Introduction
Ferroptosis is a nonapoptotic form of regulated cell
death. In recent years, numerous efforts have been made to
elucidate the underlying mechanism of ferroptosis. It is considered
that the excessive accumulation of lipid peroxides produced by the
lipoxygenase family is an important cause of ferroptosis (1,2). This
process links ferroptosis to disruption of the redox homeostasis
maintained by glutathione and glutathione peroxidase 4 (GPX4)
(2). Compounds that inhibit cystine
glutamate antiporters (system XC−) and
subsequently reduce glutathione (GSH) levels (e.g., erastin) or
GPX4 activity (e.g., RSL3) strongly induce ferroptosis (1–3).
In addition to system XC− and
GPX4, several other genes have been reported to affect cell
sensitivity to ferroptosis, including acyl-CoA synthetase long
chain family member 4 (ACSL4), tumor protein p53 (TP53) and
glutaminase 2 (GLS2) (4–6). These studies have linked ferroptosis to
a variety of cellular processes, such as iron homeostasis, redox
homeostasis, lipid metabolism and glutamine decomposition (7,8). Since
highly transformed and drug-resistant tumor cells are prone to
ferroptosis (9,10), it is important to understand the
underlying mechanism of ferroptosis and apply it to personalized
anticancer strategies.
Lung cancer is the most common cause of
cancer-related deaths in the world, with an estimated 1.8 million
deaths per year (11). Approximately
85% of patients are diagnosed with a group of histological subtypes
called non-small cell lung cancer (NSCLC), among which lung
adenocarcinoma (LUAD) is the most common subtype (12). Studies have revealed that cancer cells
grow slowly when cAMP response element-binding protein (CREB), a
transcription factor, is knocked down (13–15).
However, little is known about the role of CREB in LUAD, and the
relationship between CREB and ferroptosis remains unknown.
In the present study, we aimed to analyze CREB
expression in LUAD by immunoblotting (IB), immunohistochemistry
(IHC) and enzyme-linked immunosorbent assay (ELISA), and
investigate how CREB regulates ferroptosis by analyzing cell
viability, three-dimensional (3D) cell growth, malondialdehyde
(MDA), the generation of lipid reactive oxygen species (ROS) and
the Fe2+ concentration. Measurement of luciferase
activity and chromatin immunoprecipitation (ChIP) were used to
analyze the GPX4 promoter activity regulated by CREB. In addition,
co-immunoprecipitation (co-IP) was used to analyze CREB interaction
with other GPX4 regulators such as EP300. Targeting CREB-related
GPX4 transcription may provide new ferroptotic strategies for LUAD
treatment.
Materials and methods
Cells, vectors and patients
The cell lines used in the present study were as
follows: 293T cell line, the lung fibroblast cell lines MRC-5 and
WI-38, the lung squamous cell carcinoma (LUSC) cell lines: MES-1
and H226, and the LUAD cell lines H358, A549, H1299 and H1650. All
cell lines were purchased from Fuheng Biotechnology (Shanghai,
China), and validated by short tandem repeat (STR) analysis. All
the cell lines were cultured in Dulbecco's modified Eagle's medium
(DMEM; Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10%
fetal bovine serum (FBS; HyClone, Cytiva) and 1% penicillin and
streptomycin (Invitrogen; Thermo Fisher Scientific, Inc.).
Ferrostatin-1 (Fer-1), ZVAD-FMK, necrostatin-1 (Nec-1), erastin and
apoptozole (all from Sigma-Aldrich; Merck KGaA) were used to treat
cells. For vectors, CREB-HA and CREB-short hairpin (sh)1/sh2 were
acquired from previous studies (15,16) which
were constructed by our laboratory (Shanghai Institute of Thoracic
Oncology, Shanghai Chest Hospital, Shanghai Jiao Tong University,
Shangai, China). EP300-Myc was purchased from Addgene, Inc. Empty
vector, GFP-sh and EP300-sh1/sh2 cells were purchased from Shanghai
Biolink Co., Ltd. CREB-Del-KID, CREB-Del-bZIP, EP300-Del-KIX,
EP300-Del-Bromo and EP300-Del-CBP/p300-HAT were constructed using
overlapping PCR and cloned into the pcDNA3.1(+) vector. All the
vectors were transfected at a final concentration of 2 µg per
6-well plate using Lipofectamine™ 2000 transfection reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). 293T cells were
transfected with FBS-free DMEM for 6 h and cultured in
FBS-containing medium for another 24 h. Then, target cells were
infected with lentivirus-containing-DMEM for 24 h, and the
follow-up experiments were performed. All the aforementioned
procedures were conducted at 37°C and 5% CO2. All
primers are summarized in Table SI.
Tumorous and adjacent lung tissues of patients (mean age ± SD,
63.86±14.12 years; age range, 27.2–88.4 years; 158 males and 138
females) were recruited at the Shanghai Chest Hospital (Shanghai,
China) from September 2013 to March 2018. The diagnosis of lung
cancer was confirmed by computed tomography and histological
analyses by doctors from Shanghai Chest Hospital. Informed written
consent was obtained from all patients. The study was approved by
the institutional Ethics Committee of Shanghai Chest Hospital.
IB
IB was performed using conventional protocols that
have been previously described (17).
The primary antibodies used were: Anti-CREB (product nos. 9197 and
9104) and anti-GAPDH (product nos. 5174 and 51332) all from Cell
Signaling Technology, Inc. (CST); anti-GPX4 (product code
ab125066), anti-cysteinyl-tRNA synthetase (CARS) (product code
ab126714), anti-nuclear factor, erythroid 2 like 2 (NRF2) (product
code ab62352), anti-heat shock protein family B small member 1
(HSPB1) (also known as Hsp27; product code ab109376),
anti-spermidine/spermine N1-acetyltransferase 1 (SAT1) (product
code ab105220) all from Abcam; anti-Myc (product nos. 2276 and
2278), anti-HA (product nos. 2367 and 3724) all from CST; and
anti-EP300 (product codes ab54984 and ab275378) and anti-SET domain
bifurcated histone lysine methyltransferase 2 (SETDB2) (product
code ab5517) all from Abcam. The secondary antibodies were
anti-rabbit IgG, HRP-linked antibody (product no. 7074; CST) or
anti-mouse IgG, HRP-linked antibody (product no. 7076; CST).
IHC
IHC was performed using conventional protocols that
have been previously described (18).
The primary antibodies included anti-CREB (product no. 9197; CST).
Immunohistochemical staining was assessed by independent
pathologists.
Reverse transcription-quantitative
(RT-q) PCR
Total RNA was extracted using TRIzol (Invitrogen;
Thermo Fisher Scientific, Inc.), and the RNA was reverse
transcribed into complementary DNA using the PrimeScript RT Reagent
Kit (TaKaRa Biotechnology Co., Ltd.) according to the
manufacturer's instructions. qPCR was performed using a SYBR Premix
Ex Taq (TaKaRa Biotechnology Co., Ltd.) kit for 40 cycles (95°C for
3 sec and 60°C for 30 sec). The relative expression of mRNA level
was quantified using the 2−ΔΔCq method (19). The primers are listed in Table SI.
Measurements of cell viability, MDA,
4-HNE and Fe2+
Cells were plated at an initial density of
2×105 cells/well and cultured for 24 h at 37°C and 5%
CO2. Cell viability was measured using a Cell Titer-Glo
luminescent cell viability assay (cat. no. G7572; Promega
Corporation) according to the manufacturer's instructions. MDA,
4-HNE and Fe2+ were measured using kits from Abcam (MDA
kit, product code ab118970, 532 nm; 4-HNE kit, product code
ab238538, 450 nm; Fe2+, product code ab83366, 593 nm)
according to the manufacturer's instructions. The luminance or
absorbance was measured by a reader from BioTek Instruments,
Inc.
ChIP
ChIP was performed using a kit from Active Motif
according to the manufacturer's instructions. Cells
(2×107) were fixed using 1% formaldehyde at room
temperature for 10 min, washed with PBS and lysed using lysis
buffer from the kit. Following sonication, protein-DNA complexes
were incubated with antibody-coupled protein G beads at 4°C
overnight. The antibodies used were anti-CREB (product no. 9197;
CST), anti-IgG (product no. 3900; CST) and anti-hepatocyte nuclear
factor 4α (HNF4A; cat. no. PP-H6939-00; R&D Systems, Inc.). On
the second day, DNA was eluted in 1% SDS/0.1 M NaHCO3,
reverse crosslinked at 65°C, purified via phenol/chloroform
extraction and ethanol precipitation, and subjected to qPCR. The
primers are listed in Table SI.
Measurement of luciferase
activity
Luciferase activities were measured using a
dual-luciferase kit (Promega Corporation) according to the the
manufacturer's instructions. Wild-type (WT)- and mutant
(Mut)-GPX4-promoter luciferase reporters were constructed using the
pGL4.21 vector at our laboratory (Shanghai Institute of Thoracic
Oncology, Shanghai Chest Hospital, Shanghai Jiao Tong University,
Shanghai, China), and co-transfected into lung cancer cells with
Renilla plasmids using Lipofectamine™ 2000 transfection
reagent (Invitrogen; Thermo Fisher Scientific, Inc.). Cells were
transfected with FBS-free DMEM for 6 h and cultured in
FBS-containing medium for another 42 h in 37°C and 5%
CO2. Then, cells were harvested and then lysed in the
passive lysis buffer from the kit. The fluorescence intensity of
the luciferase reporters was then examined and normalized to the
Renilla luciferase activity.
ELISA
The concentrations of CREB were evaluated by ELISA.
The tissue samples were diluted (1:4) in a dilution buffer provided
by the manufacturer, and 50 µl of each diluted sample was added to
96-well microtiter plates for analysis. A CREB ELISA kit (cat. no.
TX11637) was purchased from Lichen Biotech, Ltd. ELISAs were
performed in strict accordance with the manufacturers' guidelines.
The signals were determined by measuring the absorbance at 450 nm
with a microplate reader (BioTek Instruments, Inc.).
Three-dimensional (3D) cell
culture
First, basement membrane extract (BME) was seeded in
a 96-well plate at 50 µl/well and warmed at 37°C for 30 min. Then,
cells were seeded on top of the plate coated with BME at a density
of 10,000 cells/well. After 7 days, cells were stained with SYTOX
Green (1 µM; Invitrogen, Thermo Fisher Scientific, Inc.) at 37°C
for 30 min. Images were collected using a light microscope, and the
relative spheroid size and amount (Φ>30 µm) were counted and
calculated.
Co-immunoprecipitation (co-IP)
For co-IP, cell lysates (containing 2×107
cells) were incubated with antibody-conjugated protein A/G magnetic
beads (Thermo Fisher Scientific, Inc.) in Western/IP lysis buffer
(Beyotime Institute of Biotechnology) at 4°C overnight.
Immunoprecipitates isolated with the magnetic beads were washed
five times with Western/IP lysis buffer before being subjected to
IB. The antibodies used for co-IP were: Anti-Myc (product no. 2276;
1:100; CST), anti-HA (product no. 2367; CST, 1:100), anti-CREB
(product no. 9104; 1:100; CST), anti-EP300 (product code ab54984;
1:100; Abcam) and anti-SETDB2 (product code ab13712; 1:50;
Abcam).
Lipid reactive oxygen species (ROS)
measurement
Lipid ROS generation was measured by adding
C11-BODIPY (Invitrogen; Thermo Fisher Scientific, Inc.) to a final
concentration of 1.5 µM for 20 min before cell harvest. Lipid
ROS-positive cells were finally assessed by a BD FACSCanto II flow
cytometer.
Bioinformatic analysis
Data concerning CREB expression in 515 LUAD and 59
normal lung specimens were obtained from The Cancer Genome Atlas
(TCGA) and further analyzed using UALCAN database (http://ualcan.path.uab.edu) (20). CREB binding motif was acquired from
JASPAR database (21). Potential
CREB-binding methyltransferase/acetyltransferase was detected using
the STRING database (22). Uniprot
database was used to analyzed the domains in CREB and EP300
proteins (23).
Statistical analysis
The differences between groups were examined using
Student's t-test, one-way ANOVA followed by Bonferroni's post hoc
test, Fisher's exact test, χ2 test and Spearman
rank-correlation analysis. P<0.05 was considered to indicate a
statistically significant difference. The statistical analysis was
performed using Graphpad Prism 8 (GraphPad Software, Inc.) or SPSS
version 21 (IBM Corp.).
Results
CREB is highly expressed in LUAD
After screening TCGA data using the UALCAN database,
it was revealed that CREB was significantly upregulated in 515 LUAD
specimens compared to 59 normal lung specimens (Fig. 1A). By using IB and IHC, it was
revealed that compared to that of the lung fibroblasts MRC-5 and
WI-38, and the LUSC cell lines MES-1 and H226, CREB was highly
expressed in LUAD cell lines, especially in A549 and H1299 cells
(Fig. 1B). After detecting the
expression of CREB in 250 paired samples of LUAD patients by
ELISAs, it was determined that the expression of CREB in the tumor
tissues was higher than that in adjacent normal tissues (Fig. 1C and D). The tissues of patients 1–10
with the most significant increase in CREB according to the ELISA
data of Fig. 1C and D, were also
selected to analyze their CREB expression. It was revealed that
CREB expression was significantly higher in the LUAD tissues than
in the adjacent normal tissues; in addition, the upregulated degree
of CREB expression in tumor tissue in Fig. 1E was more obvious than that in
Fig. 1A. A similar phenomenon was
observed in immunohistochemical analysis by measuring the tissues
of no. 1–3 patients (Fig. 1F). CREB
expression was also analyzed in squamous cell lung cancer (LUSC)
and small-cell lung cancer (SCLC) tissues by ELISAs. It was
revealed that CREB was not significantly increased in the LUSC and
SCLC tissues compared to their adjacent tissues (Fig. S1). These data indicated that CREB was
highly expressed in LUAD tissues.
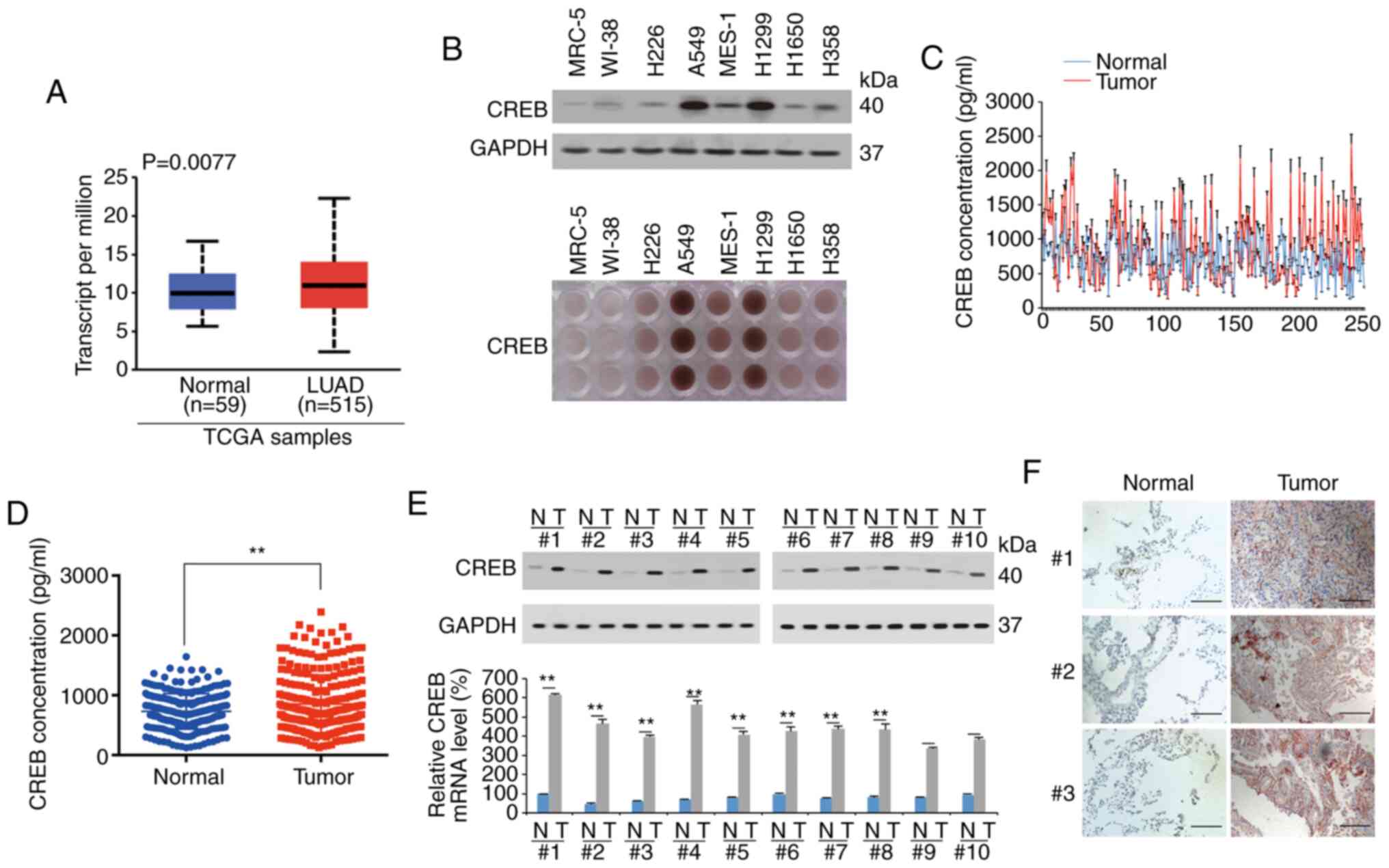 | Figure 1.CREB is upregulated in LUAD. (A)
UALCAN database was used to analyze TCGA data of CREB expression
between LUAD (n=515) and normal lung (n=59) tissues. (B) CREB
expression in lung fibroblast, LUSC and LUAD cell lines, as
measured by IB and IHC. (C and D) CREB expression in LUAD and
adjacent normal tissues was measured by ELISA, as presented in a
(C) line plot and (D) scatter plot. (E) CREB expression in 10 pairs
of LUAD tissues with the most significant increase of CREB
according to the ELISA data of C and D, as measured by IB and qPCR.
(F) CREB expression in 3 pairs of LUAD tissues, as measured by IHC
(scale bar, 800 µm). The data are presented as the mean ± SD from
three biological replicates (including IB). **P<0.01 indicates
statistical significance. Data from A were analyzed using an
independent-sample Student's t-test, D and E were analyzed using
paired Student's t-test. CREB, cAMP response element-binding
protein; LUAD, lung adenocarcinoma; TCGA, The Cancer Genome Atlas;
LUSC, lung squamous cell carcinoma; IB, immunoblotting; IHC,
immunohistochemistry; ELISA, enzyme-linked immunosorbent assay. |
CREB negatively regulates ferroptosis
in LUAD
In LUAD, CREB was reported to be a stimulator of
cell growth (24,25). However, whether CREB is a regulator of
cell death has not been investigated in detail. It was observed
that knockdown of CREB inhibited cell viability, and 3D cell
growth, whereas these effects could be reversed by the apoptotic
inhibitor ZVAD-FMK and the ferroptotic inhibitor Fer-1. However,
the CREB knockdown-induced effects could not be regulated by the
necrotic inhibitor necrostatin-1 (Nec-1) (Fig. 2A). Compared to ZVAD-FMK, Fer-1
reversed the decrease of cell viability and 3D cell growth induced
by CREB knockdown to a greater extent (Fig. 2A). It was also confirmed that CREB
knockdown upregulated the level of the lipid peroxidation product
MDA and the ferroptotic biomarkers lipid ROS and Fe2+,
and these effects could not be reversed by ZVAD-FMK and Nec-1
(Fig. 2A and C). Moreover, it was
revealed that ectopically expressed CREB could reverse the
apoptotic stimulus Apoptozole- and ferroptosis stimulus
erastin-induced decrease of cell viability and 3D cell growth
(Fig. 2B), as well as erastin-induced
MDA, Fe2+ increase and lipid ROS generation (Fig. 2B and C). These data indicated that
knockdown of CREB concurrently induced apoptosis- and
ferroptosis-like cell death.
 | Figure 2.CREB knockdown inhibits cell growth
via stimulating apoptosis- and ferroptosis-like cell death
concurrently. (A) Cell viability, 3D cell with SYTOX Green staining
and MDA were measured in CREB-knockdown cells with additional
treatment using Nec-1 (20 µM), ZVAD-FMK (20 µM) or Fer-1 (1 µM)
(scale bar, 50 µm). (B) Cell viability, 3D cell with SYTOX Green
staining and MDA were measured in cells treated with erastin (10
µM) or apoptozole (10 µM) with or without ectopically expressed
CREB (scale bar, 50 µm). (C) Fe2+ and lipid ROS
generation were measured in CREB-knockdown cells with additional
treatment using Nec-1 (20 µM), ZVAD-FMK (20 µM) or Fer-1 (1 µM) or
cells treated with erastin (10 µM) or apoptozole (10 µM) with or
without ectopically expressed CREB. The data are presented as the
mean ± SD from three biological replicates. *P<0.05, **P<0.01
indicates statistical significance. Data from A, B and C were
analyzed using a one-way ANOVA followed by Bonferroni's post hoc
test. CREB, cAMP response element-binding protein; MDA,
malondialdehyde; NEC-1, necrostatin-1; Fer-1, ferrostatin-1; ROS,
reactive oxygen species; sh, short hairpin. |
CREB specifically promotes GPX4
expression
To confirm the target of CREB in ferroptotic
regulation and to explore whether their levels were regulated by
CREB, several factors (including GPX4, ACSL4, CARS, NRF2, HSPB1 and
SAT1) (4,26–30) were
selected, which have been recently reported to exert important
roles during the ferroptotic process in cancer. It was observed
that among these factors, only the mRNA and protein levels of GPX4
were positively regulated by CREB in A549 and H1299 cells (Fig. 3A-C). In the tissues of LUAD patients
1–10, it was observed that the mRNA and protein levels of CREB and
GPX4 were upregulated in the LUAD tissues compared to the adjacent
normal tissues, and their levels in the LUAD tissues were
significantly correlated with each other (Fig. 3D-G). However, parallel experiments
revealed that the levels of SAT1 were not significantly increased
in the LUAD tissues (Fig. 3D and F).
The aforementioned data indicated that GPX4 expression was
positively regulated by CREB.
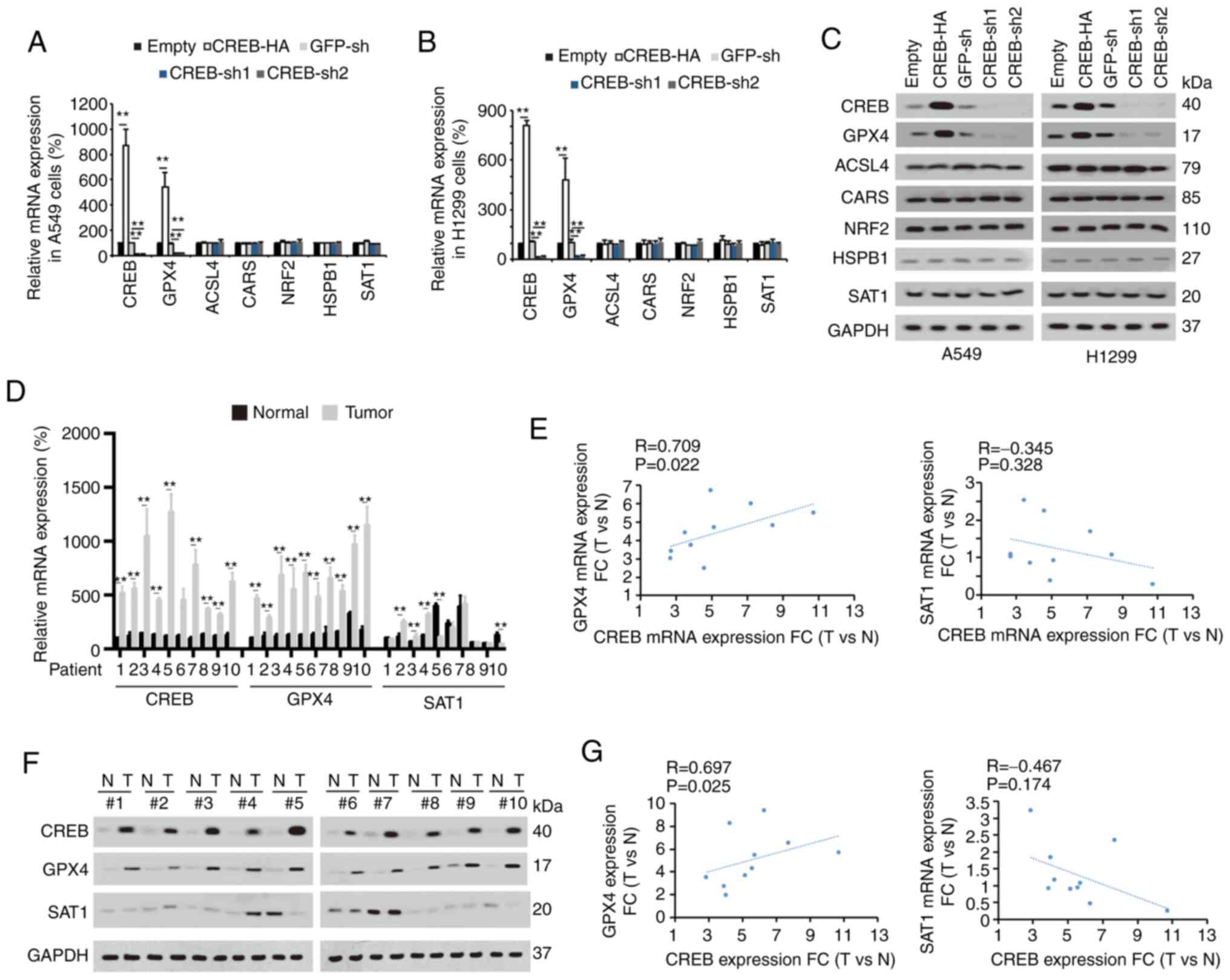 | Figure 3.CREB positively regulates GPX4. (A
and B) Indicated mRNA levels were measured in (A) A549 and (B)
H1299 cells with or without CREB overexpression or knockdown. (C)
Indicated protein expression levels were measured in A549 or H1299
cells with or without CREB overexpression or knockdown. (D) CREB,
GPX4 and SAT1 mRNA levels were measured in 10 paired of LUAD
tissues. (E) Correlation between GPX4 and CREB mRNA as well as SAT1
and CREB mRNA was calculated. (F) CREB, GPX4 and SAT1 protein
expressions were measured in 10 paired of LUAD tissues. (G)
Correlation between GPX4 and CREB protein level as well as SAT1 and
CREB protein level was calculated. The data are presented as the
mean ± SD from three biological replicates (including IB).
**P<0.01 indicates statistical significance. Data from A and B
were analyzed using a one-way ANOVA followed by Bonferroni's post
hoc test. Data from D were analyzed using a paired Student's
t-test. Data from E and G were analyzed using Spearman's rank
correlation coefficient. CREB, cAMP response element-binding
protein; GPX4, glutathione peroxidase 4, SAT1, spermidine/spermine
N1-acetyltransferase 1; LUAD, lung adenocarcinoma; IB,
immunoblotting; CARS, cysteinyl-tRNA synthetase; NRF2, nuclear
factor, erythroid 2 like 2; HSPB1, heat shock protein family B
small member 1; ACSL4, acyl-CoA synthetase long chain family member
4; sh, short hairpin; N, normal; T, tumor. |
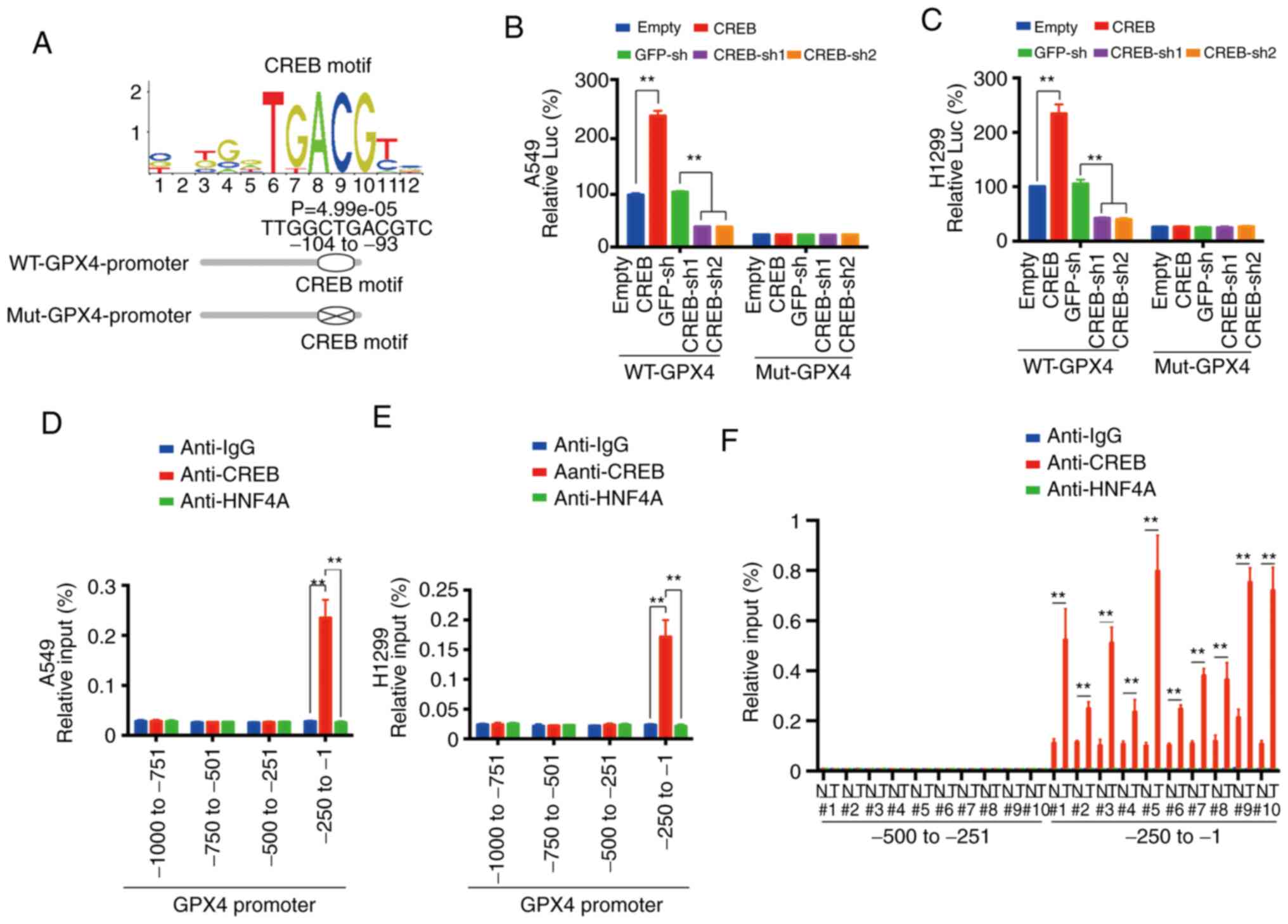
CREB directly binds the promoter of
GPX4
A CREB motif (image from JASPAR database) was
observed in the ~-104 to −93 promoter region of GPX4 and two
luciferase reporters named WT-GPX4-promoter (containing the CREB
motif) and Mut-GPX4-promoter (with the CREB motif deleted) were
constructed (Fig. 4A). Via
dual-luciferase experiments using these two reporters, it was
observed that the promoting effect of CREB for GPX4 promoter was
dependent on the CREB motif (Fig. 4B and
C). ChIP experiments revealed that CREB bound to the ~-250 to
−1 region of the GPX4 promoter (Fig. 4D
and E), and the parallel experiments indicated that CREB could
not bind to the ~-1000 to −250 region of the GPX4 promoter, and
HNF4A and control IgG could not bind to the ~-250 to −1 region of
the GPX4 promoter (Fig. 4D and E). As
for tissues from no. 1 to no. 10 patients, it was revealed that
CREB bound to the ~-250 to −1 region of the GPX4 promoter in both
the LUAD and adjacent normal tissues. However, the binding
intensity in the LUAD tissues was significantly higher than that in
the adjacent normal tissues (Fig.
4F). These data indicated that CREB directly bound to a CREB
motif in the GPX4 promoter.
EP300 enhances CREB-induced GPX4
transcription
Since histone modification, especially methylation
and acetylation, is critical for transcription (31,32), it
was further investigated whether methyltransferase or
acetyltransferase played important roles in CREB-induced GPX4
transcription. After a STRING analysis detecting potential
CREB-binding methyltransferase/acetyltransferase, it was revealed
that EP300 had the highest possibility of binding with CREB
(Fig. 5A). The co-IP experiments
confirmed that CREB could interact with EP300, whereas an obvious
interaction was not detected between CREB and SETDB2 (Fig. 5B). The UniProt database revealed
(https://www.UniProt.org) that the CREB protein
contains two important domains, KID and bZIP, and three important
domains, KIX, Bromo and CBP/300-HAT, are located in the EP300
protein (Fig. 5C). Reciprocal co-IP
experiments revealed that deletion of the bZIP or CBP/300-HAT
domain completely abolished the interaction between CREB and EP300,
suggesting that these two domains are essential for the CREB-EP300
interaction (Fig. 5D and E).
Additionally, ChIP experiments revealed that CREB or EP300
knockdown (EP300 knockdown efficiency is presented in Fig. S2) reduced the H3K27Ac levels [a
hallmark of open chromatin related to EP300 (33)] and inhibited the enrichment of CREB
and EP300 around the CREB motif in the GPX4 promoter, and these
effects could not be reversed by ectopically expressed EP300 or
CREB (Fig. 5F and G), implying that
CREB and EP300 were both critical for GPX4 transcription.
Furthermore, it was observed that ectopically expressed CREB or
EP300 increased the H3K27Ac levels, and stimulated the enrichment
of CREB and EP300 around the CREB motif in the GPX4 promoter,
whereas these effects could be blocked by deletion of the bZIP
domain and CBP/p300-HAT domain, respectively (Fig. 5H-K). In addition, EP300 was knocked
down in CREB-overexpressing cells and the cell viability and MDA
levels were analyzed. It was determined that CREB could reverse the
erastin-induced decrease in cell viability and the MDA increase;
however, these effects were abolished by further knocking down
EP300 (Fig. 5L and M). Collectively,
these data demonstrated that EP300 was essential and had a
promoting role in CREB-induced GPX4 transcription.
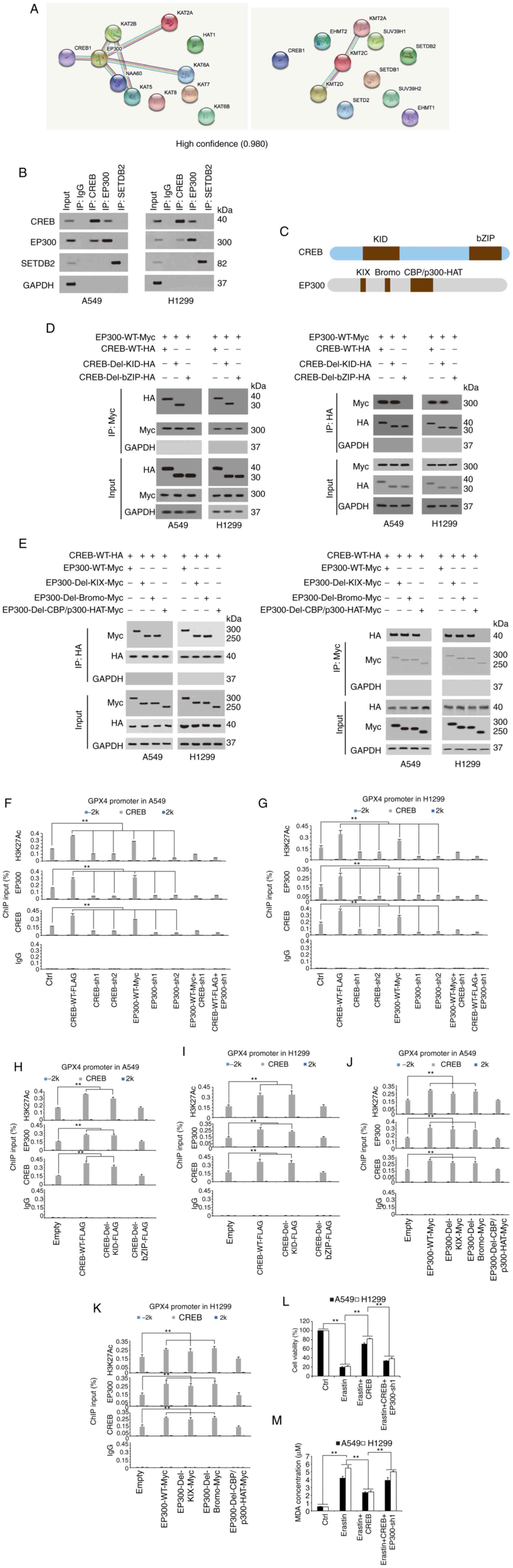 | Figure 5.EP300 stimulates CREB-dependent GPX4
transcription. (A) STRING analysis revealed CREB interacted with
methyltransferase and acyltransferase (confidence=0.980). (B)
Co-immunoprecipitation experiments performed in A549 cells using
indicated antibodies, and further analysis of CREB, EP300 and
SETDB2 expression by IB. (C) Schematic diagram shows the domains in
CREB and EP300 protein. (D and E) Co-immunoprecipitation
experiments performed using anti-HA or anti-CREB in A549 and H1299
cells with indicated CREB or EP300 plasmids overexpressed, and
further analysis of Myc and HA levels by IB. (F and G) The
enrichments of H3K27Ac, EP300 and CREB at −2 k, CREB motif or 2 k
regions of GPX4 promoter were calculated as the percentage of input
chromosomal DNA via ChIP using the corresponding antibodies in (F)
A549 and (G) H1299 cells with CREB or EP300 overexpressed or
knocked down. Anti-IgG was used as the parallel control. (H-K) The
enrichments of H3K27Ac, EP300 and CREB at −2 k, CREB motif or 2 k
regions of GPX4 promoter were calculated as the percentage of input
chromosomal DNA via ChIP using the corresponding antibodies in (H
and J) A549 and H1299 (I and K) cells with WT or mutant (H and I)
CREB or (J and K) EP300 overexpressed. Anti-IgG was used as the
parallel control. (L and M) Cell viability and MDA, respectively,
were measured in A549 and H1299 cells treated with erastin (10 µM)
with or without ectopically expressed CREB with or without EP300
knockdown. The data are presented as the mean ± SD from three
biological replicates (including IB). **P<0.01 indicates
statistical significance. Data from F-M were analyzed using a
one-way ANOVA followed by Bonferroni's post hoc test. EP300, E1A
binding protein P300; CREB, cAMP response element-binding protein;
GPX4, glutathione peroxidase 4; SETDB2, SET domain bifurcated
histone lysine methyltransferase 2; IB, immunoblotting; ChIP,
chromatin immunoprecipitation; WT, wild-type; MDA, MDA,
malondialdehyde; sh, short hairpin; Ctrl, control. |
Lipid peroxidation state may be
associated with tumor progression
Other paired LUAD tissues were selected to
investigate the correlation among CREB, GPX4, EP300 and 4-HNE, a
reactive breakdown product of the lipid peroxides that execute
ferroptosis. It was observed that the mRNA levels of CREB, GPX4,
and EP300 were significantly higher in the tumor tissues than in
the normal tissues, while the level of 4-HNE was significantly
higher in the normal tissues than in the tumor tissues (Fig. 6A-D). In addition, the level of 4-HNE
was negatively associated with the CREB, EP300 and GPX4 levels,
whereas significant positive correlations were observed between
GPX4 and CREB, EP300 and CREB, and EP300 and GPX4 (Fig. 6E-J).
 | Figure 6.Clinical significance of CREB, EP300,
GPX4 and 4-HNE. (A-D) Relative (A) CREB, (B) GPX4, (C) EP300 mRNA
levels and (D) 4-HNE concentration in 36 paired LUAD tissues. (E-G)
Correlations between 4-HNE concentration between (E) CREB, (F) GPX4
and (G) EP300 mRNA levels in 36 paired LUAD tissues. (H-J)
Correlations of mRNA levels between (H) GPX4 and CREB, (I) EP300
and CREB, as well as (J) EP300 and GPX4. (K) TCGA data of CREB and
EP300 co-expression in 515 LUAD specimens from UALCAN database. (L)
Survival in LUAD patients with CREB high (n=26) or low (n=26)
expression. (M and N) Correlations between 4-HNE levels and (M)
tumor stage and (N) tumor diameter. The data are presented as the
mean ± SD from three biological replicates. **P<0.01 indicates
statistical significance. Data from A-D were analyzed using a
paired Student's t-test. Data from E-K were analyzed using
Spearman's rank correlation coefficient. Data from L were analyzed
using the log rank analysis. Data from M were analyzed using
Fisher's exact test. Data from N were analyzed using χ2
test. CREB, cAMP response element-binding protein; EP300, E1A
binding protein P300; GPX4, glutathione peroxidase 4; 4-HNE,
4-hydroxynonenal; LUAD, lung adenocarcinoma; TCGA, The Cancer
Genome Atlas. |
Through the TCGA data from the UALCAN database, it
was revealed that the CREB level was positively associated with the
EP300 level in 515 LUAD specimens (P<0.001) (Fig. 6K). Furthermore, high expression of
CREB was significantly associated with poor prognosis in 52 LUAD
patients (P=0.008) (Fig. 6L). It was
also determined that low 4-HNE levels were associated with more
advanced tumor stages and larger tumor diameters (Fig. 6M and N), whereas high CREB, GPX4 and
EP300 levels were associated with more advanced tumor stages and
larger tumor diameters (Tables I and
II). It was also revealed that high
levels of CREB, GPX4 and EP300 were associated with advanced N
factors (Table II). Furthermore,
CREB, GPX4 and EP300 were not associated with patient age, sex or
smoking habits (Tables I and II). Collectively, CREB, GPX4, EP300 and
4-HNE, which are related to lipid peroxidation, were closely
related to tumor size and stage, and the tumors with a high degree
of malignancy were more likely to have a low degree of lipid
peroxidation.
 | Table I.Associations between mRNA levels of
CREB, EP300, GPX4 and clinicopathological parameters including age,
sex and tumor stage. |
Table I.
Associations between mRNA levels of
CREB, EP300, GPX4 and clinicopathological parameters including age,
sex and tumor stage.
|
|
|
| Sex |
| Tumor stage |
|
|---|
|
|
|
|
|
|
|
|
| mRNA levels | Age (years) | P-value
(Independent-sample Student's t-test) | Male | Female | P-value
(χ2 test) | I | II | III | IV | P-value (Fisher's
exact test) |
|---|
| CREB high | 65.44 | 0.152 | 8 | 10 | 0.180 | 0 | 7 | 8 | 3 | <0.001 |
| CREB low | 61.39 |
| 12 | 6 |
| 8 | 9 | 1 | 0 |
|
| GPX4 high | 65.94 | 0.072 | 7 | 11 | 0.044 | 1 | 6 | 8 | 3 | <0.001 |
| GPX4 low | 60.89 |
| 13 | 5 |
| 7 | 10 | 1 | 0 |
|
| EP300 high | 62.11 | 0.361 | 10 | 8 | 0.999 | 0 | 9 | 6 | 3 | <0.001 |
| EP300 low | 64.72 |
| 10 | 8 |
| 8 | 7 | 3 | 0 |
|
| Table II.Associations between mRNA levels of
CREB, EP300, GPX4 and clinicopathological parameters including
smoking habit, tumor diameter as well as N factor. |
Table II.
Associations between mRNA levels of
CREB, EP300, GPX4 and clinicopathological parameters including
smoking habit, tumor diameter as well as N factor.
|
| Smoking habit |
| Tumor diameter |
| N factor |
|
|---|
|
|
|
|
|
|
|
|
|---|
| mRNA levels | Smoking | Non-smoking | P-value
(χ2 test) | ≥3 cm | <3 cm | P-value
(χ2 test) | N0 | N1 | N2 | N3 | P-value (Fisher's
exact test) |
|---|
| CREB high | 5 | 13 | 0.725 | 15 | 3 | 0.002 | 3 | 8 | 5 | 2 | 0.001 |
| CREB low | 7 | 11 |
| 6 | 12 |
| 12 | 5 | 1 | 0 |
|
| GPX4 high | 6 | 12 | 1 | 15 | 3 | 0.002 | 3 | 8 | 5 | 2 | 0.001 |
| GPX4 low | 6 | 12 |
| 6 | 12 |
| 12 | 5 | 1 | 0 |
|
| EP300 high | 4 | 14 | 0.289 | 14 | 4 | 0.018 | 5 | 9 | 3 | 1 | <0.001 |
| EP300 low | 8 | 10 |
| 7 | 11 |
| 10 | 4 | 3 | 1 |
|
Discussion
CREB is a ubiquitous transcription factor that
activates the transcriptional activity of various promoters through
its binding (34). In NSCLC, most
studies reported that CREB directly binds to the promoters of
proto-oncogenes to exert a cancer promoting effect. For instance,
in NSCLC, loss of serine/threonine kinase 11 (LKB1) induced
CREB-regulated transcription coactivator (CRTC)-CREB complex
activation; the increased enrichment of the CRTC-CREB complex was
revealed in the promoter region of LINC00473, and this
LINC00473 was essential for the NSCLC cell growth and
survival (35). Moreover, the
CRTC-CREB complex also induced transcription of inhibitor of DNA
binding 1 (ID1), which is associated with stimulated tumor growth
and poor prognosis in NSCLC (36). In
the present study, it was revealed that CREB could directly bind to
the promoter region of GPX4, to stimulate the viability of LUAD
cells and inhibit the potential ferroptosis. In summary, CREB is an
important transcription factor in NSCLC, that can promote tumor
growth by activating a wide range of proto-oncogenes.
Transcriptional activation is regulated by histone
modifications, such as methylation and acetylation (37,38). The
interaction between CREB and EP300 has been reported previously
(39,40), yet the specific domains responsible
for the interaction in LUAD have not been elucidated. In the
present study, it was observed that the bZIP domain in CREB and the
CBP/p300-HAT were essential for the interaction between CREB and
EP300. The bZIP domain was revealed to be involved in CREB
dimerization and DNA-binding and contributed to CREB
transactivation by recruiting the coactivator TORC (41). The CBP/p300-HAT domain was revealed to
be critical for the interaction of EP300 with histones or the
transcription factor AP-2 alpha (TFAP2A) (42,43).
Therefore, the bZIP and CBP/p300-HAT domains are important for
protein interactions.
Ferroptotic therapy may be a favorable selection for
cancer treatment because ferroptosis exhibits greater induction in
certain types of tumor cells than in normal cells (44). In addition, ferroptotic treatment
specifically targets cells with a high degree of malignancy, such
as cells with a high metastatic tendency or cisplatin resistance
(9,45). In contrast, a strong antioxidant
system exists in tumor cells, which maintains ROS at an appropriate
level, stimulates the proliferation of tumor cells, and does not
cause cell death due to excessive stress (46). In LUAD, both SLC7A11 and NRF2 produce
high levels of GSH to protect tumor cells from lipid peroxidation
damage (47,48). In the present study, it was also
revealed that CREB is an important component of the antioxidant
system and plays an antioxidant role by stimulating transcription
of GPX4. According to the present data, targeting CREB inhibited
LUAD cell proliferation and promoted cell lipid peroxidation.
Therefore, CREB may be a suitable drug target in LUAD therapy.
There are two main ways to stimulate ferroptosis:
Blocking the cystine/glutamate transporter system
XC− (49) or
directly inhibiting the GSH-dependent antioxidant enzyme GPX4
(50). Studies have reported that
treating lung cancer cells using system XC inhibitors
such as sorafenib or temozolomide can inhibit cell growth and cause
cell death (51,52). However, drugs directly targeting GPX4
have not exhibited potential for clinical application. For example,
tumor cells exhibit high tolerance to RSL3 treatment in vivo
(53). In the present study, it was
observed that knockdown of CREB caused ferroptotic-like effects in
LUAD cells by inhibiting GPX4 transcription. Therefore, in the
future, a combination of drug treatment and gene knockout to
inhibit both system XC− and GPX4 may produce
improved therapeutic effects for cancer treatment.
In conclusion, it was revealed that CREB inhibited
ferroptosis by stimulating the transcription of GPX4 in the absence
of EP300. Targeting this CREB/EP300/GPX4 axis may be a new strategy
for treating LUAD.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural
Science Foundation of China (grant nos. 81822029, 81871907,
81672332, 81902869, 81902315 and 81774291), the Shanghai Municipal
Education Commission-Gaofeng Clinical Medicine (grant no.
20191834), the Shanghai Rising Star Program (grant no.
18QA1403400), the Shanghai Municipal Commission of Health and
Family Planning (grant no. 2017YQ024), the Grant Support ‘Chen
Guang’ project supported by Shanghai Municipal Education Commission
and Shanghai Education Development Foundation (grant no. 18CG16),
the Shanghai Sailing Program (grant no. 19YF1444800), and the
Nurture Projects for Basic Research of Shanghai Chest Hospital
(grant no. 2018YNJCQ06).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request. The results published and presented in Fig. 1A are in whole or part based upon data
generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Authors' contributions
ZW and XZ researched, analyzed data and wrote the
manuscript. XT constructed the plasmids. YYa and LM researched and
analyzed data. JW and YYu designed the study and revised the
manuscript for important intellectual content. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Informed written consents were obtained from all
patients. The study was approved by the institutional Ethics
Committee of Shanghai Chest Hospital (Shanghai, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Friedmann Angeli JP, Schneider M, Proneth
B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch
A, Eggenhofer E, et al: Inactivation of the ferroptosis regulator
Gpx4 triggers acute renal failure in mice. Nat Cell Biol.
16:1180–1191. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bersuker K, Hendricks JM, Li Z, Magtanong
L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al:
The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit
ferroptosis. Nature. 575:688–692. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Doll S, Proneth B, Tyurina YY, Panzilius
E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A,
et al: ACSL4 dictates ferroptosis sensitivity by shaping cellular
lipid composition. Nat Chem Biol. 13:91–98. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jiang L, Kon N, Li T, Wang SJ, Su T,
Hibshoosh H, Baer R and Gu W: Ferroptosis as a p53-mediated
activity during tumour suppression. Nature. 520:57–62. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gao M, Monian P, Quadri N, Ramasamy R and
Jiang X: Glutaminolysis and transferrin regulate ferroptosis. Mol
Cell. 59:298–308. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hassannia B, Vandenabeele P and Vanden
Berghe T: Targeting ferroptosis to iron out cancer. Cancer Cell.
35:830–849. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stockwell BR, Friedmann Angeli JP, Bayir
H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK,
Kagan VE, et al: Ferroptosis: A regulated cell death nexus linking
metabolism, redox biology, and disease. Cell. 171:273–285. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu J, Minikes AM, Gao M, Bian H, Li Y,
Stockwell BR, Chen ZN and Jiang X: Intercellular interaction
dictates cancer cell ferroptosis via NF2-YAP signalling. Nature.
572:402–406. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lee J, You JH, Kim MS and Roh JL:
Epigenetic reprogramming of epithelial-mesenchymal transition
promotes ferroptosis of head and neck cancer. Redox Biol.
37:1016972020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Melosky B, Juergens R, McLeod D, Leighl N,
Brade A, Card PB and Chu Q: Immune checkpoint-inhibitors and
chemoradiation in stage III unresectable non-small cell lung
cancer. Lung Cancer. 134:259–267. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pinto R, Petriella D, Lacalamita R,
Montrone M, Catino A, Pizzutilo P, Botticella MA, Zito FA, Del Bene
G, Zonno A, et al: KRAS-Driven lung adenocarcinoma and B Cell
infiltration: Novel insights for immunotherapy. Cancers (Basel).
11:11452019. View Article : Google Scholar
|
|
13
|
Mitton B, Chae HD, Hsu K, Dutta R,
Aldana-Masangkay G, Ferrari R, Davis K, Tiu BC, Kaul A, Lacayo N,
et al: Small molecule inhibition of cAMP response element binding
protein in human acute myeloid leukemia cells. Leukemia.
30:2302–2311. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chae HD, Mitton B, Lacayo NJ and Sakamoto
KM: Replication factor C3 is a CREB target gene that regulates cell
cycle progression through the modulation of chromatin loading of
PCNA. Leukemia. 29:1379–1389. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang J, Ma L, Weng W, Qiao Y, Zhang Y, He
J, Wang H, Xiao W, Li L, Chu Q, et al: Mutual interaction between
YAP and CREB promotes tumorigenesis in liver cancer. Hepatology.
58:1011–1020. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang J, Tang X, Weng W, Qiao Y, Lin J, Liu
W, Liu R, Ma L, Yu W, Yu Y, et al: The membrane protein melanoma
cell adhesion molecule (MCAM) is a novel tumor marker that
stimulates tumorigenesis in hepatocellular carcinoma. Oncogene.
34:5781–5795. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang X, Xu Y, Qian Z, Zheng W, Wu Q, Chen
Y, Zhu G, Liu Y, Bian Z, Xu W, et al: circRNA_104075 stimulates
YAP-dependent tumorigenesis through the regulation of HNF4a and may
serve as a diagnostic marker in hepatocellular carcinoma. Cell
Death Dis. 9:10912018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang X, Sun F, Qiao Y, Zheng W, Liu Y,
Chen Y, Wu Q, Liu X, Zhu G, Chen Y, et al: TFCP2 is required for
YAP-Dependent transcription to stimulate liver malignancy. Cell
Rep. 21:1227–1239. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chandrashekar DS, Bashel B, Balasubramanya
SA, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BV and Varambally
S: UALCAN: A portal for facilitating tumor subgroup gene expression
and survival analyses. Neoplasia. 19:649–658. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fornes O, Castro-Mondragon JA, Khan A, van
der Lee R, Zhang X, Richmond PA, Modi BP, Correard S, Gheorghe M,
Baranašić D, et al: JASPAR 2020: Update of the open-access database
of transcription factor binding profiles. Nucleic Acids Res.
48:D87–D92. 2020.PubMed/NCBI
|
|
22
|
Szklarczyk D, Gable AL, Lyon D, Junge A,
Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork
P, et al: STRING v11: Protein-protein association networks with
increased coverage, supporting functional discovery in genome-wide
experimental datasets. Nucleic Acids Res. 47:D607–D613. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
UniProt Consortium: UniProt: A worldwide
hub of protein knowledge. Nucleic Acids Res. 47:D506–D515. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yu W, Li L, Zheng F, Yang W, Zhao S, Tian
C, Yin W, Chen Y, Guo W, Zou L and Deng W: β-Catenin Cooperates
with CREB binding protein to promote the growth of tumor cells.
Cell Physiol Biochem. 44:467–478. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jin X, Di X, Wang R, Ma H, Tian C, Zhao M,
Cong S, Liu J, Li R and Wang K: RBM10 inhibits cell proliferation
of lung adenocarcinoma via RAP1/AKT/CREB signalling pathway. J Cell
Mol Med. 23:3897–3904. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lei HM, Zhang KR, Wang CH, Wang Y, Zhuang
GL, Lu LM, Zhang J, Shen Y, Chen HZ and Zhu L: Aldehyde
dehydrogenase 1A1 confers erlotinib resistance via facilitating the
reactive oxygen species-reactive carbonyl species metabolic pathway
in lung adenocarcinomas. Theranostics. 9:7122–7139. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hayano M, Yang WS, Corn CK, Pagano NC and
Stockwell BR: Loss of cysteinyl-tRNA synthetase (CARS) induces the
transsulfuration pathway and inhibits ferroptosis induced by
cystine deprivation. Cell Death Differ. 23:270–278. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R
and Tang D: Activation of the p62-Keap1-NRF2 pathway protects
against ferroptosis in hepatocellular carcinoma cells. Hepatology.
63:173–184. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sun X, Ou Z, Xie M, Kang R, Fan Y, Niu X,
Wang H, Cao L and Tang D: HSPB1 as a novel regulator of ferroptotic
cancer cell death. Oncogene. 34:5617–5625. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ou Y, Wang SJ, Li D, Chu B and Gu W:
Activation of SAT1 engages polyamine metabolism with p53-mediated
ferroptotic responses. Proc Natl Acad Sci USA. 113:E6806–E6812.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tessarz P and Kouzarides T: Histone core
modifications regulating nucleosome structure and dynamics. Nat Rev
Mol Cell Biol. 15:703–708. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kinnaird A, Zhao S, Wellen KE and
Michelakis ED: Metabolic control of epigenetics in cancer. Nat Rev
Cancer. 16:694–707. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ebrahimi A, Sevinc K, Gurhan Sevinc G,
Cribbs AP, Philpott M, Uyulur F, Morova T, Dunford JE, Göklemez S,
Arı Ş, et al: Bromodomain inhibition of the coactivators CBP/EP300
facilitate cellular reprogramming. Nat Chem Biol. 15:519–528. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hoeffler JP, Meyer TE, Yun Y, Jameson JL
and Habener JF: Cyclic AMP-responsive DNA-binding protein:
Structure based on a cloned placental cDNA. Science. 242:1430–1433.
1988. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen Z, Li JL, Lin S, Cao C, Gimbrone NT,
Yang R, Fu DA, Carper MB, Haura EB, Schabath MB, et al:
cAMP/CREB-regulated LINC00473 marks LKB1-inactivated lung cancer
and mediates tumor growth. J Clin Invest. 126:2267–2279. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rodon L, Svensson RU, Wiater E, Chun MG,
Tsai WW, Eichner LJ, Shaw RJ and Montminy M: The CREB coactivator
CRTC2 promotes oncogenesis in LKB1-mutant non-small cell lung
cancer. Sci Adv. 5:eaaw64552019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Daskalaki MG, Tsatsanis C and Kampranis
SC: Histone methylation and acetylation in macrophages as a
mechanism for regulation of inflammatory responses. J Cell Physiol.
233:6495–6507. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Thanh Nha Uyen L, Amano Y, Al-Kzayer LFY,
Kubota N, Kobayashi J, Nakazawa Y, Koike K and Sakashita K: PCDH17
functions as a common tumor suppressor gene in acute leukemia and
its transcriptional downregulation is mediated primarily by
aberrant histone acetylation, not DNA methylation. Int J Hematol.
111:451–462. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tong Q, Weaver MR, Kosmacek EA, O'Connor
BP, Harmacek L, Venkataraman S and Oberley-Deegan RE: MnTE-2-PyP
reduces prostate cancer growth and metastasis by suppressing p300
activity and p300/HIF-1/CREB binding to the promoter region of the
PAI-1 gene. Free Radic Biol Med. 94:185–194. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tung WH, Hsieh HL, Lee IT and Yang CM:
Enterovirus 71 modulates a COX-2/PGE2/cAMP-dependent viral
replication in human neuroblastoma cells: Role of the
c-Src/EGFR/p42/p44 MAPK/CREB signaling pathway. J Cell Biochem.
112:559–570. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Boer U, Eglins J, Krause D, Schnell S,
Schofl C and Knepel W: Enhancement by lithium of cAMP-induced
CRE/CREB-directed gene transcription conferred by TORC on the CREB
basic leucine zipper domain. Biochem J. 408:69–77. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Braganca J, Eloranta JJ, Bamforth SD,
Ibbitt JC, Hurst HC and Bhattacharya S: Physical and functional
interactions among AP-2 transcription factors, p300/CREB-binding
protein, and CITED2. J Biol Chem. 278:16021–16029. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu X, Wang L, Zhao K, Thompson PR, Hwang
Y, Marmorstein R and Cole PA: The structural basis of protein
acetylation by the p300/CBP transcriptional coactivator. Nature.
451:846–850. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Friedmann Angeli JP, Krysko DV and Conrad
M: Ferroptosis at the crossroads of cancer-acquired drug resistance
and immune evasion. Nat Rev Cancer. 19:405–414. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lee J, You JH, Shin D and Roh JL:
Inhibition of Glutaredoxin 5 predisposes cisplatin-resistant head
and neck cancer cells to ferroptosis. Theranostics. 10:7775–7786.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chio IIC and Tuveson DA: ROS in cancer:
The burning question. Trends Mol Med. 23:411–429. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ma L, Chen T, Zhang X, Miao Y, Tian X, Yu
K, Xu X, Niu Y, Guo S, Zhang C, et al: The m6A reader
YTHDC2 inhibits lung adenocarcinoma tumorigenesis by suppressing
SLC7A11-dependent antioxidant function. Redox Biol. 38:1018012020.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Galan-Cobo A, Sitthideatphaiboon P, Qu X,
Poteete A, Pisegna MA, Tong P, Chen PH, Boroughs LK, Rodriguez MLM,
Zhang W, et al: LKB1 and KEAP1/NRF2 pathways cooperatively promote
metabolic reprogramming with enhanced glutamine dependence in
KRAS-Mutant lung adenocarcinoma. Cancer Res. 79:3251–3267. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Song X, Zhu S, Chen P, Hou W, Wen Q, Liu
J, Xie Y, Liu J, Klionsky DJ, Kroemer G, et al: AMPK-Mediated BECN1
phosphorylation promotes ferroptosis by directly blocking system
Xc- Activity. Curr Biol. 28:2388–2399.e5. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Gaschler MM, Andia AA, Liu H, Csuka JM,
Hurlocker B, Vaiana CA, Heindel DW, Zuckerman DS, Bos PH, Reznik E,
et al: FINO2 initiates ferroptosis through GPX4 inactivation and
iron oxidation. Nat Chem Biol. 14:507–515. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Houessinon A, Francois C, Sauzay C,
Louandre C, Mongelard G, Godin C, Bodeau S, Takahashi S, Saidak Z,
Gutierrez L, et al: Metallothionein-1 as a biomarker of altered
redox metabolism in hepatocellular carcinoma cells exposed to
sorafenib. Mol Cancer. 15:382016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sehm T, Rauh M, Wiendieck K, Buchfelder M,
Eyupoglu IY and Savaskan NE: Temozolomide toxicity operates in a
xCT/SLC7a11 dependent manner and is fostered by ferroptosis.
Oncotarget. 7:74630–74647. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yang WS, SriRamaratnam R, Welsch ME,
Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji
AF, Clish CB, et al: Regulation of ferroptotic cancer cell death by
GPX4. Cell. 156:317–331. 2014. View Article : Google Scholar : PubMed/NCBI
|