Introduction
Macrophage migration inhibitory factor (MIF) is a
regulatory cytokine involved in the immune response, and as such
plays an important role in the pathogenesis of autoimmune diseases
and cancer (1,2). Findings have shown that CATT repeat
number in the MIF promoter is associated with MIF expression
level. The CATT5 repeat is the lowest expression allele
and CATT8 is the highest (3), with the clinical severity of autoimmune
inflammatory diseases and immune susceptibility being linked to
higher CATT repeats (4).
Thetranscription factor UHRF1 (90 kDa inverted CCAAT box-binding
protein) can bind to CATT-repeat polymorphisms to regulate MIF
expression, and is essential for the CATT5-8
length-dependent regulation of MIF transcription (5). UHRF1, as an epigenetic regulator, is
overexpressed in cancer and coordinates gene silencing of tumor
suppressors (6,7), potentially serving as a biomarker to
differentiate among different tumor grades (8).
Evidence suggests that HMG-box protein 1 (HBP1) can
bind to the MIF promoter and counter-regulate MIF
expression (9). HBP1 is a member of
the high mobility group (HMG) family of transcription factors and
has been demonstrated to act as a transcriptional inhibitor in
numerous cell lines, with its activation potently inhibiting the
cell cycle and regulating related genes (10); therefore, it has been suggested to
function as a tumor suppressor. Moreover, HBP1 maps to
chromosome 7q31.1, which has been reported to be frequently deleted
in myeloid and other cancers (11,12).
In the present study, we demonstrated that UHRF1
downregulates MIF expression by binding to the CATT repeat
of the MIF promoter, and decreases HBP1 expression by
promoting the interaction between MIF and HBP1 in T acute
lymphoblastic leukemia (T-ALL). In addition, HBP1 negatively
regulates MIF expression as a suppressor in T-ALL, and MIF
knockdown prolongs the life of T-ALL mice, suggesting that MIF
transcriptional regulation plays an important role in the
pathogenesis of T-ALL and is a potential treatment target and
biomarker for the prediction of prognosis in T-ALL.
Materials and methods
Study approval
The Ethics Committee of Shunde Hospital (Fo Shan)
approved the use of discarded peripheral blood from T-ALL patients
for T-cell cultivation. Informed consent for the procurement and
analysis of these samples was also obtained.
Cells and reagents
The human Jurkat T-cell line was purchased from the
American Type Culture Collection and cultured in RPMI-1640 medium
supplemented with 10% fetal bovine serum. The apoptotic stains, PI
and FITC-Annexin V (BD 556547), were obtained from BD Biosciences.
The anti-HBP1 (sc-515281), β-actin (sc-47778), MIF (sc-271631) and
anti-UHRF1 (ab57083) antibodies were purchased from Santa Cruz
Biotechnology and Abcam, respectively. The Co-IP kit (26149 Pierce)
was purchased from Pierce and the EasySep™ Human T Cell Isolation
kit was obtained from Stem Cell (cat. no. 17951, Stem Cell).
Western blot analysis
Total protein obtained from Jurkat and PBMC from
T-ALL patients was extracted using RIPA lysis and extraction buffer
(Thermo Fisher; cat. no. 89900), and equal amounts (Pierce™ BCA
Protein Assay, 23227) of 30 µg were resolved in 10% SDS-PAGE at 25
mA for 1 h on ice. The protein bands were subsequently transferred
to PVDF (polyvinylidene difluoride) membrane, and non-specific
sites were blocked with 5% BSA. The membranes were incubated
overnight at 4°C with an anti-UHRF1 (ab57083, 1/1,000) antibody,
washed three times with PBST, and incubated with an HRP-conjugated
anti-rabbit secondary (ab205719, 1/2,000) antibody for 2 h at room
temperature. ECL detection reagent (Pierce) was used to detect the
protein complexes. Densitometric analysis was performed using NIH
Image (version 1.62f). The MIF promoter CATTX
length-dependent retention of protein was detected by western
blotting of eluted binding proteins using an anti-UHRF1 or
anti-HBP1 antibody (sc-515281, 1/1,000). The following oligos for
CATT0,5-8 were used to bind UHRF1:
5′-CTTTCACCCAGCAGTATTAGTCAAT-3′
5′-CTTTCACCCATTCATTCATTCATTCATTCAGCAGTATTAGTCAAT-3′
5′-CTTTCACCCATTCATTCATTCATTCATTCATTCAGCAGTATTAGTCAAT-3′
5′-CTTTCACCCATTCATTCATTCATTCATTCATTCATTCAGCAGTATTAGTCAAT-3′
5′-CTTTCACCCATTCATTCATTCATTCATTCATTCATTCATTCAGCAGTATTAGTCAAT-3′
Flow cytometry
PI (propidium iodide) and FITC-Annexin V staining
was performed to evaluate apoptosis in Jurkat cells and primary T
cells purified from T-ALL patients by Ficoll-Hypaque and a
CD3+ T-cell isolation kit following knockdown of UHRF1
or overexpression of HBP1. Staining was analyzed using a FACS
Calibur (BD Biosciences).
Immunofluorescence confocal
microscopy
PBS-rinsed cultured cells were fixed with methanol
on ice for 2 h, blocked with 5% BSA for 1 h, and incubated with the
primary antibodies (anti-HBP1, sc-515281; MIF, sc-271631; and
anti-UHRF1, ab57083; 1/500) overnight at 4°C. The following day,
the cells were rinsed five times with PBS and incubated with a
fluorescently labeled secondary antibody in the dark for 2 h at
room temperature. After rinsing, ProLong™ Gold Antifade Mountant
with DAPI (P36931) was employed for nuclear staining. Imaging was
performed on a Leica YSCC SP5 confocal system at a magnification of
×100.
Luciferase reporter assay
analysis
MIF-794 CATT5-8-dependent transcription
was analyzed using the dual luciferase reporter assay system as
previously described (5). Each
transfection experiment was performed in triplicate and repeated at
least twice.
Co-immunoprecipitation
Cells (1×106) were transfected with empty
vector or expression plasmid using Amaxa Nucleofector™. After 24 h,
the cells were lysed in IP lysis buffer (Pierce, cat. no. 87787)
containing protease inhibitors (Roche), and lysates were
centrifuged at approximately 13,000 × g for 10 min to pellet the
cell debris at 4°C and incubated with AminoLink™ Plus coupling
resin (Pierce 26149) overnight at 4°C. The beads were washed three
times with IP wash buffer (0.025 M Tris, 0.15 M NaCl, 0.001 M EDTA,
1% NP-40, 5% glycerol, pH 7.4), and the immunoprecipitates were
eluted with elution buffer (DTT-containing SDS sample buffer) and
boiled for 5 min in SDS loading buffer. Eluates were analyzed by
western blot analysis.
Reverse transcription-quantitative
PCR
Total RNA of Jurkat and PBMC of T-ALL patients was
isolated using an RNeasy RNA extraction kit (Qiagen), and cDNA was
synthesized using a BioRad iScript cDNA synthesis kit. RT-qPCR was
carried out using the iQ SYBR-Green system (Bio-Rad). Primer
sequences were: UHRF1: (5′-ATGTGGATCCAGGTTCGGA-3′ and
5′-GAACAGCTCCTGGATCTT-3′) and HBP1:
(5′-TGAAGGCTGTGATAATGAGGAAGAT-3′ and
5′-CATAGAAAGGGTGGTCCAGCTTA′-3). MIF mRNA was determined
using the primers: 5′-CGGACAGGGTCTACATCAA-3′,
5′-CTTAGGCGAAGGTGGAGTT-3′ and 18S 5′-GCAATTATTCCCCATGAACG-3′,
5′-TGTACAAAGGGCAGGGACTT-3′. The emitted fluorescence for each
reaction was measured during the annealing/extension phase and
relative quantity values were calculated by the standard curve
method. The quantity value of 18S in each sample was used as a
normalizing control. Differences were evaluated by non-parametric
testing using the Mann-Whitney U test.
Jurkat and primary T-cell
transfection
Primary T cells were isolated by EasySep™ Human T
Cell Isolation Kit (Stem Cell, cat. no. 17951), then transfected
using Nucleofector™ solution (Lonza, cat. no. VPA-1002) and the
Nucleofector™ II system for transfecting T cells. After 24 h in an
incubator at 37°C, the cells were harvested for further
experimentation. The shRNA plasmids included UHRF1 (GI333964), HBP1
(TL312507), MIF (TR319111), or control (TR30007). The
overexpression plasmid was HBP1 (RG202260) or control (PS100010)
(Origene).
In vivo leukemia cell
transplantation
NOD-SCID-γ (NSG) mice (8–10 weeks old) were obtained
from Biocytogen (Beijing) provided with autoclaved food and clear
H2O and housed in a specific pathogen-free (SPF)
facility. Animal care was carried out in accordance with the local
Animal Welfare Act. All food, water, bedding, and cages within the
room were autoclaved or sterilized and cages were changed weekly;
the room temperature was 26–28°C, and humidity was kept at 40–60%;
with a 12-h light/dark cycle. A total of 36 mice were injected via
the tail vein with 1×106 cells per mouse (T-ALL cells
transfected with UHRF1-shRNA, HBP1-overexpression, or scrambled
shRNA control plasmids). For survival experiments, mice (n=12 mice
per group) were culled 25 days post-engraftment, or immediately
following the appearance of signs of moribund or weight loss
exceeding 10–15% of their total weight.
Statistical analysis
Results are expressed as the mean ± standard
deviation. To study the difference between two groups, the
Student's t-test and approximate calculation of normal distribution
were used for all two-tailed comparisons. One-way ANOVA followed by
Tukey's post-hoc test was used to compare more than two groups. The
similarity of expression levels in the transcriptome was assessed
by Pearson's correlation analysis. Expression heat map of genes
were selected for 1.5-fold differential expression with an FDR
<0.05 to show the different genes associated with MIF between
ALL and healthy control. NSG mice survival was assessed via
Kaplan-Meier survival curve. Analyses were performed using the
GraphPad Prism software. P<0.05 was considered statistically
significant, and P<0.01 was considered statistically very
significant.
Results
Identification of HBP1 and UHRF1
binding to the MIF promoter and regulation of MIF
transcription
To investigate whether HBP1 and UHRF1 can interact
with the MIF-794 CATT5-8 microsatellite in T-ALL, 5′
biotin-labeled oligonucleotides, including or excluding CATT
repeats of the MIF promoter (without any CATT sequences as a
control), were incubated with nuclear lysates of human T cells
followed by streptavidin beads. NaCl-eluted bound proteins were
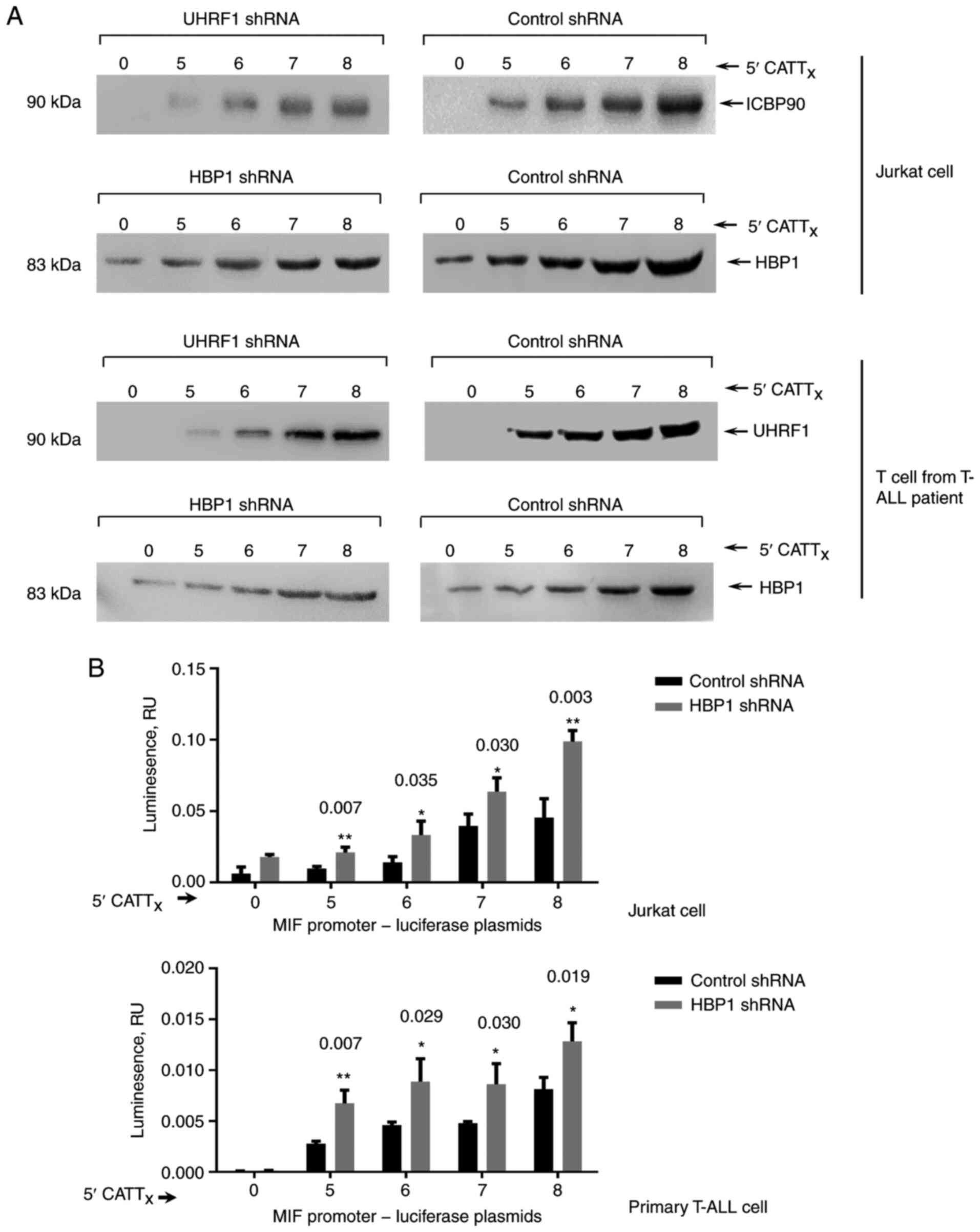
evaluated by western blot analysis (Fig.
1A). The effectiveness of this approach was verified by testing
the CATT-specific interaction of UHRF1 in T cells. As shown in
Fig. 1A, the analysis revealed
binding of HBP1 to the MIF CATT8 and control
CATT0 oligonucleotides. We previously demonstrated a
downregulatory role of UHRF1 in-794 CATT5-8-dependent
MIF expression (5); thus, we
assessed the functional role of HBP1 in MIF expression by
measuring the transcriptional activity of MIF in human T-ALL
cells using a luciferase reporter assay. The level of MIF
promoter transcript increased progressively with increasing levels
of HBP1 shRNA, with CATT5 showing the lowest gene
transcription and CATT8 showing the highest one
(Fig. 1B).
Protein crosstalk between the
transcription factor HBP1 and MIF
Co-immunoprecipitation (Co-IP) in T cells verified
the interaction between HBP1 and MIF, but not between UHRF1 and
HBP1 or between UHRF1 and MIF (Fig.
2A). Subsequently, the location of the interaction between HBP1
and MIF was confirmed in HeLa cells by confocal microscopy, showing
co-localization in the cytosol but not in the nucleus (Fig. 2B). Moreover, there was no
co-localization of UHRF1 and HBP1 (Fig.
2C).
UHRF1 downregulates both MIF and HBP1
expression
Given that UHRF1 regulates MIF expression by
binding to MIF CATT motifs, and HBP1 acts as a suppressor,
we focused further attention on defining the relationship among the
three genes. Following knockdown of UHRF1, HBP1, or both
genes simultaneously, RT-qPCR results showed that UHRF1, not only
downregulated MIF expression, but also HBP1
expression, causing loss of HBP1 repressive function in both the
T-ALL cell line and T cells from ALL patients (Fig. 3A and B). It was further confirmed by
western blotting that UHRF1 can downregulate HBP1 at the protein
level (Fig. 3C and D). Subsequently,
the effect of the overexpression of UHRF1 and HBP1 on MIF
regulation was assessed. The MIF expression level was not
significantly different following upregulation of UHRF1 or HBP1
(Fig. 4A and B), indicating that
UHRF1 cannot upregulate MIF and HBP1 cannot downregulate
MIF in T-ALL. The influence of the interaction between MIF
and HBP1 on the downregulation of HBP1 mediated by UHRF1 was
further evaluated. Knockdown of MIF by MIF shRNA increased
HBP1 expression in T-ALL cells, which was also observed
following UHRF1 knockdown (Fig. 5A
and B). Moreover, UHRF1 knockdown could not downregulate
HBP1 following inhibition of the MIF protein using an
inhibitor (Fig. 5C).
MIF silencing induces cell apoptosis
and slows leukemia progression in vivo
We also examined cell apoptosis and animal survival
following the regulation of MIF expression by UHRF1 and HBP1, which
induced apoptosis, retarded the progression of ALL, and extended
survival time. As expected, knockdown of UHRF1 or
overexpression of HBP1 in T-ALL cells reduced basal MIF expression
(Fig. 6A). MIF overexpression is
known to inhibit apoptosis in many cell types, and the two
mediators act in concert to regulate apoptotic sensitivity in the
context of inflammatory activation (10–13).
Experimental reduction of UHRF1 or upregulation of HBP1 enhanced
T-ALL cell sensitivity to apoptosis, which is consistent with the
interpretation that functional UHRF1 and HBP1 regulate MIF
expression and protect cells from apoptosis (Fig. 6B). To further assess the functions of
UHRF1 and HBP1 in the progression of T-ALL in vivo, mice
were injected with transduced Jurkat cells to observe survival
time. The results show that mice transplanted with
UHRF1-knockdown cells lived longer than those in both the
control and HBP1-overexpression groups, which is consistent with
the cell apoptosis data (Fig.
6C).
Pathogenic role of MIF
Human genetic studies indicate a high expression of
MIF and UHRF1 and low expression of HBP1 in
T-ALL, and experimental data suggest a pathogenic role of MIF in
promoting proliferation and downstream expression of chemokines in
T-ALL (Fig. 7A). Notably, a
significant correlation was observed between the mRNA expression
levels of UHRF1 and MIF (R=0.9192, P<0.0001) and
HBP1 and MIF (R=0.6977, P<0.0001) in the T-ALL
group as compared with those in the healthy control group (Fig. 7B). This correlation between the
expression levels of UHRF1 and MIF and between HBP1 and MIF
supports a functional role of UHRF1 downregulation and HBP1
upregulation with respect to MIF in T-ALL.
Discussion
Macrophage migration inhibitory factor (MIF) has
been suggested to be a pro-tumorigenic factor that promotes the
proliferation, migration, and invasion of tumor cells (13). Previous findings have shown that ALL
cells constitutively express high levels of MIF (14). Leukemic cells from most patients
express the chemokine IL-8 and the receptor CXCR1, but at lower
levels. Moreover, one report used a mouse model in which
subcutaneous ALL tumors were partially suppressed by locally
injected endothelial IL-8 (15–17). In
the present study, T cells were isolated from healthy controls and
T-ALL patients and subjected to microarray. These data are
consistent with reports that MIF expression is high and causes
increased expression of downstream chemokines such as IL-8, which
are associated with cell proliferation, suggesting that MIF plays a
pathogenic role in T-ALL. Our mechanistic understanding of the
MIF-mediated regulation of tumor cell proliferation has expanded
since the identification of MIF transcription. Compelling evidence
suggests that MIF overexpression and regulation is associated with,
and contributes to, the pathogenesis of inflammatory autoimmune and
malignant diseases (18,19); however, the mechanism underlying MIF
regulation in T-ALL has yet to be clarified.
Evidence suggests that UHRF1 positively regulates
MIF transcription (5) and HBP1
has a negative regulatory function (9). To determine the key regulatory mechanism
of MIF in T-ALL, we identified that UHRF1 and HBP1 co-regulate MIF
expression. Of note, UHRF1 can also regulate HBP1 transcription by
promoting the interaction between MIF and HBP1 proteins. We
verified a specific association between MIF and HBP1 by
co-immunoprecipitation of MIF-HBP1 complexes in vitro. To
confirm that MIF can interact with intracellular HBP1, we showed
the co-localization of endogenously expressed MIF and HBP1 in the
cytosol. We also observed that UHRF1 can only regulate MIF, but not
HBP1 without the presence of MIF. Moreover, HBP1 was able to only
upregulate MIF expression but could not downregulate
MIF expression when HBP1 expression was elevated, indicating
that UHRF1 is the key regulator in the knockdown of MIF in
T-ALL.
Furthermore, we found that the expression levels of
UHRF1 and MIF were elevated but that of HBP1
was decreased in T-ALL patients as compared with those in healthy
controls. In addition, analysis of the correlation among UHRF1,
MIF, and HBP1 expression in a gene expression dataset of
T cells from T-ALL patients shows a high correlation between
UHRF1 and MIF expression and between HBP1 and
MIF expression, supporting a positive regulatory role of
UHRF1 and a negative regulatory role of HBP1 in MIF transcription
in vivo. Moreover, UHRF1 knockdown and HBP1 overexpression
induced a greater level of apoptosis in T cells from T-ALL patients
and significantly prolonged the survival time of transplanted
mice.
Taken together, our results indicate an important
role of the UHRF1 protein in the survival and homing of malignant T
cells, which is mediated through a functional interaction between
MIF and HBP1. In conclusion, a high level of UHRF1 and a low level
of HBP1 cause MIF overexpression, resulting in tumor cell
proliferation and inhibition of cell death. The MIF/UHRF1/HBP1 axis
may represent a novel target for the therapeutic intervention of
ALL.
Acknowledgements
Not applicable.
Funding
We would like to thank the National Natural
Scientific Foundation of China (81770148, to J. Yao) for
funding.
Authors' contributions
JY contributed to the conception of the study. JY
and CZ analyzed data for the study. JY, CZ, HH, YL and XZ performed
the experiments for the study. JY and YL wrote the study. All
authors approved the study, and JY and YSL confirm the authenticity
of the data.
Availability of data and materials
All data generated or analyzed during this study are
included in this article.
Ethics approval and consent to
participate
The Ethics Committee of Shunde Hospital (Fo Shan)
approved the use of discarded peripheral blood from T-ALL patients
for T-cell cultivation. Informed consent for the procurement and
analysis of these samples was also obtained.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
UHRF1
|
inverted CCAAT box binding protein 90
kDa
|
|
HBP1
|
HMG-box protein 1
|
|
MIF
|
macrophage migration inhibitory
factor
|
|
ALL
|
acute lymphoblastic leukemia
|
|
Co-IP
|
co-immunoprecipitation
|
|
confocal
|
confocal microscopy
|
References
|
1
|
Kang I and Bucala R: The immunobiology of
MIF: Function, genetics and prospects for precision medicine. Nat
Rev Rheumatol. 15:427–437. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bloom J, Sun S and Al-Abed Y: MIF, a
controversial cytokine: A review of structural features,
challenges, and opportunities for drug development. Expert Opin
Ther Targets. 20:1463–1475. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu A, Bao F and Voravuthikunchai SP: CATT
polymorphism in MIF gene promoter is closely related to human
pulmonary tuberculosis in a southwestern China population. Int J
Immunopathol Pharmacol. May 29–2018.(Epub ahead of print). doi:
10.1177/2058738418777108.
|
|
4
|
Matia-García I, Salgado-Goytia L,
Muñoz-Valle JF, García-Arellano S, Hernández-Bello J,
Salgado-Bernabé AB and Parra-Rojas I: Macrophage migration
inhibitory factor promoter polymorphisms (−794 CATT 5–8 and −173
G>C): Relationship with mRNA expression and soluble MIF levels
in young obese subjects. Dis Markers. 2015:4612082015. View Article : Google Scholar
|
|
5
|
Yao J, Leng L, Sauler M, Fu W, Zheng J,
Zhang Y, Du X, Yu X, Lee P and Bucala R: Transcription factor
ICBP90 regulates the MIF promoter and immune susceptibility locus.
J Clin Invest. 126:732–744. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mousli M, Hopfner R, Abbady AQ, Monté D,
Jeanblanc M, Oudet P, Louis B and Bronner C: ICBP90 belongs to a
new family of proteins with an expression that is deregulated in
cancer cells. Br J Cancer. 89:120–127. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Abbady AQ, Bronner C, Trotzier MA, Hopfner
R, Bathami K, Muller CD, Jeanblanc M and Mousli M: UHRF1 expression
is downregulated in apoptosis-induced Jurkat cells. Ann N Y Acad
Sci. 1010:300–303. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ashraf W, Ibrahim A, Alhosin M, Zaayter L,
Ouararhni K, Papin C, Ahmad T, Hamiche A, Mély Y, Bronner C and
Mousli M: The epigenetic integrator UHRF1: On the road to become a
universal biomarker for cancer. Oncotarget. 8:51946–51962. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen YC, Zhang XW, Niu XH, Xin DQ, Zhao
WP, Na YQ and Mao ZB: Macrophage migration inhibitory factor is a
direct target of HBP1-mediated transcriptional repression that is
overexpressed in prostate cancer. Oncogene. 29:3067–3078. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bollaert E, Johanns M, Herinckx G, de
Rocca Serra A, Vandewalle VA, Havelange V, Rider MH, Vertommen D
and Demoulin JB: HBP1 phosphorylation by AKT regulates its
transcriptional activity and glioblastoma cell proliferation. Cell
Signal. 44:158–170. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bollaert E, de Rocca Serra A and Demoulin
JB: The HMG box transcription factor HBP1: A cell cycle inhibitor
at the crossroads of cancer signaling pathways. Cell Mol Life Sci.
76:1529–1539. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li H, Wang W, Liu X, Paulson KE, Yee AS
and Zhang X: Transcriptional factor HBP1 targets P16(INK4A),
upregulating its expression and consequently is involved in
Ras-induced premature senescence. Oncogene. 29:5083–5094. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fukaya R, Ohta S, Yaguchi T, Matsuzaki Y,
Sugihara E, Okano H, Saya H, Kawakami Y, Kawase T, Yoshida K and
Toda M: MIF maintains the tumorigenic capacity of brain
tumor-initiating cells by directly inhibiting p53. Cancer Res.
76:2813–2823. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sharaf-Eldein M, Elghannam D, Elderiny W
and Abdel-Malak C: Prognostic implication of MIF gene expression in
childhood acute lymphoblastic leukemia. Clin Lab. 64:1429–1437.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kumar S, O'Malley J, Chaudhary AK, Inigo
JR, Yadav N, Kumar R and Chandra D: Hsp60 and IL-8 axis promotes
apoptosis resistance in cancer. Br J Cancer. 121:934–943. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kuett A, Rieger C, Perathoner D, Herold T,
Wagner M, Sironi S, Sotlar K, Horny HP, Deniffel C, Drolle H and
Fiegl M: IL-8 as mediator in the microenvironment-leukaemia network
in acute myeloid leukaemia. Sci Rep. 5:184112015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vijay V, Miller R, Vue GS, Pezeshkian MB,
Maywood M, Ast AM, Drusbosky LM, Pompeu Y, Salgado AD, Lipten SD,
et al: Interleukin-8 blockade prevents activated endothelial cell
mediated proliferation and chemoresistance of acute myeloid
leukemia. Leuk Res. 84:1061802019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bilsborrow JB, Doherty E, Tilstam PV and
Bucala R: Macrophage migration inhibitory factor (MIF) as a
therapeutic target for rheumatoid arthritis and systemic lupus
erythematosus. Expert Opin Ther Targets. 23:733–744. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Noe JT and Mitchell RA: MIF-dependent
control of tumor immunity. Front Immunol. 11:6099482020. View Article : Google Scholar : PubMed/NCBI
|