Giorgio Stassi
(University of Palermo, Palermo, Italy) and Professor Ruggero De
Maria (Università Cattolica del Sacro Cuore, Rome, Italy) for
kindly gifting their cell lines and reagents. Additionally, the
authors would like to thank Dr Maria Pia Gentileschi and Dr Ilaria
Virdia from the IRCCS Regina Elena National Cancer Institute for
their technical assistance, and Mrs. Tania Merlino from the
Scientific Direction of the IRCCS Regina Elena National Cancer
Institute for language editing.
Funding
The present study was supported by grants from
Ministero della Salute (grant no. PE-2016-02361181) and Ricerca
Corrente IRE.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request
Authors' contributions
SS, AV and GDR designed the study. AV and MDS
performed the experiments. CAA performed TMA and NGS. SB performed
NGS. GDR designed and supervised the genome editing experiments.
MDS generated the CRISPR/Cas9-modified cells. IS, SB and MM
analyzed the clinical and NGS data. AV prepared the figures. AV and
SS wrote the manuscript. GDR revised the manuscript. SS and GDR
jointly supervised this work as co-last authors. All authors have
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Specimen collection was approved by the Central
Ethics Committee of IRCCS Lazio (approval no. 1058/18). All
patients signed an informed consent form for their tissues and
clinical information to be used for research purposes based on
previous approved study for tissue banking by the IFO Ethics
Committee (July 7th 2003 and subsequent amendments and
additions).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CRC
|
colorectal cancer
|
|
HIPK2
|
homeodomain-interacting protein kinase
2
|
|
5-FU
|
5-fluorouracil
|
|
OXA
|
oxaliplatin
|
|
NRF2
|
nuclear factor erythroid 2-related
factor 2
|
|
TMA
|
tissue microarray
|
|
IHC
|
immunohistochemistry
|
|
moAb
|
monoclonal antibody
|
|
NGS
|
next generation sequencing
|
|
DFS
|
disease-free survival
|
|
SE
|
standard error
|
|
XTT
|
2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide
|
References
|
1
|
Siegel RL, Miller KD, Goding Sauer A,
Fedewa SA, Butterly LF, Anderson JC, Cercek A, Smith RA and Jemal
A: Colorectal cancer statistics, 2020. CA Cancer J Clin.
70:145–164. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gunderson LL, Jessup JM, Sargent DJ,
Greene FL and Stewart AK: Revised TN categorization for colon
cancer based on national survival outcomes data. J Clin Oncol.
28:264–271. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lee JJ and Chu E: Adjuvant chemotherapy
for stage II colon cancer: The debate goes on. J Oncol Pract.
13:245–246. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Knapen DG, Cherny NI, Zygoura P, Latino
NJ, Douillard JY, Dafni U, de Vries EGE and de Groot DJ: Lessons
learnt from scoring adjuvant colon cancer trials and meta-analyses
using the ESMO-magnitude of clinical benefit scale V.1.1. ESMO
Open. 5:e0006812020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Varghese A: Chemotherapy for stage II
colon cancer. Clin Colon Rectal Surg. 28:256–261. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Guinney J, Dienstmann R, Wang X, de
Reyniès A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda
G, Angelino P, et al: The consensus molecular subtypes of
colorectal cancer. Nat Med. 21:1350–1356. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wolff RK, Hoffman MD, Wolff EC, Herrick
JS, Sakoda LC, Samowitz WS and Slattery ML: Mutation analysis of
adenomas and carcinomas of the colon: Early and late drivers. Genes
Chromosomes Cancer. 57:366–376. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
D'Orazi G, Cecchinelli B, Bruno T, Manni
I, Higashimoto Y, Saito S, Gostissa M, Coen S, Marchetti A, Del Sal
G, et al: Homeodomain-interacting protein kinase-2 phosphorylates
p53 at Ser 46 and mediates apoptosis. Nat Cell Biol. 4:11–19. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hofmann TG, Möller A, Sirma H, Zentgraf H,
Taya Y, Dröge W, Will H and Schmitz ML: Regulation of p53 activity
by its interaction with homeodomain-interacting protein kinase-2.
Nat Cell Biol. 4:1–10. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Puca R, Nardinocchi L, Givol D and D'Orazi
G: Regulation of p53 activity by HIPK2: Molecular mechanisms and
therapeutical implications in human cancer cells. Oncogene.
29:4378–4387. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Blaquiere JA and Verheyen EM:
Homeodomain-interacting protein kinases: Diverse and complex roles
in development and disease. Curr Top Dev Biol. 123:73–103. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nardinocchi L, Puca R, Givol D and D'Orazi
G: HIPK2-a therapeutical target to be (re)activated for tumor
suppression: Role in p53 activation and HIF-1α inhibition. Cell
Cycle. 9:1270–1275. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
D'Orazi G, Rinaldo C and Soddu S: Updates
on HIPK2: A resourceful oncosuppressor for clearing cancer. J Exp
Clin Cancer Res. 31:632012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hofmann TG, Glas C and Bitomsky N: HIPK2:
A tumour suppressor that controls DNA damage-induced cell fate and
cytokinesis. Bioessays. 35:55–64. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kuwano Y, Nishida K, Akaike Y, Kurokawa K,
Nishikawa T, Masuda K and Rokutan K: Homeodomain-interacting
protein kinase-2: A critical regulator of the DNA damage response
and the epigenome. Int J Mol Sci. 17:16382016. View Article : Google Scholar
|
|
16
|
Lavra L, Rinaldo C, Ulivieri A, Luciani E,
Fidanza P, Giacomelli L, Bellotti C, Ricci A, Trovato M, Soddu S,
et al: The loss of the p53 activator HIPK2 is responsible for
galectin-3 overexpression in well differentiated thyroid
carcinomas. PLoS One. 6:e206652011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nodale C, Sheffer M, Jacob-Hirsch J,
Folgiero V, Falcioni R, Aiello A, Garufi A, Rechavi G, Givol D and
D'Orazi G: HIPK2 downregulates vimentin and inhibits breast cancer
cell invasion. Cancer Biol Ther. 13:198–205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tan M, Gong H, Zeng Y, Tao L, Wang J,
Jiang J, Xu D, Bao E, Qiu J and Liu Z: Downregulation of
homeodomain-interacting protein kinase-2 contributes to bladder
cancer metastasis by regulating Wnt signaling. J Cell Biochem.
115:1762–1767. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Z, Wen P, Li F, Yao C, Wang T, Liang
B, Yang Q, Ma L and He L: HIPK2 inhibits cell metastasis and
improves chemosensitivity in esophageal squamous cell carcinoma.
Exp Ther Med. 15:1113–1118. 2018.PubMed/NCBI
|
|
20
|
Chen P, Duan X, Li X, Li J, Ba Q and Wang
H: HIPK2 suppresses tumor growth and progression of hepatocellular
carcinoma through promoting the degradation of HIF-1α. Oncogene.
39:2863–2876. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
D'Orazi G, Sciulli MG, Di Stefano V,
Riccioni S, Frattini M, Falcioni R, Bertario L, Sacchi A and
Patrignani P: Homeodomain-interacting protein kinase-2 restrains
cytosolic phospholipase A2-dependent prostaglandin E2 generation in
human colorectal cancer cells. Clin Cancer Res. 12:735–741. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nardinocchi L, Puca R, Sacchi A and
D'Orazi G: Inhibition of HIF-1alpha activity by
homeodomain-interacting protein kinase-2 correlates with
sensitization of chemoresistant cells to undergo apoptosis. Mol
Cancer. 8:12009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lin J, Zhang Q, Lu Y, Xue W, Xu Y, Zhu Y
and Hu X: Downregulation of HIPK2 increases resistance of bladder
cancer cell to cisplatin by regulating Wip1. PLoS One.
9:e984182014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Al-Beiti MA and Lu X: Expression of HIPK2
in cervical cancer: Correlation with clinicopathology and
prognosis. Aust N Z J Obstet Gynaecol. 48:329–336. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Deshmukh H, Yeh TH, Yu J, Sharma MK, Perry
A, Leonard JR, Watson MA, Gutmann DH and Nagarajan R:
High-resolution, dual-platform aCGH analysis reveals frequent HIPK2
amplification and increased expression in pilocytic astrocytomas.
Oncogene. 27:4745–4751. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cheng Y, Al-Beiti MA, Wang J, Wei G, Li J,
Liang S and Lu X: Correlation between homeodomain-interacting
protein kinase 2 and apoptosis in cervical cancer. Mol Med Rep.
5:1251–1255. 2012.PubMed/NCBI
|
|
27
|
Imberg-Kazdan K, Ha S, Greenfield A,
Poultney CS, Bonneau R, Logan SK and Garabedian MJ: A genome-wide
RNA interference screen identifies new regulators of androgen
receptor function in prostate cancer cells. Genome Res. 23:581–591.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
de la Vega L, Grishina I, Moreno R, Krüger
M, Braun T and Schmitz ML: A redox-regulated SUMO/acetylation
switch of HIPK2 controls the survival threshold to oxidative
stress. Mol Cell. 46:472–483. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Torrente L, Sanchez C, Moreno R, Chowdhry
S, Cabello P, Isono K, Koseki H, Honda T, Hayes JD, Dinkova-Kostova
AT and de la Vega L: Crosstalk between NRF2 and HIPK2 shapes
cytoprotective responses. Oncogene. 36:6204–6212. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Garufi A, Traversi G, Gilardini Montani
MS, D'Orazi V, Pistritto G, Cirone M and D'Orazi G: Reduced
chemotherapeutic sensitivity in high glucose condition: Implication
of antioxidant response. Oncotarget. 10:4691–1702. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Garufi A, Baldari S, Pettinari R,
Gilardini Montani MS, D'Orazi V, Pistritto G, Crispini A, Giorno E,
Toietta G, Marchetti F, et al: A ruthenium(II)-curcumin compound
modulates NRF2 expression balancing the cancer cell death/survival
outcome according to p53 status. J Exp Clin Cancer Res. 39:1222020.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
D'Orazi G, Garufi A and Cirone M: Nuclear
factor erythroid 2 (NF-E2) p45-related factor 2 interferes with
homeodomain-interacting protein kinase 2/p53 activity to impair
solid tumors chemosensitivity. IUBMB Life. 72:1634–1639. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vomund S, Schäfer A, Parnham MJ, Brüne B
and von Knethen A: Nrf2, the master regulator of anti-oxidative
responses. Int J Mol Sci. 18:27722017. View Article : Google Scholar
|
|
34
|
Wu S, Lu H and Bai Y: Nrf2 in cancers: A
double-edged sword. Cancer Med. 8:2252–2267. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li CQ, Kim MY, Godoy LC, Thiantanawat A,
Trudel LJ and Wogan GN: Nitric oxide activation of Keap1/Nrf2
signaling in human colon carcinoma cells. Proc Natl Acad Sci USA.
106:14547–14551. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hu T, Yao Y, Yu S, Guo H, Han L, Wang W,
Tian T, Hao Y, Liu Z, Nan K and Wang S: Clinicopathologic
significance of CXCR4 and Nrf2 in colorectal cancer. J Biomed Res.
27:283–290. 2013.PubMed/NCBI
|
|
37
|
Lee YJ, Kim WI, Bae JH, Cho MK, Lee SH,
Nam HS, Choi IH and Cho SW: Overexpression of Nrf2 promotes colon
cancer progression via ERK and AKT signaling pathways. Ann Surg
Treat Res. 98:159–167. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang Q, Deng H, Xia H, Xu M, Pan G, Mao J,
Tao S, Yamanaka K and An Y: High NF-E2-related factor 2 expression
predicts poor prognosis in patients with lung cancer: A
meta-analysis of cohort studies. Free Radic Res. Jul 24–2019.(Epub
ahead of print). doi: 10.1080/10715762.2019.1642472. View Article : Google Scholar
|
|
39
|
Wang YY, Chen J, Liu XM, Zhao R and Zhe H:
Nrf2-mediated metabolic reprogramming in cancer. Oxid Med Cell
Longev. 2018:93040912018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bawm S, Matsuura H, Elkhateeb A, Nabeta K,
Subeki, Nonaka N, Oku Y and Katakura K: In vitro antitrypanosomal
activities of quassinoid compounds from the fruits of a medicinal
plant, Brucea javanica. Vet Parasitol. 158:288–294. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhao M, Lau ST, Leung PS, Che CT and Lin
ZX: Seven quassinoids from fructus bruceae with cytotoxic effects
on pancreatic adenocarcinoma cell lines. Phytother Res.
25:1796–1800. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Panieri E, Buha A, Telkoparan-Akillilar P,
Cevik D, Kouretas D, Veskoukis A, Skaperda Z, Tsatsakis A, Wallace
D, Suzen S and Saso L: Potential applications of NRF2 modulators in
cancer therapy. Antioxidants (Basel). 9:1932020. View Article : Google Scholar
|
|
43
|
Ren D, Villeneuve NF, Jiang T, Wu T, Lau
A, Toppin HA and Zhang DD: Brusatol enhances the efficacy of
chemotherapy by inhibiting the Nrf2-mediated defense mechanism.
Proc Natl Acad Sci USA. 108:1433–1438. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cai SJ, Liu Y, Han S and Yang C: Brusatol,
an NRF2 inhibitor for future cancer therapeutic. Cell Biosci.
9:452019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Amin MB, Edge S, Greene F, Byrd DR,
Brookland RK, Washington MK, Gershenwald JE, Compton CC, Hess KR,
Sullivan DC, et al: AJCC Cancer Staging Manual. Springer; New York,
NY: pp. 252–254. 2017, PubMed/NCBI
|
|
46
|
de la Vega L, Hornung J, Kremmer E,
Milanovic M and Schmitz ML: Homeodomain-interacting protein kinase
2-dependent repression of myogenic differentiation is relieved by
its caspase-mediated cleavage. Nucleic Acids Res. 41:5731–5745.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ritter O and Schmitz ML: Differential
intracellular localization and dynamic nucleocytoplasmic shuttling
of homeodomain-interacting protein kinase family members. Biochim
Biophys Acta Mol Cell Res. 1866:1676–1686. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Manic G, Signore M, Sistigu A, Russo G,
Corradi F, Siteni S, Musella M, Vitale S, De Angelis ML, Amoreo CA,
et al: CHK1-targeted therapy to deplete DNA replication-stressed,
p53-deficient, hyperdiploid colorectal cancer stem cells. Gut.
67:903–917. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Altschul SF, Gish W, Miller W, Myers EW
and Lipman DJ: Basic local alignment search tool. J Mol Biol.
215:403–410. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-delta delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Contadini C, Monteonofrio L, Virdia I,
Prodosmo A, Valente D, Chessa L, Musio A, Fava LL, Rinaldo C, Di
Rocco G and Soddu S: p53 mitotic centrosome localization preserves
centrosome integrity and works as sensor for the mitotic
surveillance pathway. Cell Death Dis. 10:8502019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hothorn T and Lausen B: On the exact
distribution of maximally selected rank statistics. Comput Stat
Data Anal. 43:121–137. 2003. View Article : Google Scholar
|
|
53
|
Zhou L, Feng Y, Jin Y, Liu X, Sui H, Chai
N, Chen X, Liu N, Ji Q, Wang Y and Li Q: Verbascoside promotes
apoptosis by regulating HIPK2-p53 signaling in human colorectal
cancer. BMC Cancer. 14:7472014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Vousden KH and Prives C: Blinded by the
light: The growing complexity of p53. Cell. 137:413–431. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Muller PA and Vousden KH: Mutant p53 in
cancer: New functions and therapeutic opportunities. Cancer Cell.
25:304–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kukcinaviciute E, Jonusiene V,
Sasnauskiene A, Dabkeviciene D, Eidenaite E and Laurinavicius A:
Significance of Notch and Wnt signaling for chemoresistance of
colorectal cancer cells HCT116. J Cell Biochem. 119:5913–5920.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Sadeghi MR, Jeddi F, Soozangar N, Somi MH
and Samadi N: The role of Nrf2-Keap1 axis in colorectal cancer,
progression, and chemoresistance. Tumour Biol.
39:10104283177055102017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Rojo de la Vega M, Chapman E and Zhang DD:
NRF2 and the hallmarks of cancer. Cancer Cell. 34:21–43. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Feng Y, Zhou L, Sun X and Li Q:
Homeodomain-interacting protein kinase 2 (HIPK2): A promising
target for anti-cancer therapies. Oncotarget. 8:20452–20461. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Calzado MA, Renner F, Roscic A and Schmitz
ML: HIPK2: A versatile switchboard regulating the transcription
machinery and cell death. Cell Cycle. 6:139–143. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Kandoth C, McLellan MD, Vandin F, Ye K,
Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al:
Mutational landscape and significance across 12 major cancer types.
Nature. 502:333–339. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Huang Y, Liu N, Liu J, Liu Y, Zhang C,
Long S, Luo G, Zhang L and Zhang Y: Mutant p53 drives cancer
chemotherapy resistance due to loss of function on activating
transcription of PUMA. Cell Cycle. 18:3442–3455. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
No JH, Kim YB and Song YS: Targeting nrf2
signaling to combat chemoresistance. J Cancer Prev. 19:111–117.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Evans JP, Winiarski BK, Sutton PA, Jones
RP, Ressel L, Duckworth CA, Pritchard DM, Lin ZX, Fretwell VL,
Tweedle EM, et al: The Nrf2 inhibitor brusatol is a potent
antitumour agent in an orthotopic mouse model of colorectal cancer.
Oncotarget. 9:27104–27116. 2018. View Article : Google Scholar : PubMed/NCBI
|

















