Introduction
For approximately 40 years, cisplatin (CDDP) has
been approved for medical use in the treatment of a number of
cancers, including testicular, ovarian, bladder, and lung cancer,
among others (1). Recently, CDDP
has been used for the treatment of patients with acquired
resistance to molecular-targeted drugs. It has also been used in
combination with immune checkpoint inhibitors, such as
pembrolizumab (2,3). However, acquisition of CDDP
resistance leads to the failure of cancer therapy; therefore,
overcoming CDDP resistance remains an important obstacle to
successful cancer treatment. Although many researchers have
investigated the mechanism of acquired CDDP resistance, including
reduced drug accumulation (4),
increased detoxification by glutathione (5), and increased repair of CDDP-DNA
adducts (6), to date, there are
still no approved therapies for the treatment of patients with
acquired CDDP resistance.
We previously established high-grade CDDP-resistant
A549 cells (A549 cell-derived CDDP-resistant at 20 µM, ACR20 cells)
by culturing cells with 20 µM CDDP. The resistance of ACR20 cells
was 18.5-fold higher than that of A549 cells (7). We reported that CD44v overexpression
increased the expression of cystine-glutamate transporter xCT in
the plasma membrane, which led to the acquisition of CDDP
resistance (7). However, in ACR20
cells treated with an xCT inhibitor or siRNA, the sensitivity to
CDDP was not recovered to the same level as that in A549 cells
(7). Therefore, other mechanisms
for the acquisition of CDDP resistance may exist. (7). CDDP is imported into cells and
induces reactive oxygen species (ROS) generation, which causes
cytotoxicity (8). One report
showed that cytoplasmic ROS production was reduced in
CDDP-resistant cells derived from gastric cancer cells (9). Glutathione, a primary cellular
antioxidant, is composed of glutamate, cysteine, and glycine.
Therefore, we predicted that the level of glutathione was decreased
and that the levels of ROS were reduced in ACR20 cells.
Mitochondria are cellular organelles that generate
most of the energy required for biochemical reactions in cells.
Mitochondria have an oxidative phosphorylation system, which
comprises five complexes (I–V). Mitochondria possess their own
genome, mitochondrial DNA (mtDNA), which encodes mitochondrial
respiratory chain components, including seven subunits of complex I
[NADH dehydrogenase (ND)1, ND2, ND3, ND4L, ND4, ND5, and ND6], one
subunit of complex III [cytochrome b (CYTB)] and three subunits of
complex IV [cytochrome c oxidase (COX) I, II, and III], 22 tRNAs,
and 2 rRNAs. Mutations in nuclear genes or mitochondrial genes that
encode mitochondrial respiratory chain proteins result in
mitochondrial dysfunction. Numerous mtDNA mutations have been
identified in the development and progression of various
pathologies and mitochondrial dysfunction is implicated in various
human diseases, including diabetes, Alzheimer's disease, and cancer
(10). mtDNA is a critical target
of CDDP (11) and the levels of
CDDP adducts are increased in mtDNA compared with nuclear DNA
(12). This suggests that the
binding of CDDP to mtDNA may cause mtDNA damage and subsequent
mitochondrial dysfunction. In addition, in ρ° cells depleted of
mtDNA, which lose mitochondrial function, sensitivity to
doxorubicin or CDDP was found to decrease (13). Thus, mitochondrial dysfunction may
lead to the acquisition of CDDP resistance. However, how CDDP
induces mitochondrial dysfunction and how mitochondrial dysfunction
reduces cell sensitivity to CDDP is still unclear.
In the present study, the mechanism underlying CDDP
resistance was investigated in ACR20 cells. It was demonstrated
that mtDNA mutations in CDDP-resistant ACR20 cells caused loss of
mitochondrial function, upregulation of intrinsic ROS levels
(IRLs), and expression of inhibitor of apoptosis proteins (IAPs),
which contributed to the decreased sensitivity to CDDP. These
results suggest that mtDNA mutations play a role in the acquisition
of CDDP resistance.
Materials and methods
Cell culture
Human lung carcinoma cell line A549 (cat. # RCB0098,
RRID: CVCL_0023) were provided by RIKEN BioResource Research Center
through the National Bio-Resource Project of the Ministry of
Education, Culture, Sports, Science and Technology (Japan). ACR20
cells, CDDP-resistant cells, were established and cultured as
previously described (7). Both
cell lines were maintained in Dulbecco's modified Eagle's medium
(DMEM, Nacalai Tesque, Japan, cat. 08459-64) with 10% fetal bovine
serum (FBS), 100 U/ml penicillin and 100 µg/ml streptomycin in the
presence or absence of 20 µM CDDP (Wako Pure Chemical Industries,
Ltd., Osaka, Japan, cat. 033-20091) at 37°C with 5% CO2
and 95% air. Human cervical cancer HeLa cells (cat. no. CCL-2,
RRID: CVCL_0030) were obtained from American Type Culture
Collection. ρ° HeLa cells, which are deficient in mtDNA and
resistant to 6-thioguanine, were isolated as previously described
(14).
Flow cytometry analysis
Cells (1.2×105 cells) were seeded on a
60-mm dish and incubated in culture media at 37°C in 5%
CO2 and 95% air. After a 24-h incubation, the cells were
treated with 80 µM CDDP for 24 h. Annexin V/PI staining (Thermo
Fisher Scientific, Inc., cat. no. V13245) was performed following
the manufacturer's protocol. The stained cells were analyzed using
FACSCalibur (BD Biosciences) and FlowJo software 10.5.3 (Tree Star,
Inc.). Apoptotic cells were identified as Annexin
V+/PI− (early apoptosis) and Annexin
V+/PI+ (late apoptosis) cells.
Western blotting
Cells were seeded at 7×105 cells on a
60-mm dish and incubated in culture media at 37°C in 5%
CO2 and 95% air. In experiments examining cleaved
caspase-3 levels, after a 24-h incubation, the cells were treated
with 80 µM CDDP for 24 h. In experiments examining IAPs (namely,
c-IAP1, c-IAP2, and XIAP) and phosphorylation of IκB-α and NF-кB,
after a 1-h incubation, the cells were washed with PBS and treated
with 40 mM N-acetyl-L-cysteine (NAC; Sigma-Aldrich;
Merck KGaA, cat. no. A7250), an ROS inhibitor, for 24 h. Western
blot assays were performed as previously described (15). The antibodies are listed in
Table SI. After probing of
anti-phospho-IκB-α and anti-phospho-NF-кB antibodies, the blots
were stripped and reprobed with anti-IκB-α or anti-NF-кB
antibodies. After probing of anti-XIAP, anti-c-IAP1 antibody,
antic-IAP2, or anti-NDUFB8 antibodies, the blots were stripped and
reprobed with anti-GAPDH or anti-β-actin antibodies.
Cytotoxic activity assay
To evaluate whether ROS affected the sensitivity of
cells to CDDP, cells were seeded at 4.8×104 cells/well
on a 24-well plate and incubated in culture media at 37°C in 5%
CO2 and 95% air. After a 1-h incubation, the cells were
treated with 40 mM NAC for 24 h. The treated cells were washed with
PBS and cultured in culture media containing 80 µM CDDP for 48 h.
The half maximal inhibitory concentration (IC50) of CDDP
was determined following the previous study protocol (7). Cells were fixed with 4%
paraformaldehyde, stained with 0.5% crystal violet and lysed with
10% acetic acid. The optical density (O.D.) was measured at 600 nm.
The percentage of cell viability and 50% growth inhibitory
concentration was calculated as previously described (7).
ROS measurement
Cells were incubated for 48 h, washed with HBSS
(Nacalai Tesque, cat. no. 09735-75), stained with 10 µM
CM-H2DCFDA (Thermo Fisher Scientific, Inc., cat. no.
C6827) or 5 µM MitoSox (Thermo Fisher Scientific, Inc., cat. no.
M36008), and incubated for 30 min at 37°C in 5% CO2 and
95% air. Cells were washed with HBSS and observed with a
fluorescence microscope using FITC filters. The number of cells
with positive fluorescence was counted and the average number of
cells in three fields was taken as the number of ROS-producing
cells per field.
ρ A549 cell establishment
Respiration-deficient ρ A549 cells were established
by treatment of A549 cells with 50 ng/ml EtBr as previously
described (16) for 14 days
(Fig. S1A and B).
Cytoplasmic hybrid (Cybrid)
establishment
Cybrids were established as previously described
(17). In brief, A549 cells or
ACR20 cells were seeded at 1.5×106 cells or
3.0×106 cells, respectively, on a 100-mm dish for 48 h.
To prepare enucleated A549 cells or ACR20 cells (the mtDNA donor),
cells were pretreated with 10 µg/ml cytochalasin B (Sigma-Aldrich;
Merck KGaA, cat. no. 14930-96-2) for 15 min and centrifuged at
17,300 × g for 20 min. The resultant cytoplasts were fused with ρ°
HeLa cells, using polyethylene glycol. After 2 days, selective
isolation of the cybrids was performed by culture in selection
medium with 6-thioguanine, to exclude unfused A549 cells or ACR20
cells, and without uridine and pyruvate, to exclude parental ρ°
HeLa cells. The resultant cybrids contained nuclear DNA from ρ°
HeLa cells and mtDNA from enucleated A549 cells or ACR20 cells.
mtDNA extraction and mtDNA
sequencing
mtDNA extraction (Wako Pure Chemical Industries,
Ltd., cat. no. 291-55301), polymerase chain reaction (PCR) (Takara
Shuzo, Otsu, Japan, cat. no. RR001C), and purification of the PCR
products (Takara Shuzo, cat. no. 740609) were performed following
the manufacturers' protocols. Primer sequences are shown in
Table SII. DNA sequencing was
conducted by Eurofins Genomics (Tokyo, Japan).
PCR-restriction fragment length
polymorphism (PCR-RFLP)
The percentage levels of mtDNA mutations in A549
cells and ACR20 cells were examined by PCR-RFLP. In addition, to
confirm that the mtDNA derived from A549 and ACR20 cells was
incorporated into A549cyb and ACR20cyb cells,
respectively, we performed PCR-RFLP. In brief, mtDNA was extracted
from the A549 and ACR20 cells. Total DNA was extracted from
A549cyb and ACR20cyb cells. PCR amplification
was performed according to the manufacturer's protocol. Purified
PCR products were digested with the restriction enzyme. The
restriction fragments were analyzed by gel electrophoresis. PCR
primer sequences, restriction enzyme, and annealing temperature are
shown in Table SIII.
Mitochondrial activity assay
A XF24 Extracellular Flux Analyzer (Seahorse
Biosciences, USA) was used to determine the intracellular
bioenergetic profiles. A549 or ACR20 cells were seeded at
4×104 cells/well on the XF24 V7 Cell Culture microplate.
Extracellular acidification rate (ECAR) and oxygen consumption rate
(OCR) were measured as previously described (18). Mitochondria from cultured cells
were isolated using the Mitochondria Isolation Kit (BioVision,
Inc., cat. no. K288-50). Protein concentrations of isolated
mitochondrial samples were estimated using Bradford assays.
Isolated mitochondria were stored at −80°C until used.
Mitochondrial complex activities were measured using MitoCheck
Complex I Activity Assay Kit (Cayman Chemical Company, cat. no.
700930). Complex I activity was determined as activity without
inhibitor of complex I subtracted from the activity with complex I
inhibitor.
Quantitative reverse transcription
real-time PCR (RT-qPCR)
RT-qPCR was established as previously described
(7). RT-qPCR was performed using
PrimeScript™ RT Master Mix (Takara Shuzo, cat. no. RR036) and TB
Green® Premix Ex Taq™ II (Takara Shuzo, cat. no. RR820)
according to the manufacturer's protocol. Primer sequences are
shown in Table SIV. Relative
expression was calculated using the ΔΔCt method (19) with glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) as the reference gene.
Bioinformatic analysis of
NADH-ubiquinone oxidoreductase chain (ND) proteins
The structures of the mutant ND proteins were
analyzed at Altif Laboratories Inc. (Tokyo, Japan).
Statistical analysis
Data are expressed as the mean ± standard error
(SEM) of independent determinations for each experiment. Normality
was assessed by Kolmogorov-Smirnov normality test. Unpaired t-test
was used to evaluate the differences between two groups.
Comparisons of more than two groups were evaluated by one-way
analysis of variance (ANOVA), followed by the Student-Newman-Keuls
post hoc test. P-value <0.05 is indicative of a
statistically significant difference.
Results
Impairment of CDDP-induced apoptosis
in ACR20 cells
We previously established CDDP-resistant cells
derived from A549 cells, which we named ACR20 cells (7). To determine whether ACR20 cells are
resistant to CDDP-induced apoptosis, we evaluated CDDP-induced
apoptosis using Annexin V/PI staining assays. In the CDDP-treated
A549 cells, a high percentage of cells were positively stained for
Annexin V and cleaved caspase-3 expression was detected by western
blotting, thus indicating that CDDP induced apoptosis in the A549
cells (Fig. 1A-C). In contrast,
the percentage of Annexin V-stained cells was low (11%) and cleaved
caspase-3 was not detected in the CDDP-treated ACR20 cells. These
results indicate that CDDP-induced apoptosis was impaired in the
ACR20 cells.
Increase in intrinsic ROS in ACR20
cells is involved in the impairment of CDDP-induced apoptosis
To determine the mechanism underlying the impairment
of CDDP-induced apoptosis in ACR20 cells, we analyzed the
production of ROS using the ROS indicator CM-H2DCFDA.
Green fluorescence was not detected in A549 cells not treated with
CDDP but was observed after CDDP treatment (Figs. S2 and 2A). This result indicates
that CDDP induced ROS generation in the cytoplasm of the A549
cells. Unexpectedly, green fluorescence was detected in ACR20 cells
not treated with CDDP, and the number of green
fluorescence-positive cells was increased compared to that of the
A549 cells (Fig. 2A). These
results indicate that IRLs were elevated in ACR20 cells. NAC, an
inhibitor of ROS, pretreatment improved the sensitivity of ACR20
cells to CDDP (Fig. 2B). Together,
these findings suggest that the elevation of IRLs was involved in
the acquisition of CDDP resistance.
NF-κB regulates the gene expression of IAPs
(20). ROS-stimulated IκB kinase
leads to phosphorylation of IκB and NF-κB, which promotes IκB
degradation and modulates NF-κB activation (21). Thus, we hypothesized that the
elevation of IRLs in ACR20 cells would induce the expression of
IAPs and impair CDDP-induced apoptosis. The expression levels of
phosphorylated IκB-α and NF-κB in ACR20 cells were significantly
higher than those in A549 cells (P<0.01), and these levels were
suppressed in ACR20 cells by NAC pretreatment (Fig. 2C and D). Similarly, the levels of
IAPs in ACR20 cells were significantly elevated compared to those
in A549 cells (P<0.01), and the expression of IAPs in ACR20
cells was suppressed by NAC pretreatment (Fig. 2E-G). The expression of
phosphorylated IκB-α, phosphorylated NF-κB, and IAPs in A549 cells
was not affected by NAC pretreatment (data not shown). There
results suggest that elevated IRLs decreased the sensitivity of
ACR20 cells to CDDP by activating NF-κB signaling and inducing IAP
expression.
Loss of mitochondrial function in
ACR20 cells is associated with CDDP resistance
Intracellular levels of ROS are dependent on the
balance between ROS generation and ROS elimination. Increased IRLs
in cancer cells may be caused by malfunction of the mitochondrial
respiratory chain (22,23). Extracellular acidification rate
(ECAR) and oxygen consumption rate (OCR) values are important
indicators of mitochondrial respiration and glycolysis. We found
that ECAR was not changed and that OCR was decreased in the ACR20
cells when compared to the A549 cells (Fig. 3A). Mitochondrial superoxide is
generated as a byproduct of ATP production (24,25).
In A549 cells loaded with MitoSox, an indicator of mitochondrial
superoxide, red fluorescence was observed; however, in contrast,
red fluorescence was not detected in the ACR20 cells (Fig. 3B). Together, these results indicate
a loss of mitochondrial function in ACR20 cells. To investigate
whether mitochondrial dysfunction causes CDDP resistance in A549
cells, we analyzed CDDP sensitivity in mtDNA-depleted ρ A549 cells,
which showed loss of mitochondrial function. The IC50
values of CDDP in A549 and ρ A549 cells were 4.64±1.04 and
11.56±2.26 µM, respectively (Fig.
S1C). Together, these results indicate that mitochondrial
dysfunction causes CDDP-resistance in the A549 cells.
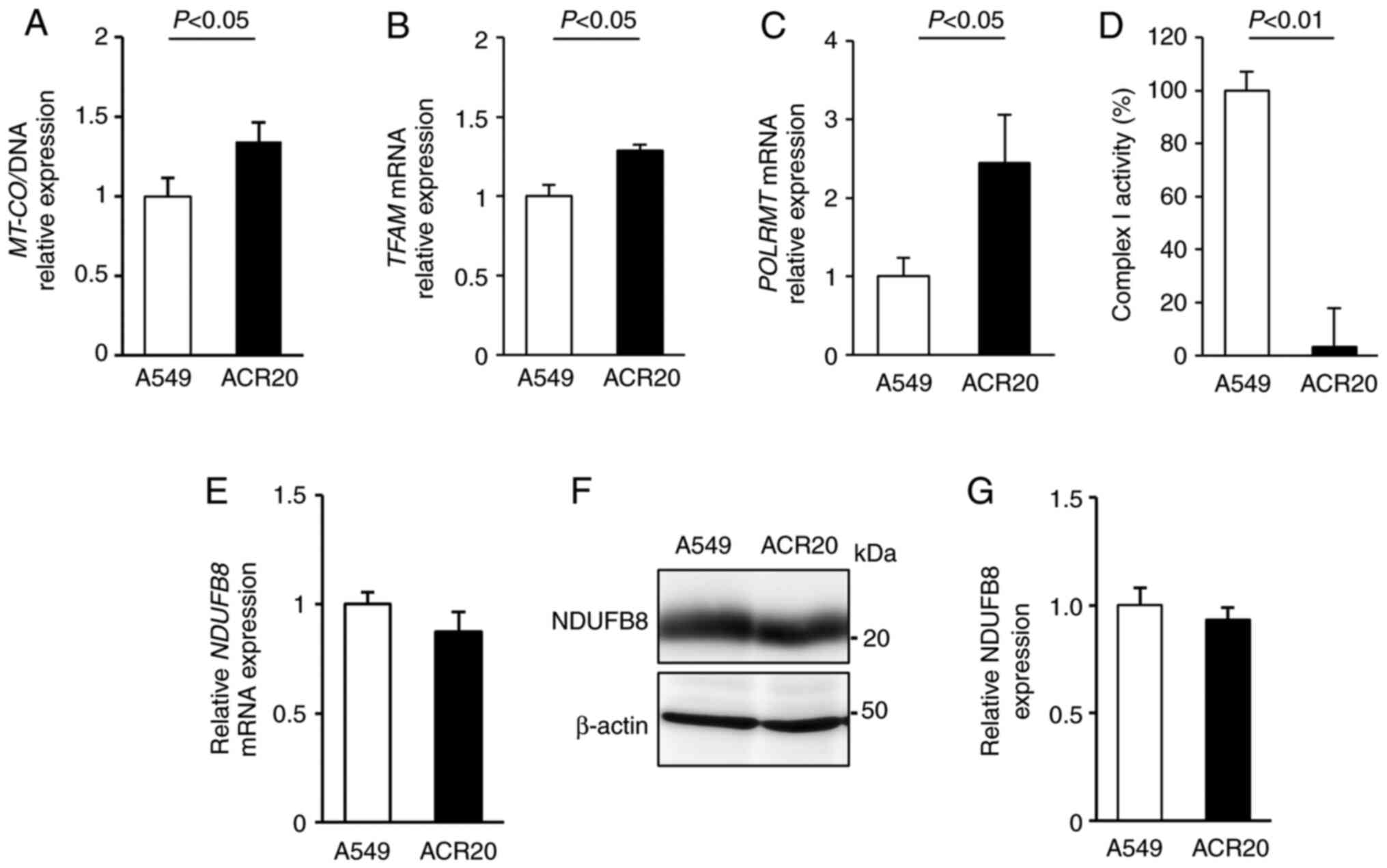
mtDNA mutations causes the decrease in
mitochondrial complex I activity in ACR20 cells
The mitochondrial genome encodes mitochondrial
respiratory chain components. Thus, decreased quality and quantity
of mtDNA lead to mitochondrial dysfunction (26–28).
To examine whether a change in the number of mtDNA copies in ACR20
cells is involved in the loss of mitochondrial function, we
evaluated the expression of mitochondria-encoded COX I (MT-COI)
levels normalized to α-tubulin levels to determine the number of
mtDNA copies (29,30). Contrary to our expectations,
MT-COI expression was increased in the ACR20 cells when
compared to the expression level in the A549 cells (Fig. 4A). The number of mtDNA copies is
regulated by transcription factor A, mitochondrial (TFAM) and RNA
polymerase mitochondrial (POLRMT) (31,32).
We found that the expression of TFAM and POLRMT mRNAs
was increased in the ACR20 cells when compared to the A549 cells
(Fig. 4B and C). These results
indicate that the number of mtDNA copies was increased in ACR20
cells, and this increase might cause the upregulation of
TFAM and POLRMT mRNA expression.
To determine the quality of mtDNA, mtDNA was
extracted from the A549 and ACR20 cells for sequencing and
identification of mtDNA mutations. Moreover, the percentage levels
of mtDNA mutations were analyzed by PCR-RFLP. 4587 T>C and 11384
C>A were included in 100% of mtDNAs in the ACR20 cells (Fig. S3A and B and Table I). 13148 C>A and 14162 G>T
were specifically detected in ACR20 cells and were included in 44.9
and 53.3% of the mtDNAs, respectively (Fig. S3C and D and Table I). Thus, four mtDNA mutations with
varying percentage levels were identified in ACR20 cells and these
mutations were located in genes encoding mitochondrial complex I.
We further found that the activity of mitochondrial complex I was
decreased in ACR20 cells compared to the A549 cells (Fig. 4D). NDUFB8 is a component of the
supernumerary subunits in mammalian complex I, which are central to
the structure, stability, and assembly (33). mRNA and proteins levels of NDUFB8
were comparable between the A549 and ACR20 cells (Fig. 4E-G). Thus, these results suggest
that the identified mtDNA mutations caused the decrease of
mitochondrial complex I activity in ACR20 cells.
 | Table I.Identified mtDNA mutations with
varying percentage levels in ACR20 cells. |
Table I.
Identified mtDNA mutations with
varying percentage levels in ACR20 cells.
|
|
|
| Percentage levels
of mutation |
|---|
|
|
|
|
|
|---|
| Mutations | Amino acid
change | Gene | A549 cells (%) | ACR20 cells
(%) |
|---|
| 4587T>C | Phe
(40)>Leu | MT-ND2 | 44.00 | 100 |
| 11384C>A | Leu
(209)>Ile | MT-ND4 | 4.28 | 100 |
| 13148C>A | Pro
(271)>Gln | MT-ND5 | 0 | 44.90 |
| 14612G>T | Ser
(21)>Tyr | MT-ND6 | 0 | 53.30 |
mtDNA mutations in ACR20 cells are
involved in CDDP resistance
To clarify whether the identified mtDNA mutations in
ACR20 cells contributed to the elevation of IRLs and the decreased
activity of complex I, cybrids (cells with the same physiological
functions such as nuclear DNA, but different only in mtDNA) were
established (17) (Fig. 5A). mtDNA derived from A549 and
ACR20 cells was incorporated into A549cyb and
ACR20cyb cells, respectively (Fig. S4). The number of ROS-producing
cells was significantly increased and the activity of complex I was
decreased in the ACR20cyb cells compared to the
A549cyb cells (P<0.01) (Fig. 5B and C). The expression of IAPs was
significantly increased in the ACR20cyb cells compared
with the A549cyb cells (XIAP and c-IAP1, P<0.05 and
P<0.01; c-IAP2; P<0.05) (Fig.
5D-F). We further examined whether the mtDNA mutations impacted
the sensitivity to CDDP. The IC50 value of CDDP in the
A549cyb and ACR20cyb cells were 0.27±0.02 and
0.69±0.02 nM, respectively (Fig.
5G). Together, these results indicate that the four identified
mtDNA mutations with varying percentage levels in ACR20 cells
caused CDDP resistance through the increased expression of IAPs via
elevation of IRLs and inactivation of complex I.
Structural analysis of proteins with
the identified mtDNA mutations
We next determined which of the four identified
mtDNA mutations may be associated with complex I inactivation using
the three-dimensional structure data of human mitochondrial complex
I. The four identified mutations (ND2 F40L, ND4 L209I, ND5 P271Q,
and ND6 S21Y) in the mitochondrial complex I were marked on the
structure of human respiratory complex I (PDB ID: 5XTB) (Fig. 6A). The mutations were located in
the transmembrane. The electric charge and polar amino acid, which
exist in proton channels affect proton translocation (34,35).
Therefore, the protein structure of the four mutations was modeled
and the effect of the mutations on polar amino acids in proton
translocation was investigated. Four mutations and key residues
were marked to three-dimensional protein structures of complex I
and the pathway of proton translocation was marked with reference
to Fiedorczuk et al (34)
and Fiedorczuk and Sazanov (35)
(Fig. 6B). ND2 F40L is located
near the charged side chains (Glu34, Lys105, and Lys135) and
hydrophilic side chains (Gln134) (Fig.
6B and C). The side chain of Phe residue contacts TMH5 and is
packed by hydrophobic interactions with the surrounding hydrophobic
residues (Fig. 6C). Although Leu
is hydrophobic, the side chain of Leu is smaller than that of Phe.
Thus, we speculated that the F40L mutation eliminated these
hydrophobic interactions and the packing between TMH2 and TMH5
(Fig. 6C). Furthermore, the
conformation of Glu134 may be changed by displacement from Phe40 to
Ile40, which may cause the structure change of the proton
translocation pathway. In contrast, the other mutations were
present at different positions of the proton translocation pathway
(Fig. 6B). These results show the
possibility that ND2 F40L affects the proton pathway, leading to
the decrease of complex I activity.
Discussion
In the present study, it was demonstrated that
elevated intrinsic ROS levels (IRLs) in ACR20 cells led to
increased phosphorylation of IκB-α and NF-κB activity, increased
inhibitors of apoptosis protein (IAP) expression and decreased cell
sensitivity to cisplatin (CDDP). NF-κB signaling is activated by
reactive oxygen species (ROS) (21) and NF-κB regulates the expression of
IAPs (20). c-IAP1, cIAP2 and XIAP
bind to caspase-3 and inhibit the activity of caspase-3 (36–40).
This suggests that ROS-induced activation of NF-κB signaling
increases the expression of IAPs and suppresses CDDP-induced
apoptosis, leading to the acquisition of CDDP resistance.
N-acetyl-L-cysteine (NAC) pretreatment improved the sensitivity of
ACR20 cells. The percentage of Annexin V/PI-positive cells in the
ACR20 cells untreated with CDDP was higher than that in the ACR20
cells treated with 80 µM CDDP. It was considered possible that
non-apoptotic cells were stained with Annexin V in ACR20 cells
under normal conditions. Annexin V forms a shield around negatively
charged phospholipid molecules. Moreover, the lipid contents in the
cytomembrane were found to be altered in cisplatin- and
doxorubicin-resistant MCF-7 cells (41). Therefore it is possible that the
lipid content of the cytomembrane in ACR20 cells may be changed and
affected by Annexin V staining; however, the detailed reason for
this is still unclear. These findings suggest that the IRLs
elevation is partly involved in the mechanism of acquisition of
CDDP resistance and that NAC administration before treatment with
CDDP may be effective for cancer patients who show resistance to
CDDP.
The elevation of IRLs in cancer cells are thought to
be caused by oncogenic signals (42,43),
oncogenic transformation (44), or
malfunction of the mitochondrial respiratory chain (22,23).
Depletion of mitrochondrial DNA (mtDNA) in hepatocarcinoma cells,
which showed a loss of mitochondrial function, was found to result
in chemoresistance to CDDP (13).
Thus, mitochondrial function may affect cell sensitivity to CDDP.
In the present study, we demonstrated that mitochondrial function
was decreased in ACR20 cells and that sensitivity to CDDP in ρ A549
cells was decreased compared to A549 cells. These findings indicate
that mitochondrial dysfunction caused the decreased sensitivity to
CDDP in A549 cells.
A previous study showed that HeLa cybrids containing
mutant mtDNA derived from the pancreatic cancer cell lines CFPAC-1
(10970 T>C in ND4; 8696 T>C and 9070 T>G in ATPase; 2905
A>G in the 16S rRNA gene) or CAPAN-2 (6267 G>A in COI; 10176
G>A in ND3) were resistant to 5-fluororacil and CDDP (45). In this study, we identified four
mutations in mtDNA (4587 T>C, 11384 C>A, 13148 C>A, and
14162 G>T) with varying percentage levels in ACR20 cells. These
mutations caused the decreased activity of complex I, elevation of
IRLs, and induction of IAP expression, which may have led to the
CDDP resistance. Furthermore, the three-dimensional structure data
suggest that the 11384 C>A mutation may be associated with the
decreased activity of complex I. Future studies will be needed to
confirm that the 1384 C>A regulates the proton translocation
pathway that affects the activity of complex I. Taken together,
11384 C>A may be a new biomarker of sensitivity to CDDP. Studies
using clinical samples may provide a clue to this possibility.
Mitochondrial function is associated with the number
of mtDNA copies (27). Although
mitochondrial function was reduced in ACR20 cells, the number of
mtDNA copies was increased. In addition, the expression levels of
TFAM and POLRMT mRNA, which encode proteins that
regulate the number of mtDNA copies, were increased in ACR20 cells.
These results suggest that upregulation of TFAM and
POLRMT mRNA caused the increased number of mtDNA copies. Our
results were consistent with a previous report, in which TFAM was
upregulated in CDDP-resistant cells (46). TFAM is not only required for both
transcription and maintenance of mtDNA but also the regulation of
anti-apoptotic genes, such as BIRC5 (47) and BCL2 (48). Expression of BIRC5 mRNA was
increased in ACR20 cells, while expression of BCL2 mRNA was
comparable between A549 and ACR20 cells (data not shown). Thus,
TFAM upregulation in ACR20 cells may be, in part, associated with
the acquisition of CDDP resistance.
In summary, we demonstrated that several newly
identified mtDNA mutations caused the elevation of IRLs, which
promoted CDDP resistance through induction of NF-кB signaling and
increased expressions of IAPs (Fig.
7). NAC treatment prior to CDDP administration may be an
effective strategy against CDDP resistance, and the newly
identified mutations in mtDNA may be potential biomarkers for
sensitivity to CDDP.
Supplementary Material
Supporting Data
Acknowledgements
We thank Gabrielle White Wolf, PhD for editing the
draft of this manuscript.
Funding
The present study was supported by a Grant-in-Aid for Scientific
Research (no. 20K07169) to SH from the Ministry of Education,
Culture, Sports, Science, and Technology of Japan.
Availability of data and materials
The datasets used and analyzed during the current
study are registered in the DDBJ under the accession number
LC655995 and LC657585 and are available from the corresponding
authors upon reasonable request.
Authors' contributions
SH designed the experiments. SH analyzed the data.
KI and KN established the cybrids. MW and NT performed and analyzed
the mitochondrial activity. SH wrote the paper. SH, SK, NS, TT and
YR evaluated and confirmed the authenticity of all the raw data.
All authors read and approved the final manuscript for
publication.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CDDP
|
cisplatin
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
IAPs
|
inhibitors of apoptosis proteins
|
|
IRLs
|
intrinsic ROS levels
|
|
NAC
|
N-acetyl-L-cysteine
|
|
ND
|
NADH-ubiquinone oxidoreductase
chain
|
|
mtDNA
|
mitochondrial DNA
|
|
ROS
|
reactive oxygen species
|
|
RT-qPCR
|
reverse transcription quantitative
real-time PCR
|
|
TMH
|
transmembrane helix
|
|
TG
|
6-thioguanine
|
References
|
1
|
Galluzzi L, Senovilla L, Vitale I, Michels
J, Martins I, Kepp O, Castedo M and Kroemer G: Molecular mechanisms
of cisplatin resistance. Oncogene. 31:1869–1883. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gandhi L, Rodriguez-Abreu D, Gadgeel S,
Esteban E, Felip E, Angelis FD, Domine M, Clingan P, Hochmair MJ,
Powell SF, et al: Pembrolizumab plus chemotherapy in metastatic
non-small-cell lung cancer. N Engl J Med. 378:2078–2092. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Paz-Ares LG, Luft A, Tafreshi A, Gumus M,
Mazieres J, Hermes B, Senler FC, Fülöp A, Rodriguez-Cid J, Sugawara
S, et al: Phase 3 study of carboplatin-paclitaxel/nab-paclitaxel
(Chemo) with or without pembrolizumab (Pembro) for patients (Pts)
with metastatic squamous (Sq) non-small cell lung cancer (NSCLC). J
Clin Oncol. 36 (15_Suppl):S105. 2018. View Article : Google Scholar
|
|
4
|
Komatsu M, Sumizawa TS, Mutoh M, Chen ZS,
Terada K, Furukawa T, Yang XL, Gao H, Miura N, Sugiyama T and
Akiyama S: Copper-transporting P-type adenosine triphosphatase
(ATP7B) is associated with cisplatin resistance. Cancer Res.
60:1312–1316. 2000.PubMed/NCBI
|
|
5
|
Lai GM, Ozols RF, Young RC and Hamilton
TC: Effect of glutathione on DNA repair in cisplatin-resistant
human ovarian cancer cell lines. J Natl Cancer Inst. 7:535–539.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zeng-Rong N, Paterson J, Alpert L, Tsao
MS, Viallet J and Alaoui-Jamali MA: Elevated DNA repair capacity is
associated with intrinsic resistance of lung cancer to
chemotherapy. Cancer Res. 55:4760–4764. 1995.PubMed/NCBI
|
|
7
|
Horibe S, Kawauchi S, Tanahashi T, Sasaki
N, Mizuno S and Rikitake Y: CD44v-dependent upregulation of xCT is
involved in the acquisition of cisplatin-resistance in human lung
cancer A549cells. Biochem Biophys Res Commun. 507:426–432. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Choi YM, Kim HK, Shim W, Anwar MA, Kwon
JW, Kwon HK, Kim HJ, Jeong H, Kim HM, Hwang D, et al: Mechanism of
cisplatin-induced cytotoxicity is correlated to impaired metabolism
due to mitochondrial ROS generation. PLoS One. 10:e01350832015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang SF, Chen MS, Chou YC, Ueng YF, Yin
PH, Yeh TS and Lee HC: Mitochondrial dysfunction enhances cisplatin
resistance in human gastric cancer cells via the ROS-activated
GCN2-eIF2α-ATF4-xCT pathway. Oncotarget. 7:74132–74151. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tuppen HA, Blakely EL, Turnbull DM and
Taylor RW: Mitochondrial DNA mutations and human disease. Biochim
Biophys Acta. 1797:113–128. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang Z, Schumaker LM, Egorin MJ, Zuhowski
EG, Guo Z and Cullen KJ: Cisplatin preferentially binds
mitochondrial DNA and voltage-dependent anion channel protein in
the mitochondrial membrane of head and neck squamous cell
carcinoma: Possible role in apoptosis. Clin Cancer Res.
12:5817–5825. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Olivero OA, Semino C, Kassim A,
Lopez-Larraza DM and Poirier MC: Preferential binding of cisplatin
to mitochondrial DNA of Chinese hamster ovary cells. Mutat Res.
346:221–230. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gonzalez-Sanchez E, Marin JJ and Perez MJ:
The expression of genes involved in hepatocellular carcinoma
chemoresistance is affected by mitochondrial genome depletion. Mol
Pharm. 11:1856–1868. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hayashi J, Ohta S, Kikuchi A, Takemitsu M,
Goto Y and Nonaka I: Introduction of disease-related mitochondrial
DNA deletions into HeLa cells lacking mitochondrial DNA results in
mitochondrial dysfunction. Proc Natl Acad Sci USA. 88:10614–10618.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Horibe S, Matsuda A, Tanahashi T, Inoue J,
Kawauchi S, Mizuno S, Ueno M, Takahashi K, Maeda Y, Maegouchi T, et
al: Cisplatin resistance in human lung cancer cells is linked with
dysregulation of cell cycle associated proteins. Life Sci.
124:31–40. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
King MP and Attardi G: Human cells lacking
mtDNA: Repopulation with exogenous mitochondria by complementation.
Science. 246:500–503. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ishikawa K, Takenaga K, Akimoto M,
Koshikawa N, Yamaguchi A, Imanishi H, Nakada K, Honma Y and Hayashi
JI: ROS-generating mitochondrial DNA mutations can regulate tumor
cell metastasis. Science. 320:661–664. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Semba H, Takeda N, Isagawa T, Sugiura Y,
Honda K, Wake M, Miyazawa H, Yamaguchi Y, Miura M, Jenkins DM, et
al: HIF-1α-PDK1 axis-induced active glycolysis plays an essential
role in macrophage migratory capacity. Nat Commun. 7:116352016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cai X, Lu W, Yang Y, Yang J, Ye J, Gu Z,
Hu C, Wang X and Cao P: Digitoflavone inhibits IκBα kinase and
enhances apoptosis induced by TNFα through downregulation of
expression of nuclear factor κB-regulated gene products in human
pancreatic cancer cells. PLoS One. 8:e771262013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Karin M: The beginning of the end: IkappaB
kinase (IKK) and NF-kappaB activation. J Biol Chem.
274:27339–27342. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Murphy MP: Mitochondrial dysfunction
indirectly elevates ROS production by the endoplasmic reticulum.
Cell Metab. 18:145–146. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tafani M, Sansone L, Limana F, Arcangeli
T, Santis ED, Polese M, Fini M and Russo MA: The interplay of
reactive oxygen species, hypoxia, inflammation, and sirtuins in
cancer initiation and progression. Oxid Med Cell Longev.
2016:39071472016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Saybaşili H, Yüksel M, Haklar G and Yalçin
AS: Effect of mitochondrial electron transport chain inhibitors on
superoxide radical generation in rat hippocampal and striatal
slices. Antioxid Redox Signal. 3:1099–1104. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Staniek K, Gille L, Kozlov AV and Nohl H:
Mitochondrial superoxide radical formation is controlled by
electron bifurcation to the high and low potential pathways. Free
Radic Res. 36:381–387. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu J, Jin Z, Zheng H and Yan LJ: Sources
and implications of NADH/NAD(+) redox imbalance in diabetes and its
complications. Diabetes Metab Syndr Obes. 9:145–153.
2016.PubMed/NCBI
|
|
27
|
Wiedemann FR, Manfredi G, Mawrin C, Beal
MF and Schon EA: Mitochondrial DNA and respiratory chain function
in spinal cords of ALS patients. J Neurochem. 80:616–625. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rusecka J, Kaliszewska M, Bartnik E and
Tońska K: Nuclear genes involved in mitochondrial diseases caused
by instability of mitochondrial DNA. J Appl Genet. 59:43–57. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cote HC, Brumme ZL, Craib KJ, Alexander
CS, Wynhoven B, Ting L, Wong H, Harris M, Harrigan PR,
O'Shaughnessy MV and Montaner JS: Changes in mitochondrial DNA as a
marker of nucleoside toxicity in HIV-infected patients. N Engl J
Med. 346:811–820. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mambo E, Gao X, Cohen Y, Guo Z, Talalay P
and Sidransky D: Electrophile and oxidant damage of mitochondrial
DNA leading to rapid evolution of homoplasmic mutations. Proc Natl
Acad Sci USA. 100:1838–1843. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kang D, Kim SH and Hamasaki N:
Mitochondrial transcription factor A (TFAM): Roles in maintenance
of mtDNA and cellular functions. Mitochondrion. 7:39–44. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kuhl I, Miranda M, Posse V, Milenkovic D,
Mourier A, Siira SJ, Bonekamp NA, Neumann U, Filipovska A, Polosa
PL, et al: POLRMT regulates the switch between replication primer
formation and gene expression of mammalian mtDNA. Sci Adv.
2:e16009632016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhu J, Vinothkumar KR and Hirst J:
Structure of mammalian respiratory complex I. Nature. 536:354–358.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fiedorczuk K, Letts JA, Degliesposti G,
Kaszuba K, Skehel M and Sazanov LA: Atomic structure of the entire
mammalian mitochondrial complex I. Nature. 538:406–410. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fiedorczuk K and Sazanov LA: Mammalian
mitochondrial complex I structure and disease-causing mutations.
Trends Cell Biol. 28:835–867. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Deveraux QL, Takahashi R, Salvesen GS and
Reed JC: X-linked IAP is a direct inhibitor of cell-death
proteases. Nature. 388:300–304. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Takahashi R, Deveraux Q, Tamm I, Welsh K,
Assa-Munt N, Salvesen GS and Reed JC: A single BIR domain of XIAP
sufficient for inhibiting caspases. J Biol Chem. 273:7787–7790.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Roy N, Deveraux QL, Takahashi R, Salvesen
GS and Reed JC: The c-IAP-1 and c-IAP-2 proteins are direct
inhibitors of specific caspases. EMBO J. 16:6914–6925. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Maier JKX, Lahoua Z, Gendron NH, Fetni R,
Johnston A, Davoodi J, Rasper D, Roy S, Slack RS, Nicholson DW and
MacKenzie AE: The neuronal apoptosis inhibitory protein is a direct
inhibitor of caspases 3 and 7. J Neurosci. 22:2035–2043. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Davoodi J, Lin L, Kelly J, Liston P and
MacKenzie AE: Neuronal apoptosis-inhibitory protein does not
interact with Smac and requires ATP to bind caspase-9. J Biol Chem.
279:40622–40628. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Todor IN, Lukyanova NY and Chekhun VF: The
lipid content of cisplatin- and doxorubicin-resistant MCF-7 human
breast cancer cells. Exp Oncol. 34:97–100. 2012.PubMed/NCBI
|
|
42
|
Vafa O, Wade M, Kern S, Beeche M, Pandita
TK, Hampton GM and Wahl GM: c-Myc can induce DNA damage, increase
reactive oxygen species, and mitigate p53 function. Mol Cell.
9:1031–1044. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hlavata L, Aguilaniu H, Pichová A and
Nyström T: The oncogenic RAS2(val19) mutation locks respiration,
independently of PKA, in a mode prone to generate ROS. EMBO J.
22:3337–3345. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lim JKM and Leprivier G: The impact of
oncogenic RAS on redox balance and implications for cancer
development. Cell Death Dis. 10:9552019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mizutani S, Miyato Y, Shidara Y, Asoh S,
Tokunaga A, Tajiri T and Ohta S: Mutations in the mitochondrial
genome confer resistance of cancer cells to anticancer drugs.
Cancer Sci. 100:1680–1687. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kidani A, Izumi H, Yoshida Y, Kashiwagi E,
Ohmori H, Tanaka T, Kuwano M and Kohno K: Thioredoxin2 enhances the
damaged DNA binding activity of mtTFA through direct interaction.
Int J Oncol. 35:1435–1440. 2009.PubMed/NCBI
|
|
47
|
Altieri DC: Survivin, cancer networks and
pathway-directed drug discovery. Nat Rev Cancer. 8:61–70. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cory S and Adams JM: The Bcl2 family:
Regulators of the cellular life-or-death switch. Nat Rev Cancer.
2:647–656. 2002. View
Article : Google Scholar : PubMed/NCBI
|