Introduction
Solid tumor blood vessels regulate the infiltration of circulating immune cells into the tumor mass. In order to reach and destroy cancer cells, circulating immune cells must first cross the tumor blood vessel barrier (1). Under physiological conditions, endothelial cells that line the luminal side of blood vessels form a continuous and non-adhesive surface for circulating immune cells. When stimulated by an inflammatory signal, endothelial cell activation triggers the rolling and adhesion of circulating leucocytes onto the endothelium, followed by their extravasation and infiltration into tissues (2). This infiltration serves a major role in tumor development. In human cancer, the infiltration of circulating immune cells, such as natural killer (NK) cells and CD8+ cytotoxic T-lymphocytes, into the tumor mass represses tumor growth (3–5). The tumor blood vessel endothelium, although imperfectly tight, actively prevents the infiltration of immune cells within the tumor mass (6), and such tumor escape from immunity is achieved, in part, by the downregulation of leukocyte adhesion receptors expressed by tumor endothelial cells (7–10).
Epidermal growth factor-like domain 7 (Egfl7) is a gene that is primarily expressed by endothelial cells during development and in adults (11–13). The endogenous expression of Egfl7 by endothelial cells represses their activation via the constitutive downregulation of the NFκB signaling pathway via the stabilization of IκBα (14). In the context of cancer, high expression levels of Egfl7 are correlated with more advanced stages of human colon cancer (15), poor survival in human hepatocarcinoma (16) and higher tumor grades in human glioma (17). Egfl7 expression has also been shown to be associated with the poor overall survival of patients with pancreatic cancer (18). Furthermore, our previous studies revealed that high Egfl7 protein scores in breast cancer cells were correlated with low vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1) expression in the tumors (19), and that high expression levels of Egfl7 in such human lesions were correlated with low endothelial cell activation in peritumoral vessels (20). Using preclinical approaches, it was also observed that Egfl7 repressed endothelial cell activation by lowering the expression levels of the leukocyte adhesion receptors ICAM-1, VCAM-1 and E-selectin at the surface of tumor blood vessels. This resulted in a reduced immune cell infiltration into the tumor mass, repression of cancer cell destruction by immune cells and promotion of tumor growth (19).
The Egfl7 protein consists of five successive domains deduced from its primary sequence, and spanning from the N- to C-terminal ends, consists of a signal peptide, an EMIlin domain, two EGF repeats and a conserved C-terminus domain. The EMIlin domain is a small, cystein-rich module initially characterized in extracellular matrix molecules, such as emilin and multimerin, that mediates protein-protein interactions (21,22). EGF repeats are common among the extracellular regions of various membrane-bound and secreted proteins, and are involved in a wide range of cellular functions, such as adhesion, coagulation and ligand/receptors interactions (23). The C-terminal sequence of Egfl7 has no significant homology with other known proteins, and part of it is markedly conserved among species, suggesting that this region has unknown roles. Despite the characterization of Egfl7 over a decade ago (11–13), there are a lack of studies examining the potential activities of its domains, except for the observation that the EMI and EGF domains interact with Notch receptor 1 (Notch1) in vitro (24). In particular, the roles of the Egfl7 domains in promoting endothelial cell anergy, limiting blood vessel transmigration of immune cells under normal conditions and promoting cancer development are yet to be determined.
The present study produced retroviruses coding for mouse Egfl7 variants with EMI, EGF or C-terminal domain deletions, and infected these into breast cancer 4T1 cells, which consequently overexpressed the variants. The contribution of these domains to cancer cell proliferation and anchorage-independent growth in vitro was analyzed. In addition, immunocompetent mice were challenged with cells expressing full-length and mutated versions of Egfl7, followed by assessment of tumor development and immune infiltration.
Materials and methods
Cell culture
Mouse mammary carcinoma 4T1 cells (cat. no. ATCC CRL-2539) were obtained from LGC Limited and were not further tested or authenticated. 4T1 cells were cultured in RPMI (Thermo Fisher Scientific, Inc.) supplemented with 10% FBS (HyClone; Cytiva), 100 U/ml penicillin and 100 µg/ml streptomycin (Thermo Fisher Scientific, Inc.) in a humidified 95% air/5% CO2 incubator at 37°C.
Jurkat immortalized T-lymphocytes cells (cat. no. ATCC TIB-152) were obtained from American Type Culture Collection and were not further characterized. Jurkat cells were cultured in RPMI supplemented with 10% FBS, 100 U/ml penicillin and 100 µg/ml streptomycin in a humidified 95% air/5% CO2 incubator at 37°C. Cells were checked for the presence of mycoplasma using the MycoAlert™ Mycoplasma Detection Kit (Lonza Group Ltd.) on a monthly basis and not used if identified as positive.
Domain database analyses
Predictions of the conserved domains of mEgfl7 were performed using National Center for Biotechnology Information (NCBI) conserved domains (https://www.ncbi.nlm.nih.gov/Structure/cdd) and the Prosite (http://smart.embl-heidelberg.de) databases using the NP_001158036 referenced sequence.
Production of cells expressing Egfl7 variants
The mouse Egfl7 cDNA or domain deletion variants corresponding to the removal of the EMI, EGF or C-terminal domains were cloned in frame with a C-terminal influenza hemagglutinin (HA)-coding sequence in the pMSCV plasmid for 4T1 cells (Clontech Laboratories, Inc.) and pLPCX vector for HUVEC (Clontech Laboratories, Inc.), allowing the production of retroviruses as described (19). Human umbilical vein endothelial cells (HUVEC) (Lonza Group, Ltd.) and 4T1 cells (7.5×103 cells/cm2) were infected with control, Egfl7- or Egfl7-deletion domain coding retroviruses for 6 h at 37°C, and whole cell populations were selected for puromycin resistance (4 µg/ml) for 7 days, after which they were used for experiments. Infected cells were used after the antibiotic selection period and not maintained further. pLPCX-mEgfl7 or pLPCX-mEgfl7-ΔEGF vectors were constructed by cloning the c-terminal HA-tagged mEgfl7 coding sequence (11) or the mEgfl7-ΔEGF deletion variant into the HindIII/EcoRI sites of the pLPCX vector. For transfection, HUVEC (4×104 cells/well) were plated in 2-cm2 well plates and transfected with 58 fmoles pLPCX-Ctrl, pLPCX-mEgfl7 or pLPCX-mEgfl7-ΔEGF using JetPEI-HUVEC (Polyplus-transfection SA) for 4 h at 37°C, before the culture medium was changed. At 48 h post-transfection, cells were lysed and RNA was extracted.
Anchorage-independent growth soft-agar assay
A basal layer of complete medium [RPMI (Thermo Fisher Scientific, Inc.) supplemented with 10% FBS (HyClone; Cytiva), 100 U/ml penicillin, 100 µg/ml streptomycin (Thermo Fisher Scientific, Inc.)] and 0.6% agar was poured into 4-cm2 well plates. A suspension of 4T1-Ctrl, -Egfl7, -ΔEMI, -ΔEGF and -ΔCt cells (2.5×103/well) in 0.45% agar was overlaid onto the basal agar layer. Complete medium was added on top of the cell-containing agar layer immediately after gelling. Cells were cultured in a humidified 95% air/5% CO2 incubator at 37°C and the culture medium was changed twice a week. The number of colonies (defined as round, white & well defined cell groups, ≥80 pixels in size) was determined at 14 days after plating by taking pictures of the illuminated plates on a dark background under a Nikon SMZ1000 binocular light microscope and analyzed with ImageJ software (version 1.53h; National Institutes of Health) (25).
Cell proliferation assay
4T1-Ctrl, -Egfl7, -ΔEMI, -ΔEGF and -ΔCt cells were seeded (1.25×103 cell/cm2) into 4-cm2 well plates and cultured in a humidified 95% air/5% CO2 incubator at 37°C. At 24 h intervals, cells were collected after trypsin digestion and counted using a Z2 Coulter counter (Beckman Coulter, Inc.).
Mouse tumor model
A total of 70 female BALB/c mice (age, 6–8 weeks; weight, 20 g) obtained from Charles River Laboratories (n=7 per experimental group in each experiment) were maintained in a pathogen-free environment (12-h light/dark cycles; 22±2°C; 55±15% humidity) with ad libitum access to water and food. Mice were housed at least one week prior to tumor grafting to allow appropriate acclimatization. 4T1 cells (5×105/50 µl) expressing HA-tagged Egfl7 or its domain variants were injected in the mammary fat-pad and tumors were measured every other day using an electronic caliper. Tumor volumes were calculated using the following formula: Volume=width × width × length × π/6 (26). The maximal tumor volume observed was 650 mm3, the maximal length was 14.8 mm and the maximal width was 8.5 mm. Mice were sacrificed via cervical dislocation. Mice were housed according to European legislation (http://data.europa.eu/eli/dir/2010/63/oj) at the Institut Pasteur de Lille Animal Facility. The protocols were approved by the French Ministry of Higher Education and Research Ethic Committee (approval no. 00294.01).
Immunohistochemistry
Tumors were either fixed in 4% paraformaldehyde at 4°C for 24 h, embedded in paraffin and sectioned (thickness, 7 µm) for H&E and NCR1 staining, or frozen in optimal cutting temperature compound in N2-cooled liquid isopentane, sectioned at −20°C (thickness, 10 µm) and post-fixed with 1% paraformaldehyde for 5 min at room temperature for CD3ε and CD31 detection. Immunostaining was performed as previously described (19). Briefly, sections were incubated in 10 mM Tris-HCl pH 7.5, 150 mM NaCl (TBS) containing 1% BSA (MilliporeSigma) and 10% goat serum (MilliporeSigma) for 2 h at room temperature, then in TBS containing 1% BSA and the following primary antibodies: CD3ε (1:100; Abcam; cat. no. ab5690), natural cytotoxicity triggering receptor 1 (NCR1; 1:100; R&D Systems, Inc.; cat. no. AF2225) and CD31 (1:100; BD Pharmingen; BD Biosciences; cat. no. 550274) for 1 h at room temperature. Subsequently, the sections were incubated with a donkey anti-rabbit Alexa fluor-488 (cat. no. A21206; 1:250; Thermo Fisher Scientific, Inc.) or a goat anti-rat Alexa Fluor 594 secondary antibody (cat. no. A11007; 1:250; Thermo Fisher Scientific, Inc.) for 1 h at room temperature. For optical microscopy, sections were H&E counterstained with Mayer's hematoxylin (MilliporeSigma) for 5 sec and then 1% eosin (MilliporeSigma) for 30 sec at room temperature. After rinsing for 10 min under tap water, the sections were dehydrated and mounted in Vectamount (Vector Laboratories). For immunofluorescence microscopy, cell nuclei were labeled by staining the sections with DAPI solution (1 µg/ml; Sigma-Aldrich; Merck KGaA) for 1 min at room temperature. Sections were analyzed and images were captured using an Axioplan-2 light microscope or an AxioImager-Z1 fluorescent microscope (Carl Zeiss AG) and ZEN software (version 2.0). Quantification was performed either by direct counting or by area fraction measurements over ≥5 separate microscopic fields using ImageJ software.
RNA extraction and reverse transcription-quantitative PCR (qPCR)
4T1 cells, HUVEC or tumors fragments were homogenized in TRIzol® (Thermo Fisher Scientific, Inc.). Total RNA was extracted and reverse transcribed using a High Capacity cDNA reverse transcription kit (Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol. qPCR was performed in duplex reactions of human or mouse FAM-labeled probes (Applied Biosystems; Thermo Fisher Scientific, Inc.; mouse Egfl7, Mm00618004_m1; human Egfl7, Hs00211952_m1; human ICAM-1, Hs00164932_m1; human VCAM-1, Hs01003372_m1; human E-selectin, Hs00950401_m1), together with a human (cat. no. 4326315E) or mouse (cat. no. 4352341E) β-actin VIC-labeled probe using TaqMan gene expression assays, reagents (Applied Biosystems; Thermo Fisher Scientific, Inc.) and conditions on a StepOne machine (Applied Biosystems; Thermo Fisher Scientific, Inc.). The following thermocycling conditions were used for qPCR: 50°C for 2 min; initial denaturation at 94°C for 10 min; followed by 40 cycles of denaturation at 94°C for 15 sec, and an annealing and extension step at 60°C for 1 min. Cq values were calculated at the upper linear range of the logarithm-2 amplification curve using StepOne software (version v2.3; Applied Biosystems; Thermo Fisher Scientific, Inc.). Expression was quantified using the 2−ΔΔCq method (27), where ΔCq = Cq of transcript of interest-Cq of β-actin measured in the same tube, and ΔΔCq = mean ΔCq experimental samples - mean ΔCq control samples of the same experiment. Relative quantity is 2−ΔΔCq and transforms the logarithmic-2 data into decimal values.
Protein extraction and western blotting
Total protein was isolated from 4T1 cells or tumor tissues using RIPA buffer containing complete protease inhibitors (Roche Diagnostics, Inc.). Proteins were quantified using a BCA protein assay kit (Pierce; Thermo Fisher Scientific, Inc.), separated (62.5 µg per lane) via 12% SDS-PAGE and transferred onto Immobilon-P membranes (Merck Millipore). The membranes were incubated for 4 h at room temperature with primary antibodies targeted against HA.11 (Clone 16B12; 1:1,500; cat. no. MMS-101P; Covance, Inc.) and actin (cat. no. sc-1615; 1:1,000; Santa Cruz Biotechnology, Inc.). Subsequently, the membranes were incubated with anti-mouse (1:10,000; cat. no. NA931-1ML; Cytiva) or anti-goat-HRP conjugated secondary antibody (1:10,000; cat. no. A8919; Sigma-Aldrich; Merck KGaA) for 1 h at room temperature. Chemiluminescence was revealed using ECL Prime western blotting kit (Thermo Fisher Scientific, Inc.) either by exposure to autoradiography films (Cytiva) or by using a Fujifilm Las3000 gel reader (FUJIFILM Wako Pure Chemical Corporation) for semi-quantification.
Immune-endothelial cell adhesion assay
Human Jurkat T-lymphocytes (1×106 cells) were incubated with DiI (2 µM; Molecular Probes; Thermo Fisher Scientific, Inc.) for 10 min at 37°C. After washing with RPMI, Jurkat cells were seeded (105 cells/cm2) onto monolayers of confluent HUVEC for 20 min at 37°C. Cells were rinsed three times with PBS and then fixed in 4% paraformaldehyde for 20 min at room temperature. Fluorescent cells were imaged with a UV-microscope. The number of adherent cells was estimated using an area fraction calculation, which was applied to images of 10–15 separate microscope fields, using ImageJ software (version 1.53h; National Institutes of Health) (25).
Statistical analysis
Each experiment was performed at least twice with at least triplicate samples per condition. Comparisons among groups were analyzed using the Kruskal-Wallis test followed by Dunn's post hoc test. Statistical analyses were performed using GraphPad Instat software v3.10 (GraphPad Software, Inc.). P<0.05 was considered to indicate a statistically significant difference. Error bars in graphs represent the calculated standard deviation.
Results
Egfl7 and its domain deletion variants have no effect on cancer cell proliferation in vitro
To analyze the roles of the Egfl7 domains in tumor growth and the escape from immunity, the present study designed retroviral expression vectors encoding either the full-length molecule or domain-deletion variants of the protein. Based on NCBI and Prosite database predictions, the N-terminal domain spans Met1-Pro27, the EMI domain is defined as the sequence containing Ser28-Ala105 and the EGF repeats span Ala106-Lys180 (Fig. 1A). Moreover, the C-terminal domain from the Glu181 to Leu275 end of Egfl7 does not contain any predicted protein feature. Vectors coding for C-terminal HA-tagged versions of the full-length protein and deletion variants corresponding to the removal of the EMIlin domain (ΔEMI), the EGF repeats (ΔEGF) or the C-terminal domain (ΔCt) (Fig. 1A) were cloned and produced into retroviruses that were then used to infect mouse mammary 4T1 carcinoma cells. Cells were subsequently selected for resistance to antibiotics as whole populations. Each construct was shown to lead to efficient infection and expression, since the selected cell populations produced a HA-tagged protein at the expected apparent size when assessed via western blotting (Fig. 1B).
 |
Figure 1.
Egfl7 and its domain variants have no effect on cancer cell proliferation in vitro. (A) Schematic representation of the full-length Egfl7 protein and its deletion variants where the EMI domain (ΔEMI), EGF repeats (ΔEGF) or C-terminal domain (ΔCt) were deleted. Numbers indicate residues in the mouse Egfl7 primary sequence. (B) Mouse breast carcinoma 4T1 cells were infected with a Ctrl retrovirus or retroviruses coding for the HA-tagged Egfl7 protein or deletion variants. Proteins were separated via 12% SDS-PAGE and analyzed by western blotting using anti-HA antibody and anti-actin antibodies. Numbers refer to the apparent molecular weights (kDa). (C) 4T1 cells expressing Ctrl retrovirus or a deletion variant retrovirus were seeded (1.25×103 cells/cm2) in culture dishes and proliferation was assessed by counting cells at indicated time points. (D) 4T1 cells expressing Ctrl retrovirus or a deletion variant (2.5×103/well) were mixed with 0.45% agar, seeded on top of a 0.6% basal agar layer and grown for 14 days. Subsequently, the (E) number of colonies and (F) average colony size were measured using ImageJ software (scale bar, 250 µm). The experiments were performed twice in triplicate conditions. Data are presented as the mean ± standard deviation. Ctrl, control; ns, not significant; Egfl7, epidermal growth factor-like domain 7; HA, hemagglutinin; Ct, C-terminal domain, SP, signal peptide.
|
Uncontrolled proliferation of cancer cells is a major hallmark of tumors (28), and Egfl7 has been shown to have significant effects on cell proliferation depending on the conditions and cell type selected (19,24,29–31). Under the conditions, overexpressing full-length Egfl7 in 4T1 cells had no effects on the cell proliferation rate compared with the 4T1-control (Ctrl) group (Fig. 1C). Similarly, deleting EMI, EGF or C-terminal domains had no effect on the proliferation rate of the 4T1-ΔEMI, -ΔEGF and -ΔCt cells (Fig. 1C), suggesting that Egfl7 and its domain variants did not participate in the regulation of 4T1 cancer cell proliferation under these conditions.
Anchorage-independent colony formation in vitro is another characteristic of cell transformation (32), and 4T1 cells are spontaneously able to form colonies under such conditions. When assessing the potential effects of Egfl7 and its domain deletion variants on colony formation by 4T1 cells, no significant differences were identified between the number of colonies emerging from 4T1-Ctrl cells and those expressing the full-length Egfl7. Variants with deletion of the EMI, EGF or the C-terminal domains also showed no differences in colony numbers (Fig. 1D and E). However, there was a tendency to a size difference of the colonies formed by 4T1-ΔEGF cells, which were on average 16% smaller compared with those formed by 4T1-Ctrl and 4T1-Egfl7 cells (Fig. 1F).
EGF repeats of Egfl7 are involved in promoting tumor growth in vivo
Despite displaying no noticeable effects on cancer cell proliferation in vitro, Egfl7 does significantly promote tumor growth in vivo (19). To identify which of its predicted domains were involved in this effect, 4T1 cells expressing either the full length Egfl7, its domain deletion variants or 4T1-Ctrl cells were inoculated into BALB/c immunocompetent mice, and tumor growth was monitored over time. The results demonstrated that 4T1-Egfl7 cells produced tumors that grew faster compared with those of 4T1-Ctrl cells, reaching 395±90 mm3 after 20 days, whereas 4T1-Ctrl tumors reached 135±55 mm3 after the same time period (Fig. 2A). Deletion of the EMI or C-terminal domain from Egfl7 did not significantly affect the growth rates of the corresponding tumors, which remained comparable to the fast growth rate of 4T1-Egfl7 tumors. On the other hand, deleting the EGF repeats significantly decreased the rate of tumor growth in mice, resulting in a ~27% smaller mean volume (290±65 mm3) compared with 4T1-Egfl7 tumors at 20 days post-inoculation (Fig. 2A). Furthermore, although the mean tumor weight at sacrifice in the 4T1-Egfl7 group was significantly higher compared with that in the 4T1-Ctrl group, a statistical difference was not observed between 4T1-Ctrl and 4T1-ΔEGF tumors, which was in line with the data obtained from the tumor growth kinetics results (Fig. 2B). As previously described, 4T1-Egfl7 formed highly necrotic tumors when compared with 4T1-Ctrl cells (19), and deleting the EGF domains significantly decreased the levels of intratumor necrosis (Fig. 2C and D).
 |
Figure 2.
Deletion of the EGF repeats from Egfl7 reduces tumor development in vivo. (A) 4T1-Ctrl or 4T1 cells expressing an Egfl7 domain deletion variant were injected (5×105 cells/50 µl PBS) into the mammary fat-pad of BALB/c mice. Tumor volume was monitored overtime. Statistics were calculated at 20 days. (B) Mean tumor weights measured at sacrifice. (C) Sections along the middle axes of representative 4T1-Ctrl, 4T1-Egfl7 and 4T1-ΔEGF tumors stained with hematoxylin and eosin and imaged using a light microscope (scale bar, 1,000 µm). (D) Necrosis was identified as unstained areas in H&E sections of tumors and quantified as a percentage of total tumor area. (E) Expression levels of Egfl7 transcripts in tumors were assessed via duplex reverse transcription-quantitative PCR. The results are expressed relative to 4T1-Ctrl values, which were set to 1. Expression levels were quantified using the 2−ΔΔCq and normalized to the internal reference gene actin. (F) Expression levels of Egfl7 protein in tumors were assessed by SDS-PAGE and western blotting for the HA-tag and actin. The experiments were performed twice with 7 animals per group each time. Data are presented as the mean ± SD. *P<0.05 and **P<0.01. Ctrl, control; ns, not significant; Egfl7, epidermal growth factor-like domain 7; HA, hemagglutinin; Ct, C-terminal domain; RQ, relative quantity.
|
To verify that variations in Egfl7 expression did not account for these differences, Egfl7 transcript levels were quantified in RNA extracted from the dissected tumors. As expected, Egfl7 expression levels were significantly higher in 4T1-Egfl7 tumors compared with those in 4T1-Ctrl tumors, whereas Egfl7 expression levels were similar between 4T1-Egfl7 and 4T1-ΔEGF tumors (Fig. 2E). Similarly, Egfl7-HA protein expression levels were significantly higher in 4T1-Egfl7 tumors compared with those in 4T1-Ctrl tumors. Moreover, Egfl7-HA protein expression levels were similar between 4T1-Egfl7 and 4T1-ΔEGF tumors (Figs. 2F and S1), and did not appear to account for the observed differences in tumor development.
EGF repeats of Egfl7 are involved in tumor escape from the immune system
The promoting effects of Egfl7 on tumor growth are due, at least in part, to the repression of blood vessel endothelial cell activation, which in turn prevents the infiltration of CD4+ and CD8+ T-lymphocytes, NK cells and other immune cells within the tumor mass (19). To assess whether the slower growth rate of 4T1-ΔEGF tumors was associated with partially restored immune infiltration, 4T1-Ctrl, -Egfl7 and -ΔEGF tumors were analyzed for T cell and NK cell infiltration using immunohistochemistry. 4T1-Egfl7 tumors were significantly less infiltrated by T-lymphocytes and NK cells compared with 4T1-Ctrl tumors, as demonstrated by staining for the CD3ε T-cell and NCR1 NK markers (Fig. 3A and B). Quantification of T-cell infiltration demonstrated that 24.3±4.2 CD3ε+ cells/field were present in 4T1-Ctrl tumor sections, whereas this number decreased to 3.2±0.7 cells/field in 4T1-Egfl7 tumors. With regards to the infiltration of NK cells, 23.2±6.2 NCR1+ cells/field were counted in 4T1-Ctrl tumor sections compared with only 8.0±0.4 cells/field in 4T1-Egfl7 tumors. By contrast, deleting the EGF repeats from Egfl7 partially restored the immune infiltration of tumors, with the number of infiltrated CD3ε+ T-lymphocytes and NCR1+ NK cells increasing by 2.82-fold (8.9±0.0 cells/field) and 1.69-fold (13.5±3.1 cells/field) in 4T1-ΔEGF tumors compared with 4T1-Egfl7 tumors (Fig. 3A and B). This increase in immune infiltration of 4T1-ΔEGF tumors suggested that the EGF repeats of Egfl7 may actively participate in preventing the recruitment of cytotoxic immune cells by tumor blood vessel endothelial cells expressing wild-type Egfl7.
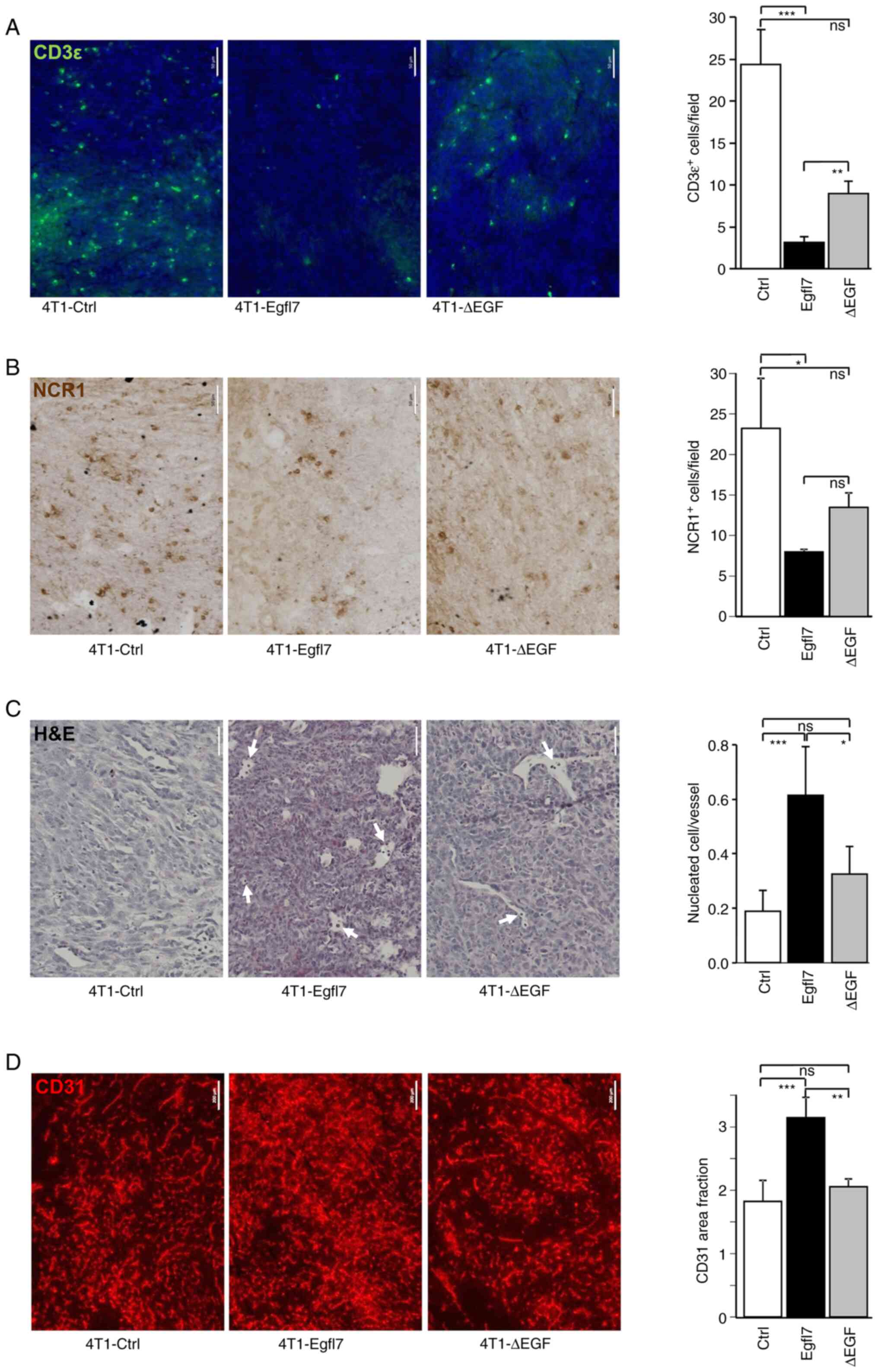 |
Figure 3.
Deletion of EGF repeats from Egfl7 decreases immune infiltration and blood vessel density in tumors. Immunostaining of 4T1-Ctrl, 4T1-Egfl7 and 4T1-ΔEGF tumor sections (A) for the pan-T cell marker CD3ε (green) and (B) for the NK cell marker NCR1 (brown) (scale bar, 50 µm). The graphs present the quantification of labelled cells. (C) H&E staining of 4T1-Ctrl, 4T1-Egfl7 and 4T1-ΔEGF tumor sections. Arrows indicate nucleated cells in blood vessels. The graph presents the quantification of nucleated cells. (D) Immunostaining of 4T1-Ctrl, 4T1-Egfl7 and 4T1-ΔEGF tumor sections for the endothelial cell marker CD31 (red) (scale bar, 200 µm). The graphs present the quantification of area fraction measurements over ≥5 separate microscopic fields of (A,B) labeled cells, (C) nucleated cells or (D) area fraction measurements. The experiments were performed twice with 7 animals per group each time. Data are presented as the mean ± SD. *P<0.05, **P<0.01 and ***P<0.001. ns, not significant. Ctrl, control; Egfl7, epidermal growth factor-like domain 7; NK, natural killer; NCR1, natural cytotoxicity triggering receptor 1.
|
Our previous study reported that immune cells that were not able to cross the tumor endothelial barrier in 4T1-Egfl7 tumors, thus remained within the lumens of these blood vessels (19). Indeed, microscopic observations of tumor sections revealed the presence of a significantly higher number of nucleated cells in the lumens of 4T1-Egfl7 tumor blood vessels compared with those in 4T1-Ctrl tumors blood vessels (Fig. 3C). Furthermore, such circulating cells were observed in significantly lower numbers in 4T1-ΔEGF tumor blood vessels compared with those in 4T1-Egfl7 tumors, but in significantly higher numbers compared with those in 4T1-Ctrl tumors, thereby confirming the role of EGF repeats in immune cell recruitment by tumor blood vessels.
Expression of Egfl7 by cancer cells promotes tumor angiogenesis in vivo (19). This effect was demonstrated in the present study by assessing the proportion of CD31+ blood vessels in tumor sections. It was found that blood vessel density was significantly increased in 4T1-Egfl7 tumors compared with that in 4T1-Ctrl tumors, whereas 4T1-ΔEGF tumors were significantly less vascularized, with blood density levels comparable to those measured in 4T1-Ctrl tumors (Fig. 3D), suggesting that the EGF repeats also actively participated in the tumor angiogenic effects of Egfl7.
EGF repeats of Egfl7 are necessary for repressing endothelial cell activation in vitro
The repression of endogenous Egfl7 activates endothelial cells in the absence of proinflammatory cytokines, which is due to an increase in the expression levels of ICAM-1, VCAM-1 and E-selectin, and a consequential spontaneous adhesion of leukocytes to endothelial cells following Egfl7 repression (19). Since HUVEC spontaneously express large amounts of human Egfl7 (hEgfl7), to evaluate whether the EGF repeats of Egfl7 were involved in endothelial cell activation in vitro, corresponding mouse Egfl7 variants were overexpressed in HUVEC. It was first examined whether overexpressing mouse Egfl7 (mEgfl7) molecules would artificially affect hEgfl7 endogenous expression. The results demonstrated that neither mEgfl7 or mEgfl7-ΔEGF transfection did so. As expected, high expression levels of mEgfl7 transcripts were detected after transfecting cells with the mouse expression vectors (Fig. 4A).
 |
Figure 4.
EGF repeats of Egfl7 participate in regulating leukocyte adhesion receptor expression and endothelial activation in vitro. (A) HUVEC were transfected with pLPCX retroviral expression plasmids encoding mEgfl7, Egfl7-ΔEGFs (mΔEGF) or Ctrl. Expression levels of human (left) and mouse (right) Egfl7 transcripts were assessed 48 h later. (B) HUVEC were infected with retroviruses and assessed for the expression of the human leukocyte adhesion receptors, ICAM-1, VCAM-1 and E-SEL, via Taqman reverse transcription-quantitative PCR. (C) HUVEC were infected with retroviruses and grown to confluence. Fluorescently-labeled Jurkat T-cells (green) were assessed for adhesion to the surface of transfected HUVEC after 20 min (scale bar, 200 µm). The graph presents the quantification of area fraction measurements of images over 10–15 separate microscopic fields of view. The experiments were performed twice in triplicate conditions. Data are presented as the mean ± SD. *P<0.05. ns, not significant; mEgfl7, mouse Egfl7; E-SEL, E-selectin; Ctrl, control; Egfl7, epidermal growth factor-like domain 7; ICAM-1, intercellular adhesion molecule-1; VCAM-1, vascular cell adhesion molecule 1; HUVEC, human umbilical vein endothelial cell; RQ, relative quantity.
|
Then, HUVEC were stably infected with retroviruses produced from the same mouse expression vectors, and whether overexpression of mEgfl7 could repress endogenous leukocyte adhesion receptors in the presence of endogenous hEgfl7 was assessed. The expression levels of ICAM-1 did not significantly vary after infecting HUVEC with either mEgfl7 or mEgfl7-ΔEGF. However, mEgfl7 overexpression did significantly repress VCAM-1 and E-selectin expression in HUVEC, and the removal of the EGF repeats of mEgfl7 partially and totally relieved this repression, respectively (Fig. 4B).
Preliminary data (data not shown) suggested that the Egfl7 protein directly interfered with the binding of leukocytes to the endothelium. To assess whether the effects would correspond to functional differences in HUVEC activation in response to the mEgfl7 forms, fluorescent Jurkat leukocytes were incubated onto confluent monolayers of HUVEC infected with Ctrl, mEgfl7 or mEgfl7-ΔEGF constructs. Under these conditions, the HUVEC-Ctrl group spontaneously supported a high rate of leukocyte adhesion (Fig. 4C) as a consequence of the retroviral infection, whereas overexpression of mEgfl7 significantly reduced the number of leukocytes adhering in the HUVEC-Egfl7 group (Fig. 4C). On the other hand and in line with the aforementioned results, the absence of the EGF repeats from mEgfl7 induced higher leukocyte adhesion levels in the HUVEC-ΔEGF group compared with those in the HUVEC-mEgfl7 group, and reached comparable levels to those observed in the HUVEC-Ctrl group.
Collectively, these results suggested that the EGF repeats of Egfl7 formed a key domain that participated in promoting tumor growth in vivo by reducing leukocyte infiltration into tumors and promoting angiogenesis. Moreover, these domains participated in regulating endothelial cell activation and their ability to interact with circulating leukocytes.
Discussion
To the best of our knowledge, the present study was the first to address the roles of the protein domains of Egfl7 in a tumoral context. In vitro, the expression of Egfl7 or the deletion domain variants had no effects on cancer cell proliferation. In vivo, the EGF repeats of Egfl7 served a significant role in the tumor-promoting effects of this protein and in reducing immune cell infiltration. These domains were also necessary to repress endothelial activation and leukocyte adhesion.
Our previous study described the mechanisms via which Egfl7 affects tumor growth. For example, overexpression of Egfl7 in cancer cells promotes tumor development by preventing the infiltration of immune cells from the blood circulation into the tumor mass (19). This is achieved in part by Egfl7-mediated downregulation of leukocyte adhesion receptors at the surface of tumor blood vessel endothelial cells, which impairs immune cell infiltration into tumors. Furthermore, in clinical samples of human cancer types, high expression levels of Egfl7 are correlated with low endothelial cell expression of ICAM-1 in the peritumoral vessels of breast lesions (19,20), thus confirming the existence of an inverse association between the expression levels of Egfl7 and tumor endothelial cell activation in patients (19,20,33). The present study demonstrated that the EGF repeats were essential to the protumoral effects of Egfl7 and to the repression of endothelial cell activation. Since the study design was aimed at deleting the Egfl7 domains without affecting the other predicted domains, it is unlikely that removing the EGF repeats profoundly disrupted the folding of the Egfl7 protein. Furthermore, earlier observations indicating that the EGF and EMI domains were necessary for the interaction between Egfl7 and Notch1 suggested that the Egfl7 proteins lacking the EGF repeats or the EMI domain were still able to interact with Notch1 (24). This suggested that the remaining domains of Egfl7 were structurally preserved in the Egfl7-ΔEGF mutants and that the loss of functions observed here were most probably due to the specific role of these domains.
Further examining the mechanistic roles of the EGF repeats of Egfl7 based on the literature is difficult as these domains do not appear to have a unique and well-defined function. EGF repeats are defined by a sequence of 30–40 amino acids found in the sequence of EGF and are present, in a more or less conserved form and in tandem repeats, in a large number of proteins, including growth factors, receptors, clotting factors and extracellular matrix proteins. These repeated domains are generally located in the extracellular portion of transmembrane proteins or in secreted proteins, and are primarily involved in protein-protein interactions (23). A subset of EGF repeats can bind calcium and serve important roles in protein folding and protein-protein interactions (34–36). In the Notch receptor family, which is involved in major regulation processes in angiogenesis and cancer (37), the extracellular domain of the receptors contains several tens of EGF repeats, some of which are required for ligand binding. Furthermore, isolated EGF repeats of Notch1 affect cancer cell adhesiveness and proliferation (38). Interestingly, Notch signaling is modified by the post-translational O-glycosylation of its EGF repeats, which appears to be a common feature of these domains (39). Developmental endothelial locus 1 (Del-1) is a glycoprotein secreted by endothelial cells that contains three EGF repeats. The second EGF repeat contains an Arg-Gly-Asp (RGD) motif that supports the association with extracellular matrix components (40). Del-1 is a potent angiogenic factor (41) that promotes tumor growth via the induction of blood vessel formation (42). In addition, Del-1 represses the interaction of circulating leukocytes with endothelial cells, and serves as an antiadhesive factor that interferes with the integrin lymphocyte function-associated antigen 1-dependent leukocyte-endothelial adhesion, thereby preventing leukocyte adhesion to the endothelium (43,44). Of note, the first domain of hEgfl7 contains an RGD sequence that is not conserved in mouse Egfl7, and the second EGF domain of Egfl7 is predicted to bind Ca2+ (11). However, whether the EGF repeats of Egfl7 are glycosylated or participate in protein-protein interactions or docking of Egfl7 into the extracellular matrix (45) remains unknown. Preliminary data (data not shown) suggested that the Egfl7 protein directly interfered with the binding of leukocytes to the endothelium, but whether the EGF repeats are involved in this interaction requires further investigation.
In the present study, the expression of Egfl7 or its deletion variants in 4T1 cancer cells did not affect the proliferation rate of the cells in vitro, which was in accordance with our previous observations using breast cancer 4T1 and lung carcinoma LLC1 cells that showed no variation in cell proliferation rates upon overexpression of Egfl7 (19). On the other hand, the repression of Egfl7 in infected HUVEC or knocking down its expression in embryonic stem cells did repress cell proliferation (29,30), whereas a similar repression in endothelial colony-forming cells promoted proliferation and migration (31). In another study, overexpression of Egfl7 repressed adult neural stem cell proliferation and self-renewal capacities (24). Collectively, these opposed observations suggest that the role of Egfl7 in cell proliferation is highly dependent on the cell type and the tissue context in which the protein is expressed.
Depending on the context, Egfl7 induces either agonistic or antagonistic effects on the Notch signaling pathway (24,30). The Notch signaling pathway is involved in regulating endothelial cell activation via a cross-talk with the NFκB signaling pathway (46–48). Our previous study examined the mechanisms via which Egfl7 regulates endothelial cell activation, and identified that Egfl7 repressed TNFα-induced activation via the active repression of the NFκB signaling pathway, thereby limiting the expression of ICAM-1, VCAM-1 and E-selectin (14). The fact that the EGF repeats participate in endothelial cell activation and that they are involved in the interaction between Egfl7 with Notch receptors suggests that Egfl7 could regulate, at least in part, the activation of endothelial cells by interfering with the cross-talk between the Notch and NFκB signaling pathways.
The present study used the 4T1 breast tumor model to evaluate the effects of Egfl7 domains on cancer growth. Our previous study showed that Egfl7 promoted tumor growth using an additional tumor model of lung adenocarcinoma LLC1 mouse tumor cells implanted in immunocompetent mice, which also demonstrated that Egfl7 did not affect LLC1 proliferation in vitro (19). Since Egfl7 acts on endothelial cell activation and not on tumor cells, it would be expected that the EGF repeats of Egfl7 have similar roles in other tumor models as well.
In conclusion, the present study demonstrated that the EGF repeats of Egfl7 may participate in the protumoral, proangiogenic and anti-inflammatory effects of Egfl7, whereas the other identified domains of the protein did not appear to be significantly involved in these functions.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr Elisabeth Werkmeister at the BioImaging Center Lille (US 41-UMS 2014-PLBS, BICeL; http://www.bicel.org/) platform.
Funding
The present study was funded by grants from the Institut National du Cancer (grant no. PLBIO 2013-100) and the Ligue contre le Cancer-Comité de l'Aisne and the Fondation ARC (grant no. SFI20111203644). SD was supported by a PhD fellowship from CNRS-Région Nord-Pas de Calais.
Availability of data and materials
The materials used during the current study are available from the corresponding author on reasonable request. Data sharing is not applicable to this article, as no datasets were generated or analyzed during the current study.
Authors' contributions
SP, SD, VM and FS performed the experiments and analyzed the data. CH and GV performed vector cloning, and gene and protein expression analyses. FS obtained funding, designed the study and wrote the manuscript. All authors read and approved the final manuscript. SP and FS confirm the authenticity of all the raw data.
Ethics approval and consent to participate
The protocols were approved by the French Ministry of Higher Education and Research Ethic Committee (approval no. 00294.01).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Authors' information
FS is Directeur de Recherche at Institut National de la Santé et de la Recherche Médicale.
Glossary
Abbreviations
Abbreviations:
|
Egfl7
|
epidermal growth factor-like domain 7
|
|
ICAM-1
|
intercellular adhesion molecule-1
|
|
NK
|
natural killer
|
|
RQ
|
relative quantity
|
|
VCAM-1
|
vascular cell adhesion molecule-1
|
References
|
1
|
Castermans K and Griffioen AW: Tumor blood vessels, a difficult hurdle for infiltrating leukocytes. Biochim Biophys Acta. 1776:160–174. 2007.PubMed/NCBI
|
|
2
|
Wettschureck N, Strilic B and Offermanns S: Passing the vascular barrier: Endothelial signaling processes controlling extravasation. Physiol Rev. 99:1467–1525. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Herberman RB, Nunn ME, Holden HT and Lavrin DH: Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int J Cancer. 16:230–139. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shrikant P and Mescher MF: Control of syngeneic tumor growth by activation of CD8+ T cells: Efficacy is limited by migration away from the site and induction of nonresponsiveness. J Immunol. 162:2858–2866. 1999.PubMed/NCBI
|
|
5
|
Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ and Schreiber RD: Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 450:903–907. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu NZ, Klitzman B, Dodge R and Dewhirst MW: Diminished leukocyte-endothelium interaction in tumor microvessels. Cancer Res. 52:4265–4268. 1992.PubMed/NCBI
|
|
7
|
Kuzu I, Bicknell R, Fletcher CD and Gatter KC: Expression of adhesion molecules on the endothelium of normal tissue vessels and vascular tumors. Lab Invest. 69:322–328. 1993.PubMed/NCBI
|
|
8
|
Piali L, Fichtel A, Terpe HJ, Imhof BA and Gisler RH: Endothelial vascular cell adhesion molecule 1 expression is suppressed by melanoma and carcinoma. J Exp Med. 181:811–816. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Griffioen AW, Damen CA, Martinotti S, Blijham GH and Groenewegen G: Endothelial intercellular adhesion molecule-1 expression is suppressed in human malignancies: The role of angiogenic factors. Cancer Res. 56:1111–1117. 1996.PubMed/NCBI
|
|
10
|
Dirkx AE, Oude Egbrink MG, Kuijpers MJ, van der Niet ST, Heijnen VV, Bouma-ter Steege JC, Wagstaff J and Griffioen AW: Tumor angiogenesis modulates leukocyte-vessel wall interactions in vivo by reducing endothelial adhesion molecule expression. Cancer Res. 63:2322–2329. 2003.PubMed/NCBI
|
|
11
|
Soncin F, Mattot V, Lionneton F, Spruyt N, Lepretre F, Begue A and Stehelin D: VE-statin, an endothelial repressor of smooth muscle cell migration. EMBO J. 22:5700–5711. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Parker LH, Schmidt M, Jin SW, Gray AM, Beis D, Pham T, Frantz G, Palmieri S, Hillan K, Stainier DY, et al: The endothelial-cell-derived secreted factor Egfl7 regulates vascular tube formation. Nature. 428:754–758. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fitch MJ, Campagnolo L, Kuhnert F and Stuhlmann H: Egfl7, a novel epidermal growth factor-domain gene expressed in endothelial cells. Dev Dyn. 230:316–324. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pinte S, Caetano B, Le Bras A, Havet C, Villain G, Dernayka R, Duez C, Mattot V and Soncin F: Endothelial cell activation is regulated by epidermal growth factor-like domain 7 (Egfl7) during inflammation. J Biol Chem. 291:24017–24028. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Díaz R, Silva J, García JM, Lorenzo Y, García V, Peña C, Rodríguez R, Muñoz C, García F, Bonilla F and Domínguez G: Deregulated expression of miR-106a predicts survival in human colon cancer patients. Genes Chromosomes Cancer. 47:794–802. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu F, Yang LY, Li YF, Ou DP, Chen DP and Fan C: Novel role for epidermal growth factor-like domain 7 in metastasis of human hepatocellular carcinoma. Hepatology. 50:1839–1850. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang CH, Li XJ, Zhou YZ, Luo Y, Li C and Yuan XR: Expression and clinical significance of EGFL7 in malignant glioma. J Cancer Res Clin Oncol. 136:1737–1743. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhou L, Li J, Zhao YP, Guo JC, Cui QC, Zhou WX, Zhang TP, Wu WM, You L and Shu H: Prognostic significance of epidermal growth factor-like domain 7 in pancreatic cancer. Hepatobiliary Pancreat Dis Int. 13:523–528. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Delfortrie S, Pinte S, Mattot V, Samson C, Villain G, Caetano B, Lauridant-Philippin G, Baranzelli MC, Bonneterre J, Trottein F, et al: Egfl7 promotes tumor escape from immunity by repressing endothelial cell activation. Cancer Res. 71:7176–7186. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pannier D, Philippin-Lauridant G, Baranzelli MC, Bertin D, Bogart E, Delprat V, Villain G, Mattot V, Bonneterre J and Soncin F: High expression levels of egfl7 correlate with low endothelial cell activation in peritumoral vessels of human breast cancer. Oncol Lett. 12:1422–1428. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Doliana R, Bot S, Bonaldo P and Colombatti A: EMI, a novel cysteine-rich domain of EMILINs and other extracellular proteins, interacts with the gC1q domains and participates in multimerization. FEBS Lett. 484:164–168. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Callebaut I, Mignotte V, Souchet M and Mornon JP: EMI domains are widespread and reveal the probable orthologs of the caenorhabditis elegans CED-1 protein. Biochem Biophys Res Commun. 300:619–623. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Campbell ID and Bork P: Epidermal growth factor-like modules. Curr Opin Struct Biol. 3:385–392. 1993. View Article : Google Scholar
|
|
24
|
Schmidt MHH, Bicker F, Nikolic I, Meister J, Babuke T, Picuric S, Müller-Esterl W, Plate KH and Dikic I: Epidermal growth factor-like domain 7 (EGFL7) modulates Notch signalling and affects neural stem cell renewal. Nat Cell Biol. 11:873–880. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Abramoff MD, Magalhães P and Ram SJ: Image processing with imageJ. Biophot Int. 11:36–42. 2004.
|
|
26
|
Tomayko MM and Reynolds CP: Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol. 24:148–154. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chiu JJ: Shear stress increases ICAM-1 and decreases VCAM-1 and E-selectin expressions induced by tumor necrosis factor-[alpha] in endothelial cells. Arterioscler Thromb Vasc Biol. 24:73–79. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hanahan D and Weinberg RA: Hallmarks of cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Durrans A and Stuhlmann H: A role for Egfl7 during endothelial organization in the embryoid body model system. J Angiogenes Res. 2:42010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nichol D, Shawber C, Fitch MJ, Bambino K, Sharma A, Kitajewski J and Stuhlmann H: Impaired angiogenesis and altered Notch signaling in mice overexpressing endothelial Egfl7. Blood. 116:6133–6143. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
D'Audigier C, Susen S, Blandinieres A, Mattot V, Saubamea B, Rossi E, Nevo N, Lecourt S, Guerin CL, Dizier B, et al: Egfl7 represses the vasculogenic potential of human endothelial progenitor cells. Stem Cell Rev Rep. 14:82–91. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Taddei ML, Giannoni E, Fiaschi T and Chiarugi P: Anoikis: An emerging hallmark in health and diseases. J Pathol. 226:380–393. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Philippin-Lauridant G, Baranzelli MC, Samson C, Fournier C, Pinte S, Mattot V, Bonneterre J and Soncin F: Expression of Egfl7 correlates with low-grade invasive lesions in human breast cancer. Int J Oncol. 42:1367–1375. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fehon RG, Kooh PJ, Rebay I, Regan CL, Xu T, Muskavitch MA and Artavanis-Tsakonas S: Molecular interactions between the protein products of the neurogenic loci Notch and delta, two EGF-homologous genes in Drosophila. Cell. 61:523–534. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rebay I, Fleming RJ, Fehon RG, Cherbas L, Cherbas P and Artavanis-Tsakonas S: Specific EGF repeats of Notch mediate interactions with delta and serrate: Implications for Notch as a multifunctional receptor. Cell. 67:687–699. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Downing AK, Knott V, Werner JM, Cardy CM, Campbell ID and Handford PA: Solution structure of a pair of calcium-binding epidermal growth factor-like domains: Implications for the Marfan syndrome and other genetic disorders. Cell. 85:597–605. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ranganathan P, Weaver KL and Capobianco AJ: Notch signalling in solid tumours: A little bit of everything but not all the time. Nat Rev Cancer. 11:338–351. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hayakawa S, Koide R, Hinou H and Nishimura SI: Synthetic human NOTCH1 EGF modules unraveled molecular mechanisms for the structural and functional roles of calcium ions and O-glycans in the ligand-binding region. Biochemistry. 55:776–787. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Taylor P, Takeuchi H, Sheppard D, Chillakuri C, Lea SM, Haltiwanger RS and Handford PA: Fringe-mediated extension of O-linked fucose in the ligand-binding region of Notch1 increases binding to mammalian Notch ligands. Proc Natl Acad Sci USA. 111:7290–7295. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hidai C, Zupancic T, Penta K, Mikhail A, Kawana M, Quertermous EE, Aoka Y, Fukagawa M, Matsui Y, Platika D, et al: Cloning and characterization of developmental endothelial locus-1: An embryonic endothelial cell protein that binds the alphavbeta3 integrin receptor. Genes Dev. 12:21–33. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhong J, Eliceiri B, Stupack D, Penta K, Sakamoto G, Quertermous T, Coleman M, Boudreau N and Varner JA: Neovascularization of ischemic tissues by gene delivery of the extracellular matrix protein Del-1. J Clin Invest. 112:30–41. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Aoka Y, Johnson FL, Penta K, Hirata Ki K, Hidai C, Schatzman R, Varner JA and Quertermous T: The embryonic angiogenic factor Del1 accelerates tumor growth by enhancing vascular formation. Microvasc Res. 64:148–161. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Choi EY, Chavakis E, Czabanka MA, Langer HF, Fraemohs L, Economopoulou M, Kundu RK, Orlandi A, Zheng YY, Prieto DA, et al: Del-1, an endogenous leukocyte-endothelial adhesion inhibitor, limits inflammatory cell recruitment. Science. 322:1101–1104. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Choi EY: Inhibition of leukocyte adhesion by developmental endothelial locus-1 (del-1). Immune Netw. 9:153–157. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Villain G, Lelievre E, Broekelmann T, Gayet O, Havet C, Werkmeister E, Mecham R, Dusetti N, Soncin F and Mattot V: MAGP-1 and fibronectin control EGFL7 functions by driving its deposition into distinct endothelial extracellular matrix locations. FEBS J. 285:4394–4412. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Verginelli F, Adesso L, Limon I, Alisi A, Gueguen M, Panera N, Giorda E, Raimondi L, Ciarapica R, Campese AF, et al: Activation of an endothelial Notch1-Jagged1 circuit induces VCAM1 expression, an effect amplified by interleukin-1β. Oncotarget. 6:43216–43229. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Nus M, Martínez-Poveda B, MacGrogan D, Chevre R, D'Amato G, Sbroggio M, Rodríguez C, Martínez-González J, Andrés V, Hidalgo A and de la Pompa JL: Endothelial Jag1-RBPJ signalling promotes inflammatory leucocyte recruitment and atherosclerosis. Cardiovasc Res. 112:568–580. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Quillard T, Coupel S, Coulon F, Fitau J, Chatelais M, Cuturi MC, Chiffoleau E and Charreau B: Impaired Notch4 activity elicits endothelial cell activation and apoptosis: Implication for transplant arteriosclerosis. Arterioscler Thromb Vasc Biol. 28:2258–2265. 2008. View Article : Google Scholar : PubMed/NCBI
|














