Introduction
Pancreatic adenocarcinoma, one of the most
aggressive human malignancies, has the propensities of difficult
diagnosis, early metastasis and resistance to treatment. Globally,
a reported 458,918 new cases and 432,242 associated deaths were
caused by pancreatic adenocarcinoma in 2018 (1). Due to a lack of effective treatments,
the estimated five-year survival rate for patients with pancreatic
adenocarcinoma has remained at <5%, which is the lowest among
all types of cancers in general (1–3). To date,
surgical resection is considered to be the most effective curative
treatment, however, <20% of patients are eligible at the time of
diagnosis (4). In addition, the
majority of patients will eventually experience recurrence, and the
five-year survival rate of patients who undergo complete surgical
resection remains <25% (4). Other
therapies for the treatment of pancreatic adenocarcinoma, including
chemotherapy and radiotherapy, are important, but remain largely
ineffective (5). Gemcitabine
(2,2-difluorodeoxycytidine, dFdC) is a standard, first-line
compound that was approved for the treatment of metastatic and
non-metastatic but locally advanced pancreatic adenocarcinoma in
1996 (6–8); however, resistance to this drug is a
considerable limitation in disease treatment. Therefore, the
investigation and application of novel targeted therapeutics that
are less susceptible to intrinsic drug resistance, and with
improved antitumor effects, are crucial to the successful treatment
of pancreatic adenocarcinoma.
Anaplastic lymphoma kinase (ALK) belongs to one of
the subfamilies of tyrosine kinases for the insulin receptor, and
initiates a number of important cancer-associated signaling
pathways. As such, ALK has been associated with the development of
anaplastic large-cell lymphoma, non-small cell lung cancer, diffuse
large B-cell lymphoma, inflammatory myofibroblastic tumors,
neuroblastoma, anaplastic thyroid cancer, rhabdomyosarcoma and
pancreatic adenocarcinoma (9–12). ALK activation promotes a variety of
functional properties such as cell survival, proliferation,
differentiation and invasiveness through the modulation of the
downstream signaling pathways of mediators including PI3K/AKT,
MEK/ERK and STAT3 (9,12–14).
Previous studies have revealed that the level of ALK
phosphorylation is higher in pancreatic tumors than in normal
pancreatic tissues; this was achieved by analyzing ALK expression
in patient tissues, and inhibiting ALK activity using crizotinib
(an inhibitor of c-MET/ALK), or ceritinib (a well-known ALK
inhibitor with antitumor effects against pancreatic adenocarcinoma)
(10,11). Combination treatment with ceritinib
and gemcitabine also significantly inhibited the growth and/or
survival of pancreatic adenocarcinoma (10). Moreover, NVP-TAE688, a well-known
inhibitor of ALK, has been revealed to induce cell death in
different types of cancer, including anaplastic large-cell
lymphoma, non-small cell lung cancer, neuroblastoma and large
B-cell lymphoma (15–20). In osteosarcoma, the inhibition of ALK
by NVP-TAE684 sensitized cell apoptosis when used in combination
with chemotherapeutic agents such as doxorubicin, paclitaxel,
docetaxel or vincristine (21).
Similar results were observed following the combined use of
NVP-TAE684 and radiotherapy in patients with non-small cell lung
cancer (22). Therefore, using
NVP-TAE684 to inhibit ALK activity may be a clinically effective
treatment option for a number of cancer types, including solid
tumors. However, the antitumor effects of NVP-TAE684 in pancreatic
adenocarcinoma have yet to be fully investigated.
Numerous cytotoxic and/or ALK-targeting agents
inhibit tumor cell survival and proliferation by promoting cell
cycle arrest at the G0/G1, S or G2/M phases (15,17,19,23–26).
The G2/M phase is one of two major checkpoints for cell cycle
regulation. Following DNA damage, cells are retained in G2/M and
prevented from entering mitosis, which provides an opportunity for
DNA repair and prevents the proliferation of damaged cells
(27). Several studies have revealed
that NVP-TAE684 and alectinib induced cell cycle arrest at the
G0/G1 phase in anaplastic large-cell lymphoma, non-small cell lung
cancer and neuroblastoma cells (15,17,19,25).
In addition, crizotinib was revealed to induce G2/M arrest in
ovarian cancer and non-small cell lung cancer cells (23,24).
However, the molecular mechanisms of NVP-TAE684 in pancreatic
adenocarcinoma cells have not been studied thus far.
In the present study, the antitumor effects of
NVP-TAE684 were investigated using human pancreatic adenocarcinoma
cells. The results indicated that NVP-TAE684 inhibited cellular
proliferation and induced G2/M arrest and apoptotic cell death by
targeting ALK in multiple human pancreatic adenocarcinoma cell
lines.
Materials and methods
Cell culture and reagents
The human pancreatic adenocarcinoma AsPC-1, Capan-1
and Colo-357 (Tissue Culture and Biobanking Shared Resource;
Georgetown University Lombardi Comprehensive Cancer Center) and
BxPC-3 cell lines (ATCC) were maintained in RPMI-1640 media
containing fetal bovine serum (FBS; 20% for AsPC-1; and 10% for
Capan-1, Colo-357 and BxPC-3 cells), 100 U/ml penicillin, 100 µg/ml
streptomycin and 1% sodium pyruvate at 37°C in a humidified
atmosphere containing 5% CO2 as previously described
(28,29). MIA PaCa-2 human pancreatic
adenocarcinoma cells (ATCC) were maintained in Dulbecco's modified
Eagles' medium (DMEM) supplemented with 10% FBS, 2.5% horse serum,
100 U/ml penicillin and 100 µg/ml streptomycin at 37°C in a
humidified atmosphere containing 5% CO2 as previously
described (28,29). The Panc-1 and CFPAC-1 human pancreatic
adenocarcinoma cell lines (ATCC) were maintained in DMEM
supplemented 10% FBS, 10 U/ml penicillin and 10 µg/ml streptomycin
at 37°C in a humidified atmosphere containing 5% CO2
(28,29). The HPDE6-C7 immortal human pancreatic
ductal epithelial cell line (provided by Dr Tsao, Montreal General
Hospital and McGill University, Montreal, Canada) was cultured in
keratinocyte serum-free medium supplemented with an epidermal
growth factor, bovine pituitary extract and 1X
antibiotic-antimycotic at 37°C in a humidified atmosphere
containing 5% CO2 as previously described (30). Serum starvation was carried out by
replacing the culture medium with fresh medium without FBS, and
incubating for 24 h. The cell culture reagents were obtained from
BioWhittaker (Lonza Group, Ltd.) and Invitrogen (Thermo Fisher
Scientific, Inc.). NVP-TAE684 was purchased from Selleck Chemicals
and dissolved in DMSO (Sigma-Aldrich; Merck KGaA).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
The human pancreatic adenocarcinoma cells (AsPC-1,
Panc-1, MIA PaCa-2, Capan-1, CFPAC-1, Colo-357 and BxPC-3) were
counted using the Luna™ Cell Counter (Logos Biosystems, Inc.) and
seeded into 96-well flat-bottom plates at a density of
2×103 cells/well. The cells were then exposed to
NVP-TAE684 alone or in combination with gemcitabine at the
indicated concentrations (0, 0.01, 0.1, 1 or 10 µM), and incubated
at 37°C for 72 h. After incubation, 10 µl MTT (1 mg/ml;
Sigma-Aldrich; Merck KGaA) in PBS was added to each well and the
cells were incubated at 37°C for a further 4 h. Following
centrifugation at 1000 × g for 2 min and removal of the medium, 150
µl DMSO was added to each well to dissolve the formazan crystals.
The absorbance was measured at 560 nm using an ELx808 Absorbance
Microplate Reader (BioTek Instruments, Inc.) as previously
described (28,29,31).
Western blot analysis
Human pancreatic adenocarcinoma cells were cultured
to ~70% confluence and NVP-TAE684 was added at the indicated
concentrations (0, 0.01, 0.1 or 1 µM). After exposure to
NVP-TAE684, the cells were lysed in cell lysis buffer (20 mM
Tris-HCl, 0.5 M NaCl, 0.25% Triton X-100, 1 mM EDTA, 1 mM EGTA, 10
mM β-glycophosphate, 10 mM NaF, 300 µM
Na3VO4, 1 mM benzamidine, 2 µM PMSF and 1 mM
DTT), and the protein concentrations were determined using a
bicinchoninic acid protein assay kit (Thermo Fisher Scientific,
Inc.). The proteins were separated by SDS-PAGE, transferred to PVDF
membranes, blocked in 1X blocking buffer at room temperature for 1
h (Sigma-Aldrich; Merck KGaA) and probed with primary antibodies at
4°C for overnight against the following: Phospho-ALK (Y1604)
(dilution 1:250; product. no. 3341S), ALK (dilution 1:1,000;
product. no. 3633S), phospho-AKT (S473) (dilution 1:500; product.
no. 4060S), AKT (dilution 1:2,000; product. no. 4685S),
phospho-ERK1/2 (Y202/T204) (dilution 1:1,000; product. no. 4370S),
ERK1/2 (dilution 1:2,000; product. no. 9102S), phospho-STAT3 (Y705)
(dilution 1:250; product. no. 9145S) (all Cell Signaling
Technology, Inc.) and α-tubulin (dilution 1:2,000; product. no.
T6074) (Sigma-Aldrich; Merck KGaA). The membranes were then
incubated with horseradish peroxidase (HRP)-conjugated goat
anti-mouse (dilution 1:2,000; product. no. A9917) or anti-rabbit
(dilution 1:2,000; product. no. 12-348) secondary antibodies
(Sigma-Aldrich; Merck KGaA) at room temperature for 1 h and
visualized using a chemiluminescence kit (Santa Cruz Biotechnology,
Inc.) according to the manufacturer's protocol. The membranes were
subsequently exposed to X-ray film (American X-ray and Medical
Supply, Inc.) and developed as previously described (28,29,31).
Flow cytometry
Human pancreatic adenocarcinoma cells were treated
with NVP-TAE684 and harvested by trypsinization. The cells were
washed with PBS and fixed overnight in 70% ethanol at −20°C. The
cells were then incubated with 20 µg/ml propidium iodide (BD
Biosciences) and 40 µg/ml RNase A (BD Biosciences) in 1X PBS, and
analyzed using a FACSCalibur flow cytometer (BD Biosciences) as
previously described (28,29,31).
Caspase-3/7 activity assay
Caspase-3/7 activity was determined using the
Caspase-Glo® 3/7 Assay System (Promega Corporation)
according to the manufacturer's protocol. MIA PaCa-2 and Colo-357
cells were treated with NVP-TAE684 alone or in combination with
gemcitabine at the indicated concentrations (0, 0.1, 1 or 10 µM),
and the caspase-3/7 activity of the cell lysates was determined.
Luminescence was measured at 490 nm using a VICTOR X multilabel
plate reader (PerkinElmer, Inc.) as previously described (32).
Small interfering RNA (siRNA)
transfection
For the RNA interference experiments, 100 nM of each
ALK siRNA (#1) (5′-AAUACUGACAGCCACAGGCAAUGUC-3′), ALK siRNA (#2)
(5′-UUAGGUGGGACAGUACAGCUUCCCU-3′) and the control siRNA
(5′-GACGAGCGGCACGUGCACA-3′) were purchased from Bioneer
Corporation. MIA PaCa-2 cell transfection was conducted using
Lipofectamine® 2000 at 37°C for 6 h (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
After 48 h the transfected cells were processed for western
blotting, cell proliferation, cell cycle analysis, and caspase-3/7
activity assay as previously described (28,29).
Trypan Blue exclusion assay
The cell monolayers were harvested by
trypsinization, resuspended in complete medium, stained at room
temperature for 3 min with trypan blue and counted. The number of
viable cells was calculated using the Luna™ Cell Counter as
previously described (28).
Statistical analysis
The two-tailed Student's t-test was used for two
group comparisons and the one-way ANOVA with post hoc Tukey's
honest significant difference (HSD) test was used to analyze the
significance of the differences for more than two group
comparisons. Data are expressed as the mean ± standard deviation
(SD), and values of P<0.05 and P<0.01 were considered to
indicate a statistically significant and highly statistically
significant differences, respectively (33).
Results
ALK activity is induced in human
pancreatic adenocarcinoma cells and immortal human pancreatic
ductal epithelial cells
Firstly, the phosphorylation levels of ALK at Y1604
in human pancreatic adenocarcinoma cells (AsPC-1, Panc-1, MIA
PaCa-2, Capan-1, CFPAC-1, Colo-357 and BxPC-3) and immortal human
pancreatic duct epithelial cells (HPDE6-C7) was assessed, following
culture in complete or serum-free medium (serum starvation) to
identify whether serum affects the phosphorylation levels of ALK at
Y1604. The western blot results revealed that all cells cultured in
normal culture condition with the addition of serum possessed high
levels of ALK phosphorylation at Y1604 (Fig. 1). Furthermore, it was observed that
all cells cultured in serum-starved condition also possessed high
levels of ALK phosphorylation at Y1604 (Fig. 1). These data indicated that the high
phosphorylation levels of ALK are not influenced by the presence of
serum. In order to investigate the antitumor effects of NVP-TAE684
in pancreatic adenocarcinoma, all of the aforementioned human
pancreatic adenocarcinoma cell lines were used for further
experimentation.
NVP-TAE684 induces apoptotic cell
death
NVP-TAE684 is a potent and selective inhibitor of
ALK. In order to investigate the antitumor effects of NVP-TAE684 in
pancreatic adenocarcinoma, AsPC-1, Panc-1, MIA PaCa-2, Capan-1,
CFPAC-1, Colo-357 and BxPC-3 cells were treated with various
concentrations of NVP-TAE684 (0, 0.01, 0.1, 1 and/or 10 µM) for 72
h, and cell viability was assessed using an MTT assay. NVP-TAE684
significantly reduced the number of viable cells in all cell lines
in a dose-dependent manner (Fig. 2).
In addition, the IC50 of NVP-TAE684 was determined to be
0.85±0.005, 0.81±0.01, 0.29±0.002, 0.86±0.012, 0.44±0.007,
0.66±0.009 and 0.25±0.006 in AsPC-1, Panc-1, MIA PaCa-2, Capan-1,
CFPAC-1, Colo-357 and BxPC-3 cells, respectively (Table I). Furthermore, MIA PaCa-2, BxPC-3 and
CFPAC-1 cells were highly sensitive to NVP-TAE684, while Colo-357,
AsPC-1, Panc-1 and Capan-1 cells appeared to be less sensitive
(Fig. 2).
 | Table I.Antitumor effects of NVP-TAE684 in
human pancreatic adenocarcinoma cells. |
Table I.
Antitumor effects of NVP-TAE684 in
human pancreatic adenocarcinoma cells.
|
| IC50
(µM) |
|---|
|
|
|
|---|
| Human pancreatic
adenocarcinoma cells | AsPC-1 | Panc-1 | MIA PaCa-2 | Capan-1 | CFPAC-1 | Colo-357 | BxPC-3 |
|---|
| NVP-TAE684 | 0.85±0.005 | 0.81±0.01 | 0.29±0.002 | 0.86±0.012 | 0.44±0.007 | 0.66±0.009 | 0.25±0.006 |
To further investigate the mechanisms by which
NVP-TAE684 promotes apoptosis and identify the molecular mechanisms
of sensitivity and/or resistance of pancreatic adenocarcinoma cells
to NVP-TAE684, MIA PaCa-2 cells (which were found to be highly
sensitive to NVP-TAE684) and Colo-357 cells (which were less
sensitive to NVP-TAE684) were treated with various concentrations
of NVP-TAE684 (0, 0.1, 1 and/or 10 µM) for 24 h. Apoptosis was
detected using caspase-3/7 activity analysis, and a significant
increase in caspase-3/7 activity was observed in both cell lines
cells following treatment with 1 and 10 µM NVP-TAE684 (Fig. 3). Collectively, these data indicated
that NVP-TAE684 significantly reduced proliferation and induced
apoptosis in human pancreatic adenocarcinoma cells.
NVP-TAE684 induces G2/M and sub-G1
arrest
Next, the NVP-TAE684-induced inhibitory effects on
ALK on cell cycle progression were investigated. MIA PaCa-2 and
Colo-357 cells were treated for 24 h with the indicated
concentrations of NVP-TAE684, and their cell cycle profiles were
flow cytometrically assessed. NVP-TAE684 significantly promoted
cell cycle arrest at the G2/M phase [from 17.5 to 74.7% in MIA
PaCa-2 cells (1 µM), and from 14.1 to 73.2% in Colo-357 cells (10
µM)] and significantly decreased the number of cells in the G0/G1
phase (from 49.6 to 22.5% in MIA PaCa-2 cells, and from 56.8 to
22.4% in Colo-357 cells) and S phase (from 32.9 to 2.8% in MIA
PaCa-2 cells, and from 29.1 to 4.4% in Colo-357) (Fig. 4A). An increase in the sub-G1
population was also observed following the administration of
NVP-TAE684 (at various concentrations), though this was more
apparent in MIA PaCa-2 than Colo-357 cells (Fig. 4B). Collectively, these data indicated
that NVP-TAE684 significantly promoted cell cycle arrest at the
G2/M phase in human pancreatic adenocarcinoma cells.
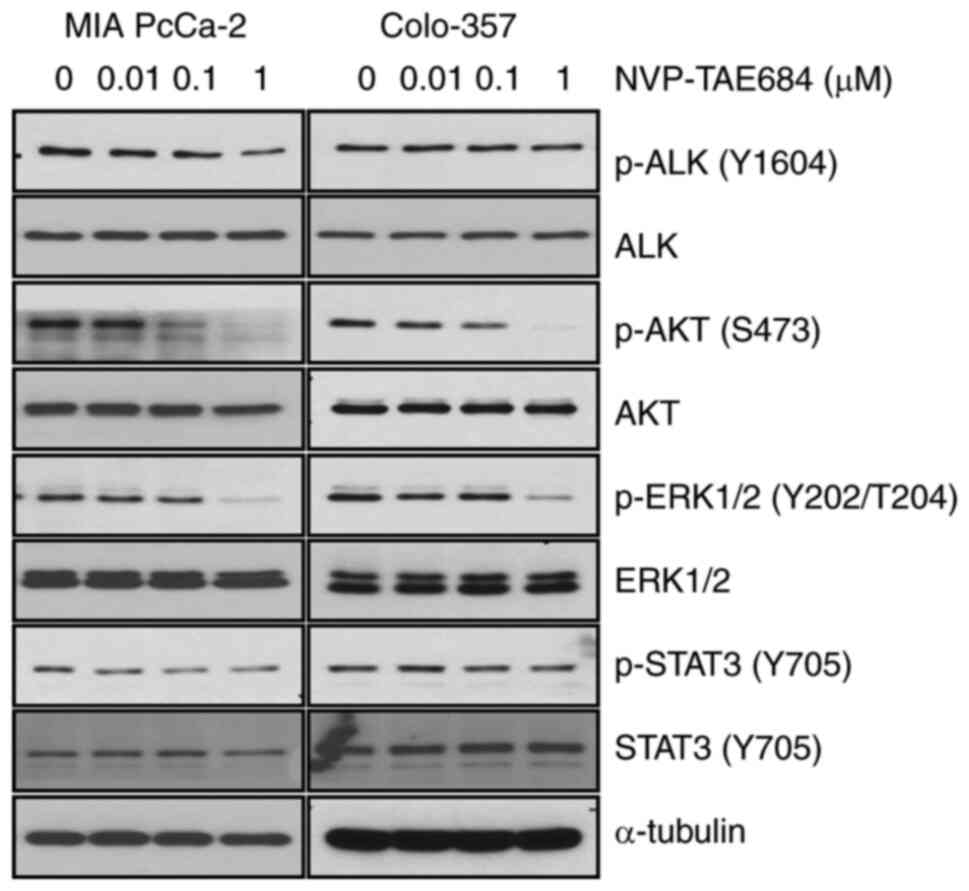
NVP-TAE684 decreases ALK activity
In order to identify whether the decrease in cell
proliferation, and the increase in apoptosis and G2/M arrest were
associated with the inhibition of ALK phosphorylation, MIA PaCa-2
and Colo-357 cells were treated with NVP-TAE684 (0, 0.01, 0.1
and/or 1 µM) for 8 h. NVP-TAE684 markedly reduced the levels of ALK
phosphorylation at Y1604 in both cell lines (Fig. 5). Furthermore, the phosphorylation
levels of downstream mediators of the ALK signaling pathway, such
as AKT, ERK1/2 and STAT3, were also determined following NVP-TAE684
treatment. Under these conditions, NVP-TAE684 also markedly reduced
the phosphorylation levels of AKT (at S473) and ERK1/2 (at
Y202/T204), and to a lesser degree, STAT3 (at Y705) in both cell
lines (Fig. 5). Collectively, these
findings demonstrated that the antitumor effects of NVP-TAE684 in
human pancreatic adenocarcinoma cells were closely associated with
the inhibition of ALK phosphorylation and mostly through
significant reduction of AKT and ERK1/2 phosphorylation.
ALK-knockdown decreases cellular
proliferation and induces apoptotic death and G2/M arrest
Since the NVP-TAE684-induced inhibition of ALK
inhibited cellular proliferation, and induced apoptotic cell death
and G2/M arrest, MIA PaCa-2 cells were transfected with either ALK
siRNA (#1 or #2) or the control siRNA to compare the effects of
NVP-TAE684 and ALK-knockdown, and to confirm the antitumor effects
of NVP-TAE684. At a concentration of 0.1 µM ALK siRNA (#1 or #2),
ALK-knockdown decreased the levels of total and phosphorylated ALK
in MIA PaCa-2 cells (Fig. 6A). Under
these conditions, the phosphorylation levels of AKT (S473) and
ERK1/2 (Y202/T204) but not STAT3 (Y705) were also decreased in
these cells (Fig. 6A). In addition,
knocking down ALK decreased cell survival (Fig. 6B) and induced apoptotic cell death, as
indicated by the induction of caspase-3/7 activity (Fig. 6C) and the accumulation of cells in the
sub-G1 phase (from 8.4 in the control group to 25.2% in
the ALK siRNA (#1), and 17.1% in the ALK siRNA (#2) groups
(Fig. 6E). Furthermore, ALK-knockdown
promoted cell cycle arrest at the G2/M phase from 14.5 in the
control group to 47.7% in the ALK siRNA (#1), and 31.9% in the ALK
siRNA (#2) groups. The cell population in the G0/G1 phase was also
decreased from 39.6% in the control group to 24.6 and 24.3% in the
ALK siRNA (#1) and (#2) groups, respectively, and the proportion of
cells in the S phase was reduced from 45.9% in the control group to
24.6 and 43.8% in the ALK siRNA (#1) and (#2) groups, respectively
(Fig. 6D). Collectively, targeting
ALK with either NVP-TAE684 or ALK siRNA reduced survival, induced
apoptosis and promoted G2/M arrest in human pancreatic
adenocarcinoma cells.
Synergistic cytotoxic effects of
NVP-TAE684 and gemcitabine
In order to investigate the potential beneficial
effects of NVP-TAE684 and gemcitabine combination therapy, MIA
PaCa-2, Colo-357, AsPC-1 and BxPC-3 cells were treated with
NVP-TAE684 and gemcitabine for 72 h at the indicated
concentrations. The combination of NVP-TAE684 and gemcitabine
synergistically inhibited cellular proliferation (Fig. 7A). To further investigate the
synergism between NVP-TAE684 and gemcitabine, both compounds were
used to assess the induction of apoptotic cell death in MIA PaCa-2
and Colo-357 cells. The cells were treated with either NVP-TAE684
or gemcitabine alone, or a combination of both drugs, at the
indicated concentrations for 24 h, and a caspase-3/7 assay was
performed to evaluate apoptosis. Compared with cells treated with
either drug alone, the combination of NVP-TAE684 and gemcitabine
synergistically increased apoptosis in both cell lines by
significantly inducing caspase-3/7 activity (Fig. 7B). Furthermore, in order to confirm
whether inhibiting ALK enhanced sensitivity to gemcitabine, MIA
PaCa-2 cells pretreated with either ALK siRNA (#1 or #2) or the
control siRNA were incubated with the indicated concentrations of
gemcitabine for 72 h. The results revealed that a combination of
ALK siRNA and gemcitabine more effectively reduced proliferation
than a combination of the control-siRNA and gemcitabine (Fig. 7C). Collectively, these results
indicated that targeting ALK with NVP-TAE684 or ALK siRNA enhanced
gemcitabine-induced cell death in human pancreatic adenocarcinoma
cells by inducing apoptosis.
Discussion
In the present study, the mechanisms underlying the
antitumor effects of NVP-TAE684, a potent and selective inhibitor
of ALK, were investigated in human pancreatic adenocarcinoma cells.
The results indicated that the levels of ALK phosphorylation were
increased in human pancreatic adenocarcinoma cell lines (AsPC-1,
Panc-1, MIA PaCa-2, Capan-1, CFPAC-1, Colo-357 and BxPC-3) and
immortal human pancreatic duct epithelial cells (HPDE6-C7), and
that NVP-TAE684 inhibited cell survival in all of the pancreatic
adenocarcinoma cell lines investigated. Furthermore, NVP-TAE687
significantly induced G2/M phase cell cycle arrest and apoptotic
cell death, and decreased the phosphorylation of ALK and downstream
members of the ALK signaling pathway. To further confirm the
effects of NVP-TAE687, ALK siRNA-knockdown also reduced cell
survival and induced G2/M arrest and apoptotic cell death;
additionally, the inhibition of ALK with NVP-TAE684 or siRNA
enhanced gemcitabine-induced apoptosis. To the best of our
knowledge, the present study is the first to report that
NVP-TAE684-induced ALK inhibition reduced cell viability and
induced apoptosis and G2/M phase cell cycle arrest in human
pancreatic adenocarcinoma cells.
A number of small molecular kinase inhibitors have
been developed to target ALK and its downstream signaling-pathway
proteins, the effects of which have been confirmed in various
cancer types, including anaplastic large-cell lymphoma, non-small
cell lung cancer, neuroblastoma, large B-cell lymphoma and
pancreatic adenocarcinoma (10,11,15–20).
Notably, using NVP-TAE684 to retard ALK activity exerted
significant antitumor effects in anaplastic large-cell lymphoma,
non-small cell lung cancer, neuroblastoma and large B-cell lymphoma
(15–20). In addition, NVP-TAE684-associated ALK
inhibition decreased cell survival and induced apoptosis in
osteosarcoma, which was enhanced by combination treatment with
chemotherapeutic drugs such as doxorubicin, paclitaxel, docetaxel
and vincristine (21), and with
radiotherapy in non-small cell lung cancer (22). Furthermore, targeting ALK activity
with crizotinib or ceritinib resulted in significant antitumor
effects against pancreatic adenocarcinoma (10,11), and
inhibiting ALK with ceritinib significantly enhanced the
sensitivity of pancreatic adenocarcinoma cells to gemcitabine
(10). However, there are currently
no studies focused on the antitumor effects and molecular
mechanisms of NVP-TAE684 in pancreatic adenocarcinoma, or
comparisons with effective ALK inhibitors such as NVP-TAE 684,
crizotinib or ceritinib in the treatment of other types of cancer
as previously reported (10,11,15–20). In
the present study, seven human pancreatic adenocarcinoma cell lines
with relatively high ALK phosphorylation levels at Y1604 were
revealed to be sensitive to NVP-TAE684 treatment. Among them, MIA
PaCa-2, BxPC-3 and CFPAC-1 cells exhibited high sensitivity to
NVP-TAE684, and Colo-357, AsPC-1, Panc-1 and Capan-1 cells were
relatively less sensitive to NVP-TAE684. Notably,
NVP-TAE684-induced ALK inhibition also significantly enhanced the
antitumor effects of gemcitabine in pancreatic adenocarcinoma
cells. Therefore, these developments indicated the possible
clinical significance of targeted therapy with well-known ALK
inhibitors such as NVP-TAE684, ceritinib and/or crizotinib for the
effective treatment of various cancers with high ALK activity,
including pancreatic adenocarcinoma.
The G2/M checkpoint is an important regulatory point
of the cell cycle (27), and cell
cycle arrest at this phase indicates that damaged DNA is difficult
to repair (34,35). Previously, ALK inhibition using
crizotinib was revealed to promote G2/M arrest in A2780 and SKOV3
ovarian cancer cells (23) and
non-small cell lung cancer cells including HCC78, SPC-A1 and PC-9
(24,36). On the other hand, inhibition of ALK
with NVP-TAE684 induced G1 arrest in H2228 and HCC78 non-small cell
lung cancer cells (17,36), Karpas-299 anaplastic large-cell
lymphoma cells (15) and LM1 diffuse
large B-cell lymphoma cells (19). In
the present study, NVP-TAE684 induced cell cycle arrest at the G2/M
phase in MIA PaCa-2 and Colo-357 pancreatic adenocarcinoma cells.
However, the molecular mechanisms of NVP-TAE684-associated cell
cycle arrest could not be demonstrated, which may be due to the
increased downregulation of CD30 and off-target inhibition of
aurora kinase (15,36,37).
It is well known that ALK activates various
downstream signaling pathways including those of AKT, ERK1/2 and
STAT3 which regulate cellular proliferation, survival, division and
invasion (9,12–14).
Therefore, to determine the ability of NVP-TAE684 to target
downstream signaling pathway proteins, western blot analysis was
performed using pancreatic adenocarcinoma cells treated with
various concentrations of NVP-TAE684. Among the proteins of these
downstream signaling pathways, NVP-TAE684 effectively inhibited the
phosphorylation of AKT (S473) and ERK1/2 (Y202/T204), and to a
lesser degree, STAT3 (Y705) in pancreatic adenocarcinoma cells,
indicating that NVP-TAE684-induced ALK inhibition effectively
reduced the activities of these signaling pathways. Overall, it was
hypothesized that the antitumor effects of NVP-TAE684 were closely
associated with the inhibition of ALK signaling, which ultimately
resulted in a reduction in cell survival and the induction of
apoptotic cell death and G2/M arrest in pancreatic adenocarcinoma
cells.
In conclusion, the findings of the present study
demonstrated that inhibiting ALK activity with NVP-TAE684 reduced
cell survival and induced apoptotic cell death and G2/M arrest in
pancreatic adenocarcinoma cells. However, there are still some
limitations to our research. First, the effect(s) of NVP-TAE684 on
cell survival of normal cell lines was not investigated; second,
western blot images of cleaved caspase-3 expression are required to
examine the induction of apoptotic cell death by NVP-TAE684; third,
animal model study/patient-derived xenograft experiments need to be
performed to better investigate the important role of NVP-TAE684 in
pancreatic adenocarcinoma cells; last the molecular mechanisms of
the antitumor effects of NVP-TAE684, alone or in combination with
other chemotherapeutic drugs such as gemcitabine, require further
investigation, NVP-TAE684 may be a novel compound for the treatment
of patients with pancreatic adenocarcinoma.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Foundation for Science and Technology Development (NAFOSTED; grant
no. 108.06-2020.03) (HQD).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HQD, VTT, THD and YSS conceived and designed the
study. HQD, VTT, HTN, PTN, HTHT, TNHB, VPLD, TTD and KSY conducted
the experiments and performed the statistical analysis. HQD, VTT
and YSS wrote the manuscript. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A, Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ryan DP, Hong TS and Bardeesy N:
Pancreatic adenocarcinoma. N Engl J Med. 371:1039–1049. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kamisawa T, Wood LD, Itoi T and Takaori K:
Pancreatic cancer. Lancet. 388:73–85. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Oberstein PE and Olive KP: Pancreatic
cancer: Why is it so hard to treat? Therap Adv Gastroenterol.
6:321–327. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Moore M: Activity of gemcitabine in
patients with advanced pancreatic carcinoma. A review. Cancer.
78:633–638. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Carmichael J, Fink U, Russell RC, Spittle
MF, Harris AL, Spiessi G and Blatter J: Phase II study of
gemcitabine in patients with advanced pancreatic cancer. Br J
Cancer. 73:101–105. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Casper ES, Green MR, Kelsen DP, Heelan RT,
Brown TD, Flombaum CD, Trochanowski B and Tarassoff PG: Phase II
trial of gemcitabine (2,2-difluorodeoxycytidine) in patients with
adenocarcinoma of the pancreas. Invest New Drugs. 12:29–34. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Roskoski R Jr: Anaplasmic lymphoma kinase
(ALK): Structure, oncogenic activation and pharmacological
inhibition. Pharmacol Res. 68:68–94. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yan HH, Jung KH, Son MK, Fang Z, Kim SJ,
Ryu YL, Kim J, Kim MH and Hong SS: Crizotinib exhibits antitumor
activity by targeting ALK signaling not c-MET in pancreatic cancer.
Oncotarget. 5:9150–9168. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jamshed MB, Munir F, Shahid N, Sadiq U,
Muhammad SA, Ghanem NB, Zhong H, Li X and Zhang Q: Antitumor
activity and combined inhibitory effect of ceritinib with
gemcitabine in pancreatic cancer. Am J Physiol Gastrointest Liver
Physiol. 318:G109–G119. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Y, Wang L..Guan S, Cao W, Wang H,
Chen Z, Zhao Y, Yu Y, Zhang H, Pang JC, et al: Novel ALK inhibitor
AZD3463 inhibits neuroblastoma growth by overcoming crizotinib
resistance and inducing apoptosis. Sci Rep. 6:194232016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chiarle R, Voena C, Ambrogio C, Piva R and
Inghirami G: The anaplastic lymphoma kinase in the phathogenesis of
cancer. Nat Rev Cancer. 8:11–23. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mossé YP, Wood A and Maris JM: Inhibition
of ALK signaling for cancer therapy. Clin Cancer Res. 15:5609–5614.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Galkin AV, Melnick JS, Kim S, Hood TL, Li
N, Li L, Xia G, Steensma R, Chopiuk G, Jiang J, et al:
Identification of NVP-TAE684, a potent, selective and efficacious
inhibitor of NPM-ALK. Proc Natl Acad Sci USA. 104:270–275. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
McDermott U, Iafrate AJ, Gray NS, Shioda
T, Classon M, Maheswaran S, Zhou W, Choi HG, Smith SL, Dowell L, et
al: Genomic alternations of anaplastic lymphoma kinase may
sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer
Res. 68:3389–3395. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li Y, Ye X, Liu J, Zha J and Pei L:
Evaluation of EML4-ALK fusion proteins in non-small cell lung
cancer using small molecule inhibitors. Neoplasia. 13:1–11. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Van Roosbroeck K, Cools J, Dierickx D,
Thomas J, Vandenberghe P, Stul M, Delabie J, De Wolf-Peeters C,
Marynen P and Wlodarska I: ALK-Positive large B-cell lymphomas with
cryptic SEC31A-ALK and NPM1-ALK fusions. Haematologica. 95:509–513.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cerchietti L, Damm-Welk C, Vater I,
Klapper W, Harder L, Pott C, Yang SN, Reiter A, Siebert R, Melnick
A and Woessmann W: Inhibition of anaplastic lymphoma kinase (ALK)
activity provides a therapeutic approach for CLTC-ALK-positive
human diffuse large B cell lymphomas. PLoS One. 6:e184362011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Duijkers FA, Gaal J, Meijerink JP,
Admiraal P, Pieters R, de Krijger RR and van Noesel MM: Anaplastic
lymphoma kinase (ALK) inhibitor response in neuroblastoma is highly
correlated with ALK mutation status, ALK mRNA and protein levels.
Cell Oncol (Dordr). 34:409–417. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ye S, Zhang J, Shen J, Gao Y, Li Y, Choy
E, Cote G, Harmon D, Mankin H, Gray NS, et al: NVP-TAE684 reverses
multidrug resistance (MDR) in human osteosarcoma by inhibiting
P-glycoprotein (PGP1) function. Br J Pharmacol. 173:613–626. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dai Y, Wei Q, Schwager C, Hanne J, Zhou C,
Herfarth K, Rieken S, Lipson KE and Debus J: Oncogene addition and
radiation oncology: effect of radiotherapy with photons and carbon
ions in ALK-EML4 translocated NSCLC. Radiat Oncol. 13:12018.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Huang XX, Xie FF, Hou LJ, Chen XX, Ou RY,
Yu JT, Qiu JG, Zhang WJ, Jiang QW, Yang Y, et al: Crizotinib
synergizes with cisplatin in preclinical models of ovarian cancer.
Am J Transl Res. 9:1667–1679. 2017.PubMed/NCBI
|
|
24
|
Liu Z, Jiang L, Li Y, Xie B, Xie J, Wang
Z, Zhou X, Jiang H, Fang Y, Pan H and Han W: Cyclosporine A
sensitizes lung cancer cells to crizotinib through inhibition of
the Ca2+/calcineurin/erk pathway. EBioMedicine.
42:326–339. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Alam MW, Borenäs M, Lind DE,
Cervantes-Madris D, Umapathy G, Palmer RH and Hallberg B:
Alectinib, an anaplastic lymphoma kinase inhibitor, abolishes ALK
activity and growth in ALK-positive neuroblastoma cells. Front
Oncol. 9:5792019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Osawa T, Davies D and Hartley JA:
Mechanism of cell death resulting from DNA interstrand
cross-linking in mammalian cells. Cell Dealth Dis. 2:e1872011.
View Article : Google Scholar
|
|
27
|
Stark GR and Taylor WR: Analyzing the G2/M
checkpoint. Methods Mol Biol. 280:51–82. 2004.PubMed/NCBI
|
|
28
|
Duong HQ, Hwang JS, Kim HJ, Kang HJ, Seong
YS and Bae I: Aldehyde dehydrogenase 1A1 confers intrinsic and
acquired resistance to gemcitabine in human pancreatic
adenocarcinoma MIA PaCa-2 cells. Int J Oncol. 41:855–861. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Duong HQ, Hwang JS, Kim HJ, Seong YS and
Bae I: BML-275, an AMPK inhibitor, induces DNA damage, G2/M arrest
and apoptosis in human pancreatic cancer cells. Int J Oncol.
41:2227–2236. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Furukawa T, Duquid WP, Rosenberg L,
Viallet J, Galloway DA and Tsao MS: Long-term culture and
immortalization of epithelial cells from normal adult human
pancreatic ducts transfected vy the E6E7 gene of human papilloma
virus 16. Am J Pathol. 148:1763–1770. 1996.PubMed/NCBI
|
|
31
|
Duong HQ, Kim HJ, Kang HJ, Seong YS and
Bae I: ZSTK474, a PI3K inhibitor, suppresses proliferation and
sensitizes human pancreatic adenocarcinoma cells to gemcitabine.
Oncol Rep. 27:182–188. 2012.PubMed/NCBI
|
|
32
|
Duong HQ, Hong YB, Kim JS, Lee HS, Yi YW,
Kim YJ, Wang A, Zhao W, Cho CH, Seong YS and Bae I: Inhibition of
checkpoint kinase 2 (CHK2) enhances sensitivity of pancreatic
adenocarcinoma cells to gemcitabine. J Cell Mol Med. 17:1261–1270.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
You KS, Yi YW, Kwak SJ and Seong YS:
Inhibition of RPTOR overcomes resistance to EGFR inhibition in
triple-negative breast cancer cells. Int J Oncol. 52:828–840.
2018.PubMed/NCBI
|
|
34
|
Laiho M and Latonen L: Cell cycle control,
DNA damage checkpoints and cancer. Ann Med. 35:391–397. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lobrich M and Jeggo PA: The impact of a
negligent G2/M checkpoint on genomic instability and cancer
induction. Nat Rev Cancer. 7:861–869. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Davies KD, Le AT, Theodoro MF, Skokan MC,
Aisner DL, Berge EM, Terracciano LM, Cappuzzo F, Incarbone M,
Roncalli M, et al: Identifying and targeting ROS1 gene fusions in
non-small cell lung cancer. Clin Cancer Res. 18:4570–4579. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lovly CM, Heuckmann JM, de Stanchina E,
Chen H, Thomas RK, Liang C and Pao W: Insights into ALK-driven
cancers revealed through development of novel ALK tyrosine kinase
inhibitors. Cancer Res. 71:4920–4931. 2011. View Article : Google Scholar : PubMed/NCBI
|