Introduction
Hepatocellular carcinoma (HCC) accounts for ~80% of
primary liver cancers and is the third leading cause of
cancer-related mortality worldwide (1,2). In
patients with early-stage liver cancer, surgery resection and
thermal ablation are the most common available treatment options
(3,4). However, the majority of patients with
liver cancer are diagnosed at an advanced stage, and are therefore
unlikely to respond to these treatment options due to the rapid
development and distant metastases of HCC (5). Over the past decade, treatment
options for HCC have progressed and include multi-kinase inhibitors
(sorafenib and lenvatinib) and immune checkpoint inhibitors.
However, treatment of advanced HCC remains unsatisfactory, as the
overall survival rates of patients with liver cancer are marginally
extended following treatment (6).
Therefore, further investigations are required for the development
of novel potential therapies for liver cancer including HCC.
Protein neddylation is an important
post-translational modification that adds the ubiquitin-like
molecule NEDD8 to substrate proteins (7). This is carried out via a series of
processes that are successively catalyzed by E1 NEDD8-activating
enzyme (NAE1), E2 neddylation conjugation enzymes and E3
neddylation ligases. Previous studies have demonstrated that
neddylation regulates protein function and stabilization.
Well-established substrates of the neddylation pathway are the
Cullin family of proteins that are the scaffold components of
cullin-RING ligases (CRLs). Neddylation of Cullin is indispensable
for the activation of CRLs and the subsequent ubiquitination and
degradation of CRL substrates (8).
Results of a previous study revealed that protein neddylation is
upregulated in numerous human cancers, including liver cancer, and
inhibition of neddylation exhibits anticancer potential (9).
MLN4924, also known as pevonedistat, is a specific
inhibitor of NAE. MLN4924 suppresses the entire protein neddylation
modification (10). Results of a
previous study demonstrated that MLN4924 exhibits suppressive
activity against a variety of human cancer cells. Moreover, MLN4924
has been advanced into several Phase II/III clinical trials against
solid tumors and hematologic malignancies (11–14).
Mechanistic studies that focus on the role of MLN4924 in cancer
suppression revealed that MLN4924 effectively induced the DNA
damage response, nonhomologous end-joining repair, DNA
re-replication stress and oxidative stress at the biochemical
level, and induced cell cycle arrest, apoptosis, autophagy and
senescence at the cellular level (15). However, results of a previous study
demonstrated that the anticancer effects of MLN4924, particularly
in solid tumors, remain unsatisfactory due to drug resistance
(16). This factor limits the
clinical use of MLN4924; thus, exploring the mechanisms of drug
resistance of cancer cells to MLN4924 may help overcome
chemoresistance and improve the associated clinical outcomes.
Chemoresistance is a major obstacle to curing
cancer. Previous research has demonstrated that inflammation is an
important factor that attenuates the chemosensitivity of cancer
cells to anticancer agents. For example, doxorubicin activates the
NF-κB signaling pathway, resulting in transactivation of
inflammatory factors and potent anti-apoptosis genes, which leads
to the chemoresistance of cancer cells (17). In addition, multiple previous
studies have demonstrated that the NF-κB signaling pathway is
aberrantly activated in multiple cancer types, such as HCC
(18), cervical cancer (19) and lung cancer (20), providing a rationale for overcoming
chemoresistance by targeting the NF-κB pathway. Notably, NF-κB
inhibitor α (IκBα), the upstream molecule of NF-κB, plays an
important role by inhibiting NF-κB activity (21). In the resting state, IκBα combines
with the NF-κB dimer to retain its existence in the cytoplasm. On
the other hand, IκBα is triggered by diverse extracellular signals
in the activating state, such as lipopolysaccharide, tumor necrosis
factor (TNF) and growth factors. Subsequently, IκBα is
phosphorylated by the IκB kinase complex and recognized by an E3
ubiquitin ligase for ubiquitination-dependent degradation (21). Moreover, IκBα is considered a
tumor-suppressing factor in different types of cancer, such as
breast (22), ovarian (23), gastric (24), colorectal (25) and liver cancer (26), mainly due to its inhibition of
NF-κB activity. However, it remains unclear whether the levels of
IκBα can be regulated by MLN4924 in liver cancer cells.
Results of the present study demonstrated that
downregulation of IκBα and the subsequent inflammation in response
to MLN4924 limits the antitumor potential of MLN4924 in liver
cancer cells. Mechanistic studies revealed that MLN4924 stabilized
β-transducin repeat-containing protein (β-TrCP), promoting the K48
linkage of ubiquitin to the IκBα protein, leading to the
degradation of IκBα via proteasomes. In addition, β-TrCP knockdown
sensitized liver cancer cells to MLN4924. Collectively, the results
of the present study revealed that the β-TrCP/IκBα/inflammation
axis limited the sensitivity of liver cancer cells to neddylation
inhibition, suggesting that interference of this pathway may act as
a novel target for developing new sensitizing strategies of MLN4924
to liver cancer treatment.
Materials and methods
Reagents
MLN4924 and MG132 (a proteasome inhibitor) were
purchased from Selleck Chemicals. Opti-MEM was purchased from
Invitrogen (Thermo Fisher Scientific, Inc.). Cycloheximide (CHX)
was purchased from Chengdu XiYa Chemical Technology Co., Ltd.
RNAiso Plus reagent, SYBR Green Mix and Reverse Transcription kit
were purchased from Takara Bio, Inc. X-tremeGENE HP DNA
Transfection Reagent, X-tremeGENE siRNA Transfection Reagent and
Cocktail were purchased from Roche Diagnostics, GmbH. Cell Counting
Kit-8 (CCK-8) was purchased from Dojindo Laboratories, Inc. Small
interfering (si)RNAs targeting β-TrCP and control siRNA were
synthesized by Shanghai GenePharma Co., Ltd.
Cell lines and culture
The human liver cancer cell line LM3 was purchased
from BeNa Culture Collection (cat. no. BNCC342335), the HepG2 cell
line (cat. no. HB-8065) was obtained from the American Type Culture
Collection and the Huh7 cell line (cat. no. SCSP-526) was purchased
from the National Collection of Authenticated Cell Cultures of the
Chinese Academy of Sciences (https://www.cellbank.org.cn/). All cells were cultured
in DMEM containing 10% fetal bovine serum at 37°C in a 5%
CO2 incubator.
Plasmid construction
The DNA fragment encoding IκBα (NM_020529) was
synthesized by Shanghai GeneChem Co., Ltd., and inserted into the
vector pcDNA-3.1. The resulting plasmid was named pcDNA-IκBα or
overexpression (OE)-IκBα. The His-ub (His-ubiquitin) plasmids and
the corresponding mutant plasmids were constructed by Tsingke
Biotechnology Co., Ltd.
Reverse transcription-quantitative
(RT-q)PCR
Total RNA was isolated from cultured cells using
RNAiso Plus reagent, and the first-strand cDNA was synthesized
using the PrimeScript RT Reagent Kit with gDNA Eraser (Takara Bio,
Inc.) according to the manufacturer's instructions. qPCR was
performed using a One-Step RT-PCR kit (ComWin) and SYBR Green Mix.
The PCR cycle parameters were as follows: 95°C for 30 sec, and 40
cycles at 95°C for 5 sec and 60°C for 1 min. Results were
calculated based on the quantification cycle (Cq), and the relative
fold change was determined using the 2−ΔΔCq method
(27). Primers used for RT-qPCR
are listed as follows: IL-6-forward (F), 5′-TACCCCCAGGAGAAGATTCC-3′
and IL-6-reverse (R), 5′-TTTTCTGCCAGTGCCTCTTT-3′; IL-8-F,
5′-CGGAAGGAACCATCTCACTGTG-3′ and IL-8R,
5′-AGAAATCAGGAAGGCTGCCAAG-3′; TNF-α-F, 5′-AACCTCCTCTCTGCCATCAA-3′
and TNF-α-R, 5′-CCAAAGTAGACCTGCCCAGA-3′; β-TrCP-F,
5′-CAGTTCTGCACTTGCGTTTC-3′ and β-TrCP-R,
5′-CTCACTACCAGCCTGTCCCT-3′; IκBα-F, 5′-AAGTGATCCGCCAGGTGAAG-3′ and
IκBα-R, 5′-CTGCTCACAGGCAAGGTGTA-3′; and β-actin-F,
5′-GTGAAGGTGACAGCAGTCGGTT-3′ and β-actin-R,
5′-GAAGTGGGGTGGCTTTTAGGA-3′.
Transient transfection
Liver cancer cells were seeded and cultured in
6-well plates overnight at 37°C in an incubator, and small
interfering RNAs (siRNAs, 100 pmol) or plasmids (2 µg) were
transfected into liver cancer cells using X-tremeGENE siRNA
Transfection Reagent or X-tremeGENE HP DNA Transfection Reagent for
48 h in 37°C incubator, respectively, according to the
manufacturer's instructions. The sequences of siRNAs are listed as
follows: si-β-TrCP (100 pmol), 5′-AAGUGGAAUUUGUGGAACAUC-3′ and
si-negative control (si-NC; 100 pmol),
5′-UUCUCCGAACGUGUCACGUTT-3′.
MG132 treatment protocol
Following treatment with MLN4924 for 48 h, LM3 and
Huh7 cells were incubated with or without 20 µM MG132 for 5 h at
37°C. Subsequently, western blotting was employed to determine the
protein levels of IκBα.
Western blot analysis
Cells were lysed with RIPA lysis buffer (Beyotime
Institute of Biotechnology) supplemented with a protease inhibitor
mixture (Roche Diagnostics), and then the lysates were centrifuged
at 16,000 × g for 15 min at 4°C. The protein concentrations were
then detected using a BCA kit (Beyotime Institute of
Biotechnology), according to the manufacturer's instructions.
Following denaturing, samples (100 µg/lane) were separated by 10%
SDS-PAGE (Beyotime Institute of Biotechnology) and then transferred
to NC membranes (Cytiva). The NC membranes were then blocked in 5%
nonfat milk in Tris-buffered saline containing 0.1% Tween-20 (TBST)
for 1 h at room temperature and then incubated with the indicated
primary antibodies. Primary antibodies against ubiquitin (cat. no.
sc-271289; 1:1,000 dilution), β-TrCP (cat. no. sc-390629; 1:1,000
dilution) and IκBα (cat. no. sc-373893; 1:1,000 dilution) were
purchased from Santa Cruz Biotechnology, Inc. Anti-cleaved
caspase-3 was purchased from Cell Signaling Technology (product no.
9664; 1:1,000 dilution). Primary antibodies against GAPDH (cat. no.
AG019; 1:5,000 dilution) and tubulin (cat. no. AT819; 1:5,000
dilution) were purchased from Beyotime Institute of Biotechnology.
All primary antibodies were incubated at 4°C overnight. Secondary
antibodies, HRP-labeled goat anti-rabbit (cat. no. 7074S) and
anti-mouse (cat. no. 7076S) immunoglobulin G, were purchased from
Cell Signaling Technology, Inc. The secondary antibodies were
diluted in 5% skim milk at a ratio of 1:5,000 and incubated at room
temperature for 1 h, and then the protein signals were measured
using chemiluminescence detection reagent (cat. no. WBKLS0500;
Millipore; Merck KGaA) and the Bio-Rad Imaging System (Image Lab
4.1; Bio-Rad Laboratories, Inc.).
Immunoprecipitation (IP)
Liver cancer cells were lysed using IP lysis buffer
purchased from Beyotime Institute of Biotechnology, and then the
lysates were centrifuged at 10,000 × g for 10 min at 4°C.
Subsequently, the supernatant was collected into another EP tube.
The anti-IκBα antibody (cat. no. sc-373893) was then added to the
lysate in a ratio of 2:1 mg total protein. Following overnight
incubation at 4°C, 40 ml protein A/G agarose beads (Santa Cruz
Biotechnology, Inc.) were added to each sample and incubated at
room temperature for 2 h. The immunoprecipitates were washed five
times with ice-cold PBS and isolated by centrifugation (600 × g, 3
min, 4°C). Finally, the precipitates were mixed with 1X SDS-PAGE
loading buffer and boiled at 100°C for 10 min, and then assessed by
western blot analysis.
In vitro ubiquitination assay
To examine the ubiquitination of IκBα by MLN4924,
liver cancer cells were co-transfected with His-ub (His-ubiquitin)
and HA-IκBα, and subsequently incubated with MLN4924 (0.4 µM) for
48 h at 37°C. MG132 (20 µM) was added to the medium for 5 h, cells
were harvested and subsequently split into two aliquots; one for
direct western blotting and one for in vitro ubiquitination.
Briefly, cells were lysed in buffer A [6 mol/l guanidinium-HCl, 0.1
mol/l Na2HPO4/NaH2PO4, 10 mmol/l Tris-HCl (pH
8.0), 5 mmol/l imidazole and 10 mmol/l β-mercaptoethanol] on ice,
sonicated to reduce viscosity and mixed with 50 µl
Ni2+-NTA-agarose beads (Qiagen, Inc.) overnight. Beads
were successively washed with buffer A with 10 mM
β-mercaptoethanol, buffer B [8 mM urea, 0.1 M
Na2HPO4/NaH2PO4, 10 mM Tris/HCl (pH, 8.0),
and 10 mM β-mercaptoethanol], buffer C [8 mM urea, 0.1 M
Na2HPO4/NaH2PO4, 10 mM Tris/HCl (pH, 6.3), 10
mM β-mercaptoethanol and 0.2% Triton X-100], and buffer C with 10
mM β-mercaptoethanol and 0.1% Triton X-100. Subsequently,
His6-tagged ubiquitinated proteins were eluted with buffer D [200
mM imidazole, 0.15 M Tris-HCl (pH, 6.7), 30% glycerol, 0.72 M
β-mercaptoethanol and 5% SDS]. The polyubiquitination of IκBα was
detected using western blot analysis, using an anti-HA antibody
(cat. no. 11867423001, 1:5,000; Roche Diagnostics).
Cell proliferation assays
Liver cancer cells were seeded in 96-well plates at
3,000 cells per well overnight at 37°C, and transiently transfected
with the indicated plasmids or siRNAs, and then incubated with
MLN4924 for 48 h. Subsequently, the CCK-8 agent was added to each
well at a volume of 10 µl per well, and then incubated at 37°C for
30–60 min, followed by detection of the absorption value at 450 nm
with a microplate reader (Thermo Scientific, Inc.).
Colony formation assays
Liver cancer cells were seeded in 6-well plates at
500 cells per well overnight at 37°C, and transiently transfected
with the indicated plasmids or siRNAs or incubated with MLN4924
(0.4 µM) for 10 days. Subsequently, the cells were fixed with 4%
formaldehyde (15 min at room temperature) and stained with 0.1%
crystal violet at room temperature for 10 min. During colony
growth, the culture medium was replaced every 3 days. Colonies with
>50 cells were counted manually, 10–14 days after plating.
Bioinformatics analysis
UALCAN and GEPIA online databases were utilized to
determine the mRNA level of IκBα (NFKBIA). Briefly, NFKBIA was
selected after logging in the website (UALCAN, http://ualcan.path.uab.edu/analysis.html).
Subsequently, liver hepatocellular carcinoma (LIHC) was selected as
the subject of analysis. Thereafter, the button ‘Explore’ was
selected and the result for the expression of NFKBIA in LIHC based
on sample tapes was obtained. For the usage of GEPIA online
database (|Log2FC| Cutoff: 1; P-value Cutoff: 0.01), NFKBIA was
typed into the box under ‘Enter gene/isoform name’ after logging in
the website (GEPIA, http://gepia2.cancer-pku.cn/#index), and then the
‘Boxplots’ button was selected followed by LIHC. Thereafter, the
button ‘Plot’ was selected and the result of the mRNA level of
NFKBIA in 160 normal liver tissues and 369 liver cancer tissues was
obtained. For the analysis of the protein level of IκBα in
different human cancers and corresponding normal tissues, Clinical
Proteomic Tumor Analysis Consortium (CPTAC, http://ualcan.path.uab.edu/analysis-prot.html)
online database was employed. NFKBIA was typed into the box under
‘Enter gene names’ column after logging in the website, and then
LIHC was selected, followed by the analysis of the protein level of
NFKBIA between normal and liver cancer tissues. For the analysis of
normal and primary cancer of pan-cancer, the ‘Pan-cancer view’ link
was selected. Z-values represent standard deviations from the
median across samples for the given cancer type in analyzing the
protein level of IκBα.
Statistical analysis
GraphPad Prism 8.0 (GraphPad Software, Inc.) was
used to analyze data. Data are presented as the mean ± standard
deviation. Comparisons between two groups were performed using a
two-tailed unpaired Student's t-test. Comparisons between multiple
groups were performed using one-way ANOVA, followed by Tukey's post
hoc test. The data are presented as the mean ± SD of at least three
independent experiments. P<0.05 was considered to indicate a
statistically significant difference.
Results
MLN4924 suppresses the survival of
liver cancer cells and downregulates IκBα
A previous study demonstrated that MLN4924 exhibits
antitumor potential (28). To
confirm this phenomenon, the effects of MLN4924 on the growth of
liver cancer cells were investigated in the present study. As
revealed in Fig. 1A, 0.8 µM
MLN4924 increased the natant cells compared with the control group,
while 0.4 µM MLN4924 exhibited no notable effect on cell morphology
(Fig. 1A). Therefore, this
concentration (0.4 µM) of MLN4924 was selected to investigate the
chemoresistance of liver cancer cells in the subsequent
experiments. Moreover, the results of the CCK-8 assay demonstrated
that MLN4924 suppressed the growth of liver cancer cells in a
dose-dependent manner. However, the half maximal inhibitory
concentration (IC50) values of MLN4924 in LM3 and Huh7
cells were 13.01 and 12.36 µM, respectively (Fig. 1B and C), indicating that MLN4924
markedly suppressed the growth of liver cancer cells at high
concentrations. As IκBα is considered a tumor-suppressing factor in
various cancers due to its inhibition of NF-κB activity, the
expression levels of IκBα in cancer tissues were further
investigated using bioinformatics analysis. The results obtained
from the UALCAN database demonstrated that the IκBα mRNA expression
levels exhibited no significant difference between 50 healthy liver
tissues and 371 LIHC tissues (Fig.
S1A). These results were further confirmed using an alternative
database, GEPIA, containing 160 healthy liver tissues and 369 HCC
tissues (Fig. S1B). Notably,
protein expression analysis using data from the Clinical Proteomic
Tumor Analysis Consortium (CPTAC) revealed that the IκBα protein
expression levels were markedly reduced in HCC (Fig. S1C), ovarian cancer, uterine corpus
endometrial carcinoma and lung adenocarcinoma (Fig. S1D). These results suggested that
the regulation of IκBα during liver carcinogenesis and development
may occur at a post-translational level. Subsequently, the effects
of MLN4924 on IκBα expression were explored. As shown in Fig. 1D and E, MLN4924 significantly
decreased IκBα protein expression levels in a dose-dependent
manner; however, it exerted no effect on the mRNA expression.
Furthermore, it was determined whether MLN4924 inhibited the
protein level of IκBα in a time-dependent manner. As revealed in
Fig. S2, the protein level of
IκBα was markedly decreased following treatment with MLN4924 for 48
and 72 h, and thus 48 h was selected in the subsequent experiments.
As IκBα plays a key role in anti-inflammation (29), mRNA expression levels of
interleukin (IL)-6, IL-8 and TNF-α, downstream molecules of IκBα,
were explored in the present study. As shown in Fig. 1F and G, MLN4924 significantly
increased the mRNA expression levels of IL-6, IL-8 and TNF-α.
Collectively, these findings indicated that MLN4924 possesses an
antitumor role in liver cancer cells; however, decreased IκBα
expression may impact its antitumor potential.
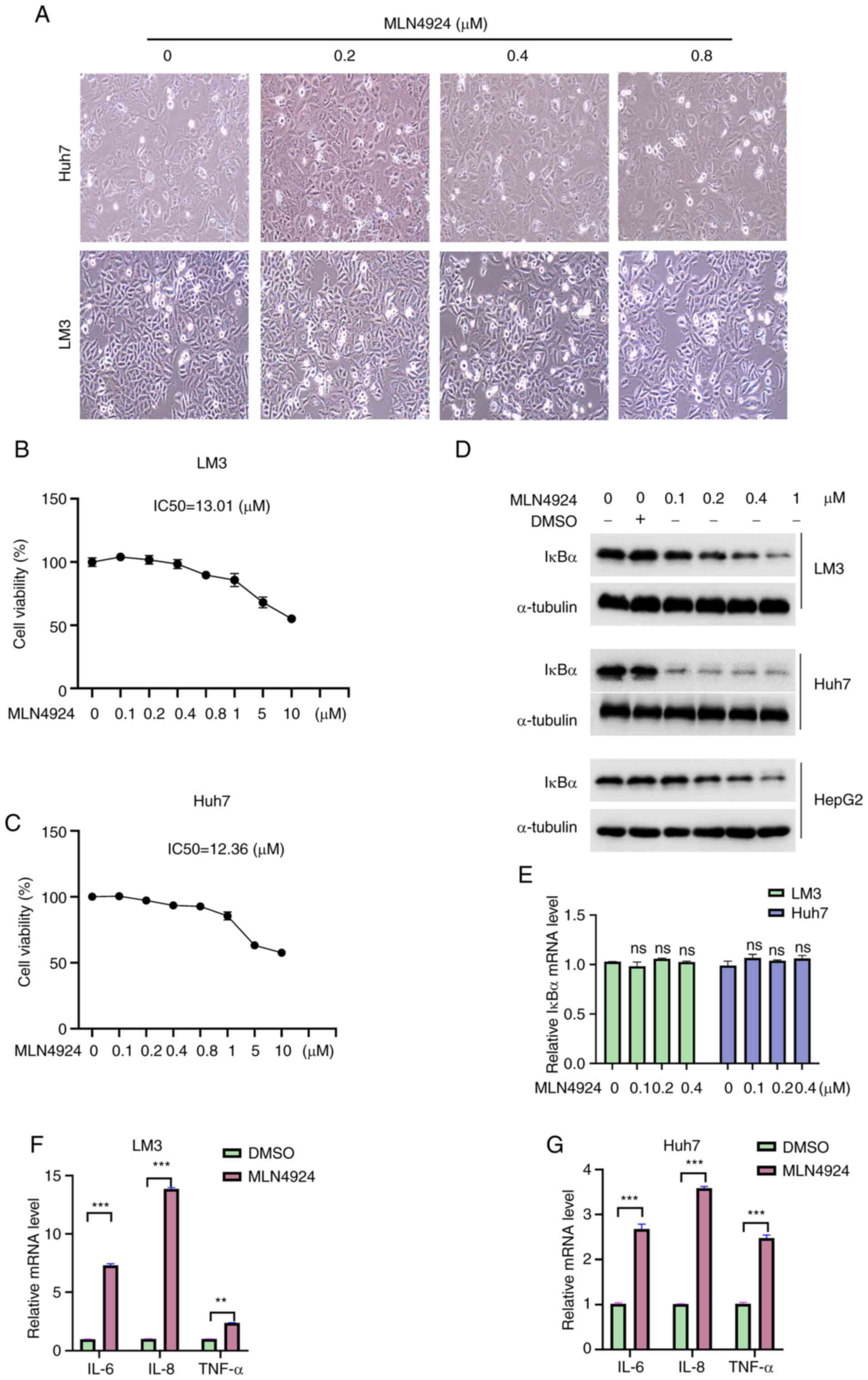 | Figure 1.MLN4924 suppresses the survival of
liver cancer cells and downregulates IκBα expression. (A) LM3 and
Huh7 cells were treated with the indicated concentrations of
MLN4924 for 48 h, and cells were imaged under a phase-contrast
microscope (magnification, ×200). (B and C) LM3 and Huh7 cells were
treated with the indicated concentrations of MLN4924 for 48 h, and
a Cell Counting Kit-8 assay was utilized to evaluate the
cytotoxicity of MLN4924. (D) Following treatment with the indicated
concentrations of MLN4924 for 48 h, the protein expression levels
of IκBα were determined in LM3, Huh7 and HepG2 cells using western
blot analysis. α-Tubulin was used as a loading control. (E) LM3 and
Huh7 cells were treated as previously described, and the mRNA
expression levels of IκBα were determined using RT-qPCR. (F and G)
LM3 and Huh7 cells were treated with 0.4 µM MLN4924 for 24 h, and
RT-qPCR was used to assess the mRNA expression levels of IL-6, IL-8
and TNF-α. β-Actin was used as a loading control. **P<0.01 and
***P<0.001. IκBα, NF-κB inhibitor α; RT-qPCR, reverse
transcription-quantitative PCR; IL, interleukin; TNF, tumor
necrosis factor; DMSO, dimethyl sulfoxide. |
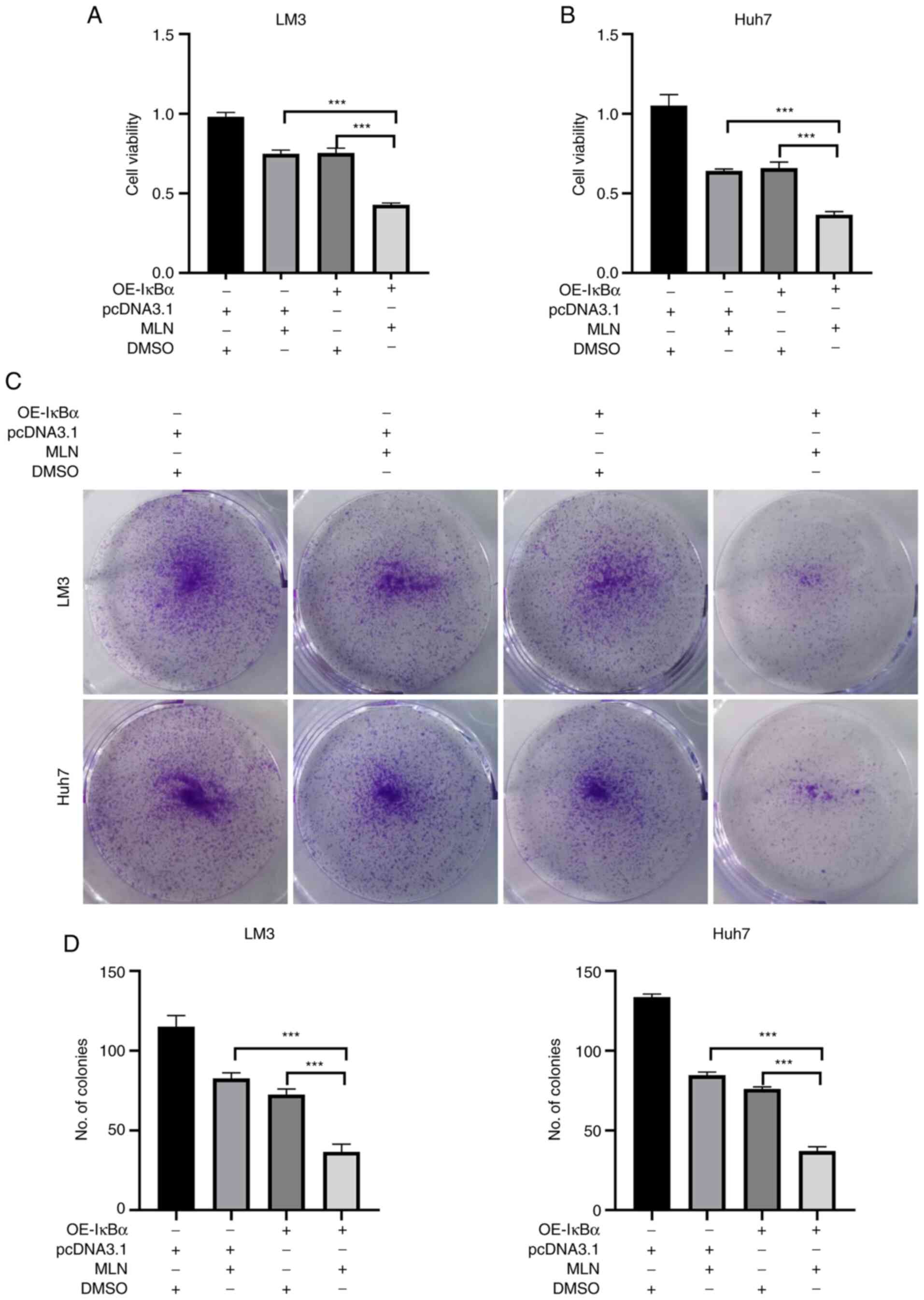
IκBα overexpression sensitizes MLN4924
to inhibit liver cancer cells
To clarify whether MLN4924-induced IκBα
downregulation is involved in the insensitivity of liver cancer
cells to MLN4924 treatment, LM3 and Huh7 cells were treated with
MLN4924 in the presence or absence of OE-IκBα. Firstly, the
efficacy of the plasmid transfection (OE-IκBα) was determined using
qPCR and it was determined that the mRNA level of IκBα was
significantly increased after transfection with OE-IκBα plasmid,
suggesting that transfection with OE-IκBα successfully
overexpressed IκBα in both LM3 and Huh7 cells (Fig. S3). As revealed in Fig. 2A and B, cotreatment with MLN4924
and OE-IκBα significantly suppressed the growth of liver cancer
cells compared with MLN4924 or OE-IκBα treatment alone. Moreover,
the results of the colony formation assay revealed that cotreatment
with MLN4924 and OE-IκBα significantly inhibited the number of
colonies, compared with MLN4924 or OE-IκBα treatment alone
(Fig. 2C and D). These results
indicated that downregulation of IκBα is a novel resistance factor
of MLN4924, and overexpression of IκBα sensitizes MLN4924 to
inhibit liver cancer cells.
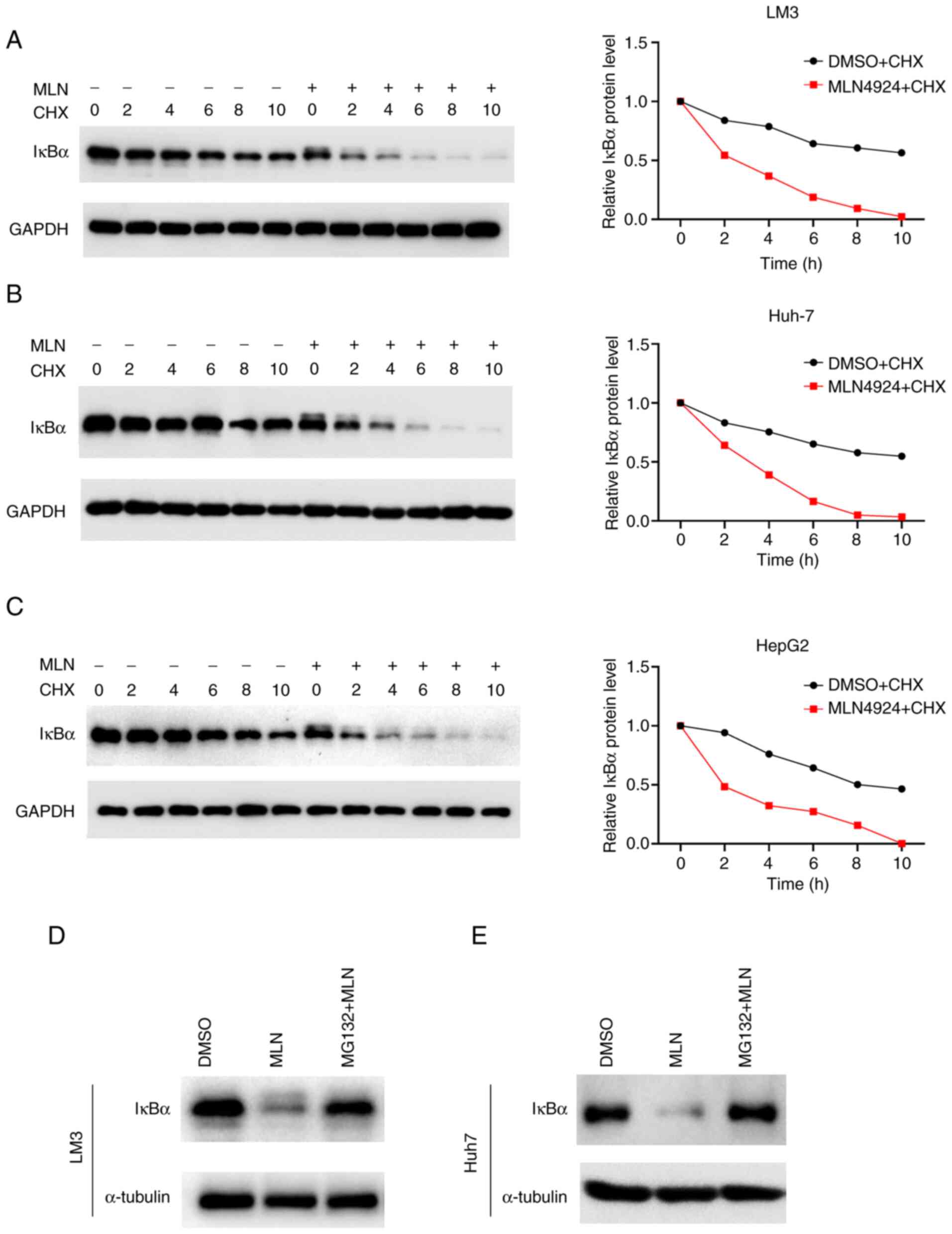
MLN4924 decreases the protein
stability of IκBα
The mechanism by which MLN4924 decreases the
expression of IκBα was explored in the present study. The results
of the RT-qPCR analysis revealed that IκBα mRNA expression levels
exhibited no significant change following MLN4924 treatment
(Fig. 1E), suggesting that MLN4924
downregulated IκBα at a post-translational level. Thus, IκBα
protein stability was determined following the addition of CHX in
the presence or absence of MLN4924. As demonstrated in Fig. 3A-C, MLN4924 treatment markedly
decreased the protein stability of IκBα and reduced the half-life
of IκBα protein in LM3, Huh7 and HepG2 cells. The results of
previous research demonstrated that proteasomes play an important
role in protein degradation (30).
To verify whether the proteasome is involved in MLN4924-mediated
IκBα degradation, the proteasome inhibitor MG132 was utilized. A
previous study demonstrated that MG132 (20 µM) always exerts its
function to inhibit protein degradation at 4–6 h (31), thus, MG132 (20 µM) was added and
incubated with LM3 and Huh7 cells at 37°C for 5 h before
harvesting. As shown in Fig. 3D and
E, MG132 treatment markedly attenuated MLN4924-induced IκBα
downregulation. These results indicated that MLN4924 promoted IκBα
protein degradation via a proteasome-associated pathway.
MLN4924 promotes the K48 linkage of
ubiquitin to IκBα protein
A previous study demonstrated that the
ubiquitin–proteasome pathway is key in controlling the degradation
of proteins in eukaryocytes (30).
Thus, the role of MLN4924 in IκBα protein degradation via the
ubiquitin-proteasome pathway was explored. As revealed in Fig. 4A and B, MLN4924 markedly enhanced
the ubiquitination of endogenous IκBα, and this was further
confirmed using exogenous overexpression of IκBα (Fig. 4C and D). These results indicated
that MLN4924 promoted the polyubiquitination of IκBα protein. To
determine the type of ubiquitin chains involved in MLN4924-mediated
IκBα polyubiquitination, six ubiquitin mutants (K63, K63R, K48,
K48R, K11 and K11R) were used along with the wild-type (WT)
ubiquitin. Results of the in vitro ubiquitination assay
indicated that K63 and K11 ubiquitin mutants attenuated the
formation of the polyubiquitin chain to IκBα, compared with the K48
ubiquitin mutant and WT ubiquitin. Moreover, the results of the
present study demonstrated that the K48R ubiquitin mutant
significantly attenuated the formation of the polyubiquitin chain
to IκBα, compared with WT, and the K63R and K11R mutant forms of
ubiquitin (Fig. 4E). Collectively,
these results indicated that MLN4924 promoted the K48-linked
ubiquitination of IκBα.
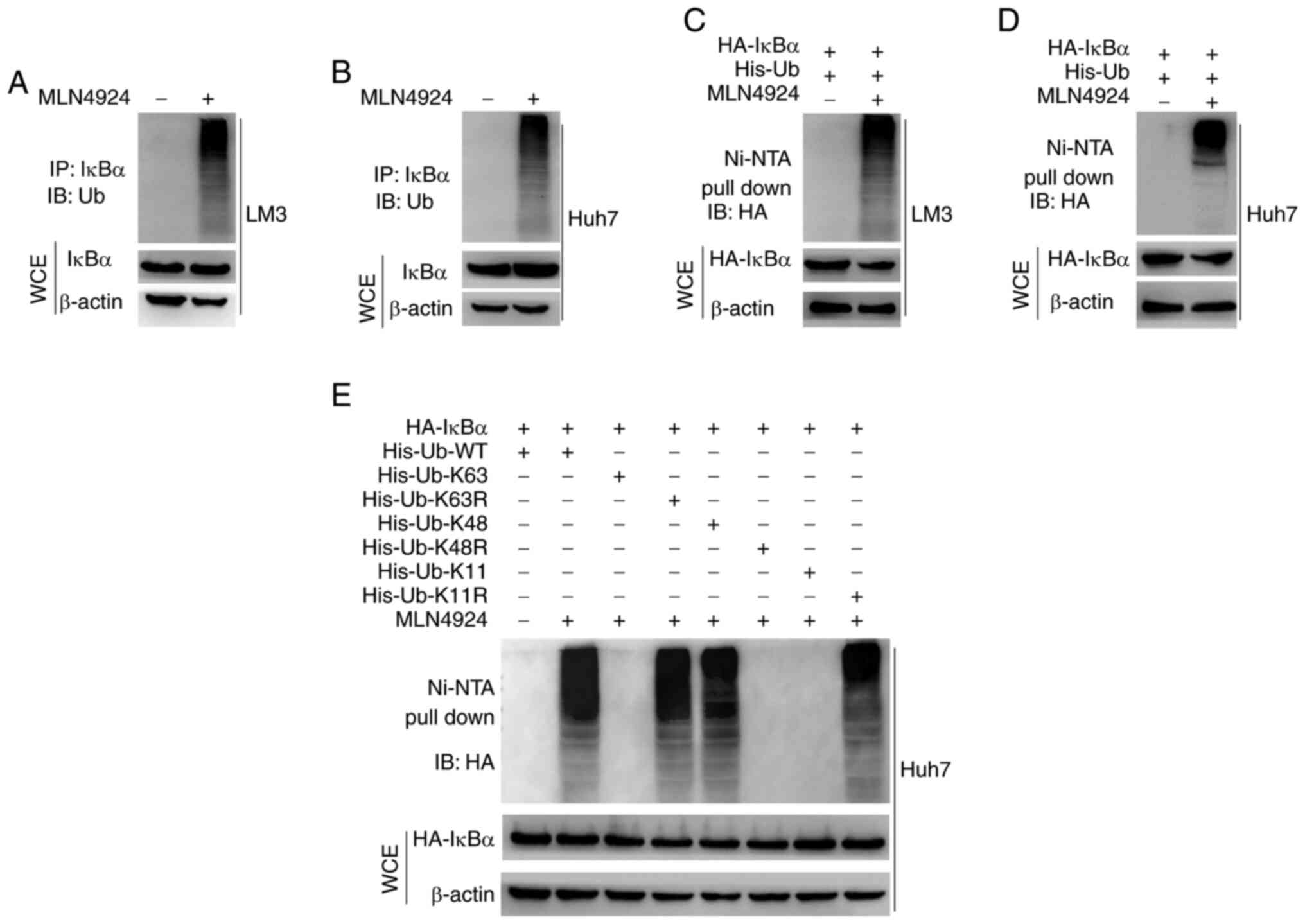 | Figure 4.MLN4924 promotes the K48 linkage of
ubiquitin to IκBα protein. (A and B) LM3 and Huh7 cells were
treated with 0.4 µM MLN4924 or DMSO for 48 h, and 20 µM MG132 was
subsequently added for 5 h at 37°C. IκBα protein was
immunoprecipitated using anti-IκBα. Moreover, ubiquitination of
IκBα in the immunoprecipitated fraction was assessed by western
blot analysis, using anti-ubiquitin. (C and D) Following
co-transfection with HA-IκBα and His-ub plasmids for 24 h, LM3 and
Huh7 cells were treated with 0.4 µM MLN4924 or DMSO for 24 h, and
20 µM MG132 was subsequently added for 5 h at 37°C. Ni-NTA beads
were used to pull down the His-ub tagged proteins. Ubiquitination
of HA-IκBα in the pull-down fraction was assessed by western blot
analysis, using anti-HA. Whole cell lysates were subjected to
western blot analysis. (E) Huh7 cells were transfected with the
indicated plasmids for 48 h, incubated with 20 µM MG132 for 5 h at
37°C, lysed with 6 M guanidine solution, pulled down with Ni-NTA
beads and western blot analysis was carried out to determine the
levels of HA-IκBα. The whole cell lysates were subjected to western
blot analysis. IκBα, NF-κB inhibitor α; DMSO, dimethyl sulfoxide;
His-ubiquitin, His-ub; WCE, whole cell extract. |
MLN4924 promotes the degradation of
IκBα protein via stabilizing β-TrCP
The mechanism underlying MLN4924-induced degradation
of IκBα was further explored in the present study. The results of a
previous study demonstrated that β-TrCP, also named FBXW1, is an
important E3 ligase for the ubiquitin-proteasome pathway-dependent
degradation of IκBα (32). Thus,
the role of MLN4924 in IκBα degradation via β-TrCP was explored.
The results of the western blot analysis demonstrated that MLN4924
markedly enhanced the protein expression levels of β-TrCP in LM3,
Huh7 and HepG2 cells compared with the controls (Fig. 5A). In addition, LM3 and Huh7 cells
were transfected with si-β-TrCP or si-NC in the presence or absence
of MLN4924. As shown in Fig. 5B and
C, β-TrCP knockdown markedly reduced MLN4924-mediated IκBα
downregulation. These results indicated that MLN4924 promoted the
degradation of IκBα via upregulation of β-TrCP. Moreover, the
protein stability of β-TrCP was evaluated by adding translation
inhibitor CHX in the presence or absence of MLN4924. The results of
the present study demonstrated that MLN4924 significantly enhanced
the protein stability of β-TrCP and prolonged the half-life of
β-TrCP in LM3, Huh7 and HepG2 cells (Fig. 5D-F). Collectively, these results
indicated that MLN4924 induces the degradation of IκBα via
stablizing β-TrCP.
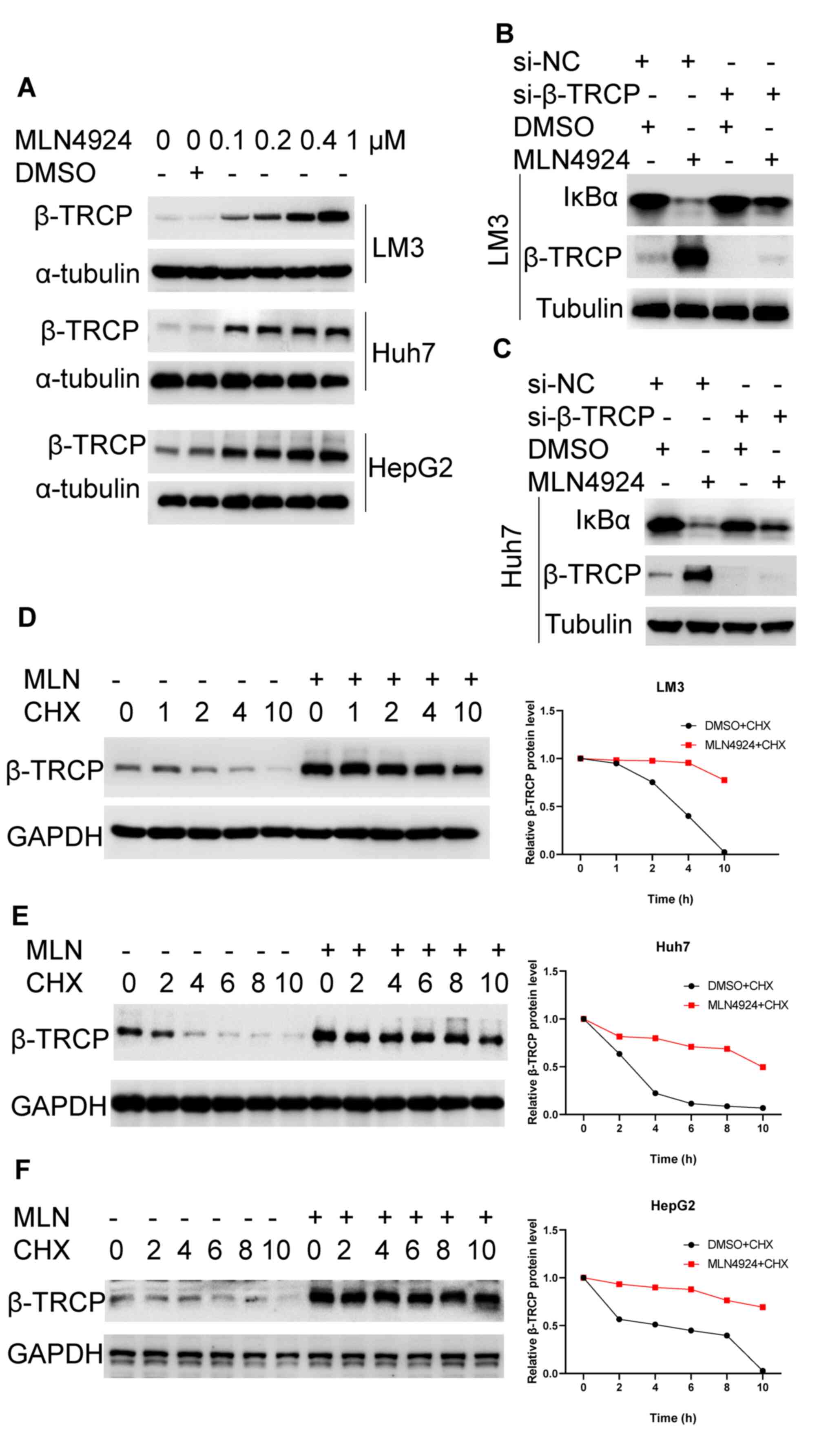 | Figure 5.MLN4924 promotes the degradation of
IκBα protein via stabilizing β-TrCP. (A) Following treatment with
the indicated concentrations of MLN4924 for 48 h, the protein
expression levels of β-TrCP were determined in LM3, Huh7 and HepG2
cells using western blot analysis. (B and C) Following transfection
with si-β-TrCP or si-NC for 24 h, LM3 and Huh7 cells were treated
with 0.4 µM MLN4924 or DMSO for 24 h. Subsequently, the protein
expression levels of IκBα and β-TrCP were determined using western
blot analysis. (D-F) Following treatment with 0.4 µM MLN4924 or
DMSO for 48 h, LM3, Huh7 and HepG2 cells were incubated with 10
µg/ml cycloheximide for the indicated time-points. Subsequently,
the protein expression levels of β-TrCP were determined using
western blot analysis and quantified using Image Lab. IκBα, NF-κB
inhibitor α; β-TrCP, β-transducin repeat-containing protein; siRNA,
small interfering RNA; si-NC, negative control siRNA; DMSO,
dimethyl sulfoxide; CHX, cycloheximide; MLN, MLN4924. |
β-TrCP knockdown enhances the
antitumor potential of MLN4924 in liver cancer cells via inhibiting
inflammation
To clarify whether knockdown of β-TrCP enhanced the
antitumor potential of MLN4924 in liver cancer cells via inhibiting
inflammation, LM3 and Huh7 cells were treated with MLN4924 in the
presence or absence of si-β-TrCP. As revealed in Fig. 6A and B, transfection with si-β-TrCP
significantly reduced the mRNA expression of β-TrCP. Moreover,
MLN4924 treatment notably enhanced the mRNA expression levels of
IL-6, IL-8 and TNF-α, which was significantly attenuated following
β-TrCP knockdown in LM3 and Huh7 cells. Moreover, the results of
the CCK-8 assay demonstrated that cotreatment with si-β-TrCP and
MLN4924 significantly suppressed the growth of liver cancer cells,
compared with MLN4924 or si-β-TrCP treatment alone (Fig. 6C and D). In addition, the results
of the western blot analysis demonstrated that β-TrCP knockdown
markedly enhanced MLN4924-induced caspase-3 upregulation compared
with the controls (Fig. 6E and F).
Collectively, these results demonstrated that β-TrCP knockdown
enhanced the antitumor potential of MLN4924 in liver cancer cells
via inhibiting inflammation.
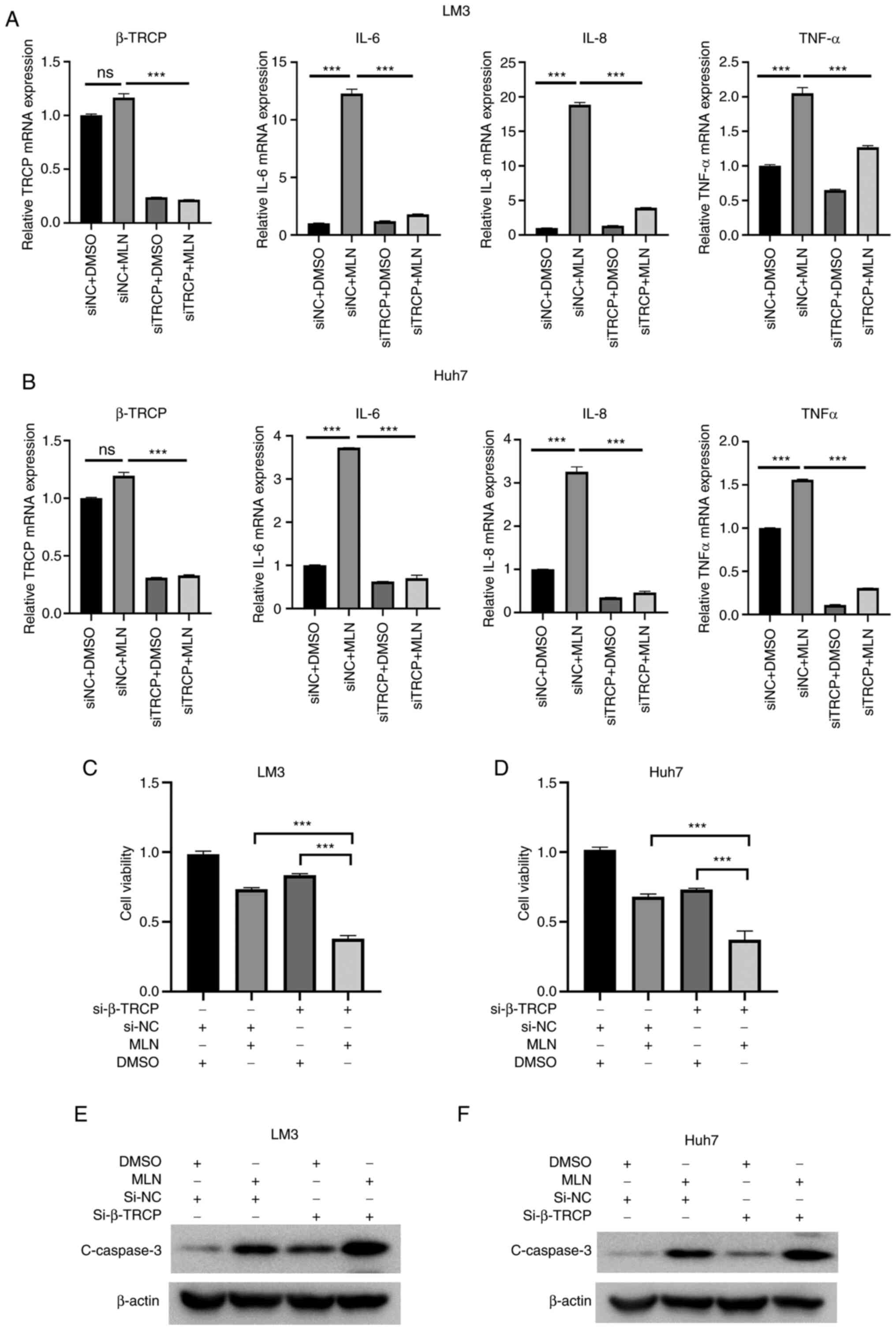 | Figure 6.β-TrCP knockdown enhances the
antitumor potential of MLN4924 in liver cancer cells via inhibiting
inflammation. (A and B) Following transfection with si-β-TrCP or
si-NC for 24 h, LM3 and Huh7 cells were treated with 0.4 µM MLN4924
or DMSO for 24 h. Subsequently, the mRNA expression levels of IL-6,
β-TrCP, IL-8 and TNF-α were assessed using reverse
transcription-quantitative PCR. (C and D) Following transfection
with si-β-TrCP or si-NC for 24 h, LM3 and Huh7 cells were treated
with 0.4 µM MLN4924 or DMSO for 48 h. A Cell Counting Kit-8 assay
was used to determine the growth of liver cancer cells. (E and F)
LM3 and Huh7 cells were treated as previously described, and the
protein expression levels of caspase-3 were determined using
western blot analysis. ***P<0.001. β-TrCP, β-transducin
repeat-containing protein; siRNA, small interfering RNA; si-NC,
negative control siRNA; DMSO, dimethyl sulfoxide; IL, interleukin;
TNF, tumor necrosis factor; ns, not significant. |
Discussion
The results of a previous study (28) demonstrated that MLN4924 exhibits
high suppressive activity against a variety of human cancer cells.
However, further investigations into the specific mechanisms
underlying MLN4924 as an anticancer treatment are required. To the
best of our knowledge, the results of the present study were the
first to demonstrate that downregulation of IκBα and subsequent
inflammation is key for attenuating the antitumor potential of
MLN4924 in liver cancer cells. The mechanistic study of
MLN4924-induced IκBα downregulation revealed that MLN4924
stabilized β-TrCP, promoting the ubiquitination of IκBα; thus,
enhancing the degradation of IκBα and subsequent inflammation. This
therefore attenuated the antitumor potential of MLN4924 in liver
cancer cells. Moreover, the results of the present study
demonstrated that β-TrCP knockdown enhanced the antitumor potential
of MLN4924 in liver cancer cells via inhibiting inflammation.
Collectively, the afore mentioned results may provide a novel
potential strategy for sensitizing MLN4924 in the treatment of
liver cancer.
The results of previous studies have demonstrated
that inflammation could lead to chemoresistance of cancer cells
(17,33). Zhong et al revealed that the
NF-κB-IL6-STAT3 axis activated by intratumoral LPS promoted the
expression of cyclin D1, c-Myc, Bcl-2 and survivin, facilitating
prostate cancer proliferation and docetaxel chemoresistance
(33). Neddylation-inhibitor
MLN4924 exhibits broad anticancer potential and may be used as a
chemotherapeutic agent in the future. However, it remains to be
fully elucidated whether inflammation is involved in the
chemoresistance of liver cancer cells in response to MLN4924
treatment. The results of the present study demonstrated that
MLN4924 promoted IκBα degradation and increased the expression of
inflammatory factors (IL-6, IL-8 and TNF-α), leading to the
chemoresistance of liver cancer cells to MLN4924. Notably, the
results of a previous study demonstrated that MLN4924 inhibited
activation of the NF-κB pathway via inhibiting CRL-mediated IκBα
degradation. However, the results of this study did not indicate
that treatment with MLN4924 decreased the protein expression levels
of IκBα (34). Moreover, IκBα
knockdown significantly increased the MLN4924-mediated
proliferation inhibition in esophageal cancer cells (34). Therefore, the contrasting results
of the two studies may be ascribed to the different cancer cell
lines, as liver cancer is one of several cancers that is often
associated with chronic inflammation.
MLN4924, a neddylation inhibitor, suppresses CRLs
via inhibiting NAE-mediated cullin neddylation (28). Notably, the results of the present
study demonstrated that MLN4924 promoted the proteasome-dependent
degradation of IκBα and shortened the half-life of IκBα, following
treatment with MG132 and translation inhibitor CHX. Moreover, the
K48-linkage of ubiquitin to IκBα was significantly increased
following treatment with MLN4924, which reflects that some E3
ligases may be involved in MLN4924-mediated IκBα degradation. The
expression of β-TrCP, a well-established E3 ligase for IκBα
degradation (35), was markedly
increased following MLN4924 treatment in liver cancer cells.
However, the molecular mechanism by which MLN4924 stabilizes β-TrCP
remains unclear. Further studies are warranted to clarify which
protein is targeted by MLN4924 directly and participates in the
regulation of β-TrCP stability. Moreover, in the present study it
was determined that MLN4924 promoted IκBα degradation via
stabilizing β-TrCP, despite a previous study which reported that
β-TrCP is inhibited by MLN4924 (36). Therefore, it was hypothesized that
increase of β-TrCP may be a resistance response of liver cancer
cells when treated with neddylation inhibitor MLN4924.
β-TrCP acts as a substrate receptor and constitutes
an active SCFβ−TrCP ligase with a scaffold protein
cullin 1 (CUL1), a RING protein RBX1 and an adaptor protein SKP1
(37). β-TrCP plays a critical
role in the regulation of various physiological and pathological
processes, including signal transduction, cell cycle progression,
cell migration, DNA damage response and tumorigenesis, by governing
large amounts of key regulators for ubiquitination and proteasomal
degradation (38). The results of
previous studies have demonstrated that β-TrCP plays a vital role
in carcinogenesis via promoting the degradation of its substrates,
including oncoproteins and tumor suppressors (38). Specifically, the results of
previous studies demonstrated that β-TrCP is upregulated in liver
cancer tissues (39,40). Previous research has also
demonstrated that β-TrCP promotes HCC progression and metastasis
via degrading leucine zipper tumor suppressor 2 (LZTS2) in HCC
cells (41). These results
indicated that β-TrCP acts as an oncoprotein in liver cancer, which
provides clear rationale for targeting β-TrCP to overcome
chemoresistance. The results of the present study demonstrated that
β-TrCP was increased following MLN4924 treatment, and β-TrCP
knockdown enhanced the antitumor potential of MLN4924 in liver
cancer cells. Moreover, MLN4924 enhanced the protein expression
levels of β-TrCP via inhibiting its degradation and prolonging its
half-life. However, the mechanism by which MLN4924 stabilizes
β-TrCP remains to be fully elucidated. Previous investigations
demonstrated that β-TrCP1 is ubiquitinated and degraded by
SCFSKP2 ubiquitin ligase, SAG-CUL5 ubiquitin ligase with
UBCH10 and UBE2S E2s, as well as the SMURF2:UBCH5 complex (38). Moreover, members of the
de-ubiquitinating enzyme (DUB) family, such as USP24 (42) and USP47 (43,44),
enhance the stability of β-TrCP. Further investigations into the
role of E3 ligases and DUBs in MLN4924-mediated β-TrCP upregulation
are required.
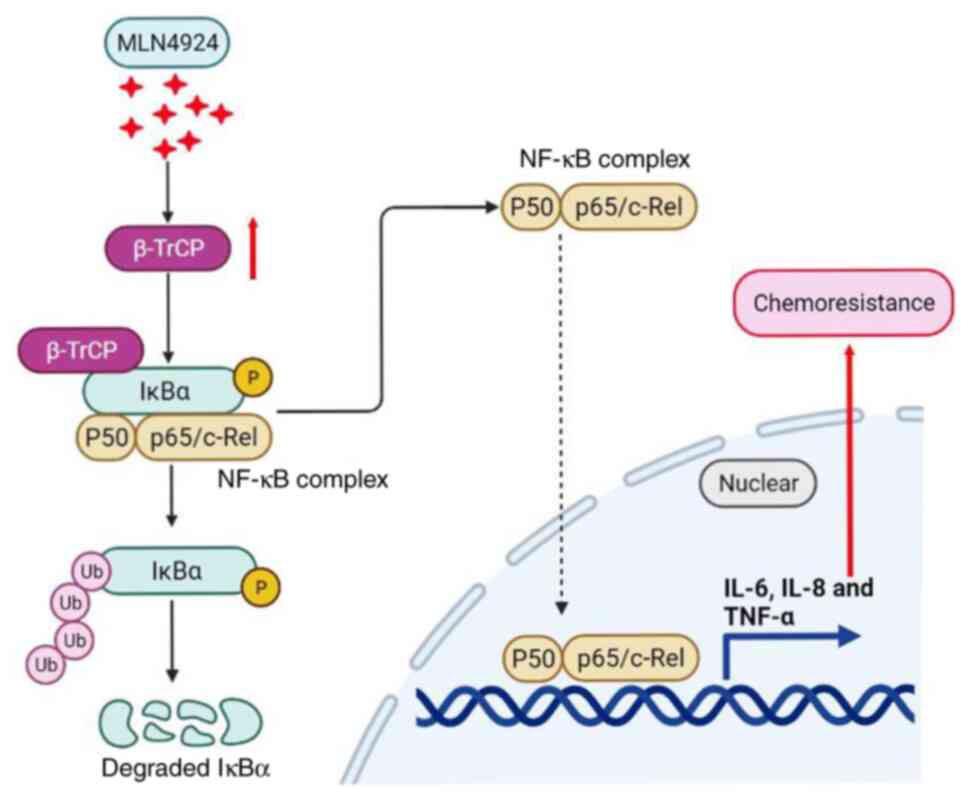
In conclusion, the results of the present study
demonstrated that downregulation of IκBα and the subsequent
inflammation attenuate the antitumor potential of MLN4924 in liver
cancer cells. Mechanistic studies demonstrated that MLN4924
enhanced the protein stability of β-TrCP, promoting the
ubiquitination of IκBα protein, leading to the ubiquitin–mediated
degradation of the IκBα protein, which is summarized in a working
diagram (Fig. 7). In addition, the
results of the present study demonstrated that β-TrCP knockdown
markedly sensitized MLN4924 in suppressing the growth of liver
cancer cells via attenuating MLN4924-mediated IκBα downregulation
and inflammation. The results of the present study not only provide
novel insights into the molecular mechanisms by which MLN4924
promotes the degradation of IκBα and subsequent inflammation, but
also offer a potential therapeutic strategy for sensitizing MLN4924
in the treatment of liver cancer.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZhixiangY and ZhenzhouY supervised the present
study. HX designed the research, performed the experiments and
drafted the manuscript. HX, DZ, YL and LMa contributed to the
acquisition of data. HX, DZ and YL analyzed and interpreted the
data. HX, YL, LMa and LMeng performed the statistical analysis. HX
and ZhixiangY confirm the authenticity of all the raw data. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Forner A, Reig M and Bruix J:
Hepatocellular carcinoma. Lancet. 391:1301–1314. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Forner A, Llovet JM and Bruix J:
Hepatocellular carcinoma. Lancet. 379:1245–1255. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Llovet JM, Zucman-Rossi J, Pikarsky E,
Sangro B, Schwartz M, Sherman M and Gores G: Hepatocellular
carcinoma. Nat Rev Dis Primers. 2:160182016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Llovet JM, Kelley RK, Villanueva A, Singal
AG, Pikarsky E, Roayaie S, Lencioni R, Koike K, Zucman-Rossi J and
Finn RS: Hepatocellular carcinoma. Nat Rev Dis Primers. 7:62021.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Enchev RI, Schulman BA and Peter M:
Protein neddylation: Beyond cullin-RING ligases. Nat Rev Mol Cell
Biol. 16:30–44. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yu Q, Jiang Y and Sun Y: Anticancer drug
discovery by targeting cullin neddylation. Acta Pharm Sin B.
10:746–765. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhou L, Zhang W, Sun Y and Jia L: Protein
neddylation and its alterations in human cancers for targeted
therapy. Cell Signal. 44:92–102. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Soucy TA, Smith PG, Milhollen MA, Berger
AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP,
Critchley S, et al: An inhibitor of NEDD8-activating enzyme as a
new approach to treat cancer. Nature. 458:732–736. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Luo Z, Yu G, Lee HW, Li L, Wang L, Yang D,
Pan Y, Ding C, Qian J, Wu L, et al: The Nedd8-activating enzyme
inhibitor MLN4924 induces autophagy and apoptosis to suppress liver
cancer cell growth. Cancer Res. 72:3360–3371. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Soucy TA, Dick LR, Smith PG, Milhollen MA
and Brownell JE: The NEDD8 conjugation pathway and its relevance in
cancer biology and therapy. Genes Cancer. 1:708–716. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Milhollen MA, Traore T, Adams-Duffy J,
Thomas MP, Berger AJ, Dang L, Dick LR, Garnsey JJ, Koenig E,
Langston SP, et al: MLN4924, a NEDD8-activating enzyme inhibitor,
is active in diffuse large B-cell lymphoma models: Rationale for
treatment of NF-{kappa}B-dependent lymphoma. Blood. 116:1515–1523.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sarantopoulos J, Shapiro GI, Cohen RB,
Clark JW, Kauh JS, Weiss GJ, Cleary JM, Mahalingam D, Pickard MD,
Faessel HM, et al: Phase I study of the investigational
NEDD8-activating enzyme inhibitor pevonedistat (TAK-924/MLN4924) in
patients with advanced solid tumors. Clin Cancer Res. 22:847–857.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhou Q, Li H, Li Y, Tan M, Fan S, Cao C,
Meng F, Zhu L, Zhao L, Guan MX, et al: Inhibiting neddylation
modification alters mitochondrial morphology and reprograms energy
metabolism in cancer cells. JCI Insight. 4:e1215822019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ferdosi SR, Taylor B, Lee M, Tang N, Peng
S, Bybee R, Reid G, Hartman L, Garcia-Mansfield K, Sharma R, et al:
PTEN loss drives resistance to the neddylation inhibitor MLN4924 in
glioblastoma and can be overcome with TOP2A inhibitors. Neuro
Oncol. 19:noac0672022. View Article : Google Scholar
|
|
17
|
Taniguchi K and Karin M: NF-κB,
inflammation, immunity and cancer: Coming of age. Nat Rev Immunol.
18:309–324. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Elsharkawy AM and Mann DA: Nuclear
factor-kappaB and the hepatic inflammation-fibrosis-cancer axis.
Hepatology. 46:590–597. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shostak K, Zhang X, Hubert P, Göktuna SI,
Jiang Z, Klevernic I, Hildebrand J, Roncarati P, Hennuy B, Ladang
A, et al: NF-κB-induced KIAA1199 promotes survival through EGFR
signalling. Nat Commun. 5:52322014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen CY, Chang JT, Ho YF and Shyu AB:
MiR-26 down-regulates TNF-α/NF-κB signalling and IL-6 expression by
silencing HMGA1 and MALT1. Nucleic Acids Res. 44:3772–3787. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hoffmann A, Levchenko A, Scott ML and
Baltimore D: The IkappaB-NF-kappaB signaling module: Temporal
control and selective gene activation. Science. 298:1241–1245.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu B, Sun L, Liu Q, Gong C, Yao Y, Lv X,
Lin L, Yao H, Su F, Li D, et al: A cytoplasmic NF-κB interacting
long noncoding RNA blocks IκB phosphorylation and suppresses breast
cancer metastasis. Cancer Cell. 27:370–381. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ataie-Kachoie P, Badar S, Morris DL and
Pourgholami MH: Minocycline targets the NF-κB Nexus through
suppression of TGF-β1-TAK1-IκB signaling in ovarian cancer. Mol
Cancer Res. 11:1279–1291. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cao L, Zhu S, Lu H, Soutto M, Bhat N, Chen
Z, Peng D, Lin J, Lu J, Li P, et al: Helicobacter pylori-induced
RASAL2 through activation of nuclear factor-κB promotes gastric
tumorigenesis via beta-catenin signaling axis. Gastroenterology.
162:1716–1731. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lu YX, Ju HQ, Wang F, Chen LZ, Wu QN,
Sheng H, Mo HY, Pan ZZ, Xie D, Kang TB, et al: Inhibition of the
NF-κB pathway by nafamostat mesilate suppresses colorectal cancer
growth and metastasis. Cancer Lett. 380:87–97. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xie C, Zhang LZ, Chen ZL, Zhong WJ, Fang
JH, Zhu Y, Xiao MH, Guo ZW, Zhao N, He X and Zhuang SM: A
hMTR4-PDIA3P1-miR-125/124-TRAF6 regulatory axis and its function in
NF kappa B signaling and chemoresistance. Hepatology. 71:1660–1677.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhou L, Jiang Y, Luo Q, Li L and Jia L:
Neddylation: A novel modulator of the tumor microenvironment. Mol
Cancer. 18:772019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gamble C, McIntosh K, Scott R, Ho KH,
Plevin R and Paul A: Inhibitory kappa B Kinases as targets for
pharmacological regulation. Br J Pharmacol. 165:802–819. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Narayanan S, Cai CY, Assaraf YG, Guo HQ,
Cui Q, Wei L, Huang JJ, Ashby CR Jr and Chen ZS: Targeting the
ubiquitin-proteasome pathway to overcome anti-cancer drug
resistance. Drug Resist Updat. 48:1006632020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fiedler MA, Wernke-Dollries K and Stark
JM: Inhibition of TNF-alpha-induced NF-kappaB activation and IL-8
release in A549 cells with the proteasome inhibitor MG-132. Am J
Respir Cell Mol Biol. 19:259–268. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Winston JT, Strack P, Beer-Romero P, Chu
CY, Elledge SJ and Harper JW: The SCFbeta-TRCP-ubiquitin ligase
complex associates specifically with phosphorylated destruction
motifs in IkappaBalpha and beta-catenin and stimulates IkappaBalpha
ubiquitination in vitro. Genes Dev. 13:270–283. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhong W, Wu K, Long Z, Zhou X, Zhong C,
Wang S, Lai H, Guo Y, Lv D, Lu J and Mao X: Gut dysbiosis promotes
prostate cancer progression and docetaxel resistance via activating
NF-κB-IL6-STAT3 axis. Microbiome. 10:942022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liang Y, Jiang Y, Jin X, Chen P, Heng Y,
Cai L, Zhang W, Li L and Jia L: Neddylation inhibition activates
the protective autophagy through NF-κB-catalase-ATF3 Axis in human
esophageal cancer cells. Cell Commun Signal. 18:722020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yaron A, Hatzubai A, Davis M, Lavon I,
Amit S, Manning AM, Andersen JS, Mann M, Mercurio F and Ben-Neriah
Y: Identi-fication of the receptor component of the
IkappaBalpha-ubiquitin ligase. Nature. 396:590–594. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tanaka T, Nakatani T and Kamitani T:
Inhibition of NEDD8-conjugation pathway by novel molecules:
Potential approaches to anticancer therapy. Mol Oncol. 6:267–275.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang Z, Liu P, Inuzuka H and Wei W: Roles
of F-box proteins in cancer. Nat Rev Cancer. 14:233–247. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bi Y, Cui D, Xiong X and Zhao Y: The
characteristics and roles of β-TrCP1/2 in carcinogenesis. FEBS J.
288:3351–3374. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Koch A, Waha A, Hartmann W, Hrychyk A,
Schüller U, Waha A, Wharton KA Jr, Fuchs SY, von Schweinitz D and
Pietsch T: Elevated expression of Wnt antagonists is a common event
in hepatoblastomas. Clin Cancer Res. 11:4295–4304. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Perra A, Kowalik MA, Ghiso E,
Ledda-Columbano GM, Tommaso LD, Angioni MM, Raschioni C, Testore E,
Roncalli M, Giordano S and Columbano A: YAP activation is an early
event and a potential therapeutic target in liver cancer
development. J Hepatol. 61:1088–1096. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lu Y, Li X, Liu H, Xue J, Zeng Z, Dong X,
Zhang T, Wu G, Yang K and Xu S: β-Trcp and CK1δ-mediated
degradation of LZTS2 activates PI3K/AKT signaling to drive
tumorigenesis and metastasis in hepatocellular carcinoma. Oncogene.
40:1269–1283. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang YC, Wu YS, Hung CY, Wang SA, Young
MJ, Hsu TI and Hung JJ: USP24 induces IL-6 in tumor-associated
microenvironment by stabilizing p300 and beta-TrCP and promotes
cancer malignancy. Nat Commun. 9:39962018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Peschiaroli A, Skaar JR, Pagano M and
Melino G: The ubiquitin-specific protease USP47 is a novel
beta-TRCP interactor regulating cell survival. Oncogene.
29:1384–1393. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bufalieri F, Infante P, Bernardi F,
Caimano M, Romania P, Moretti M, Severini LL, Talbot J, Melaiu O,
Tanori M, et al: ERAP1 promotes Hedgehog-dependent tumorigenesis by
controlling USP47-mediated degradation of betaTrCP. Nat Commun.
10:33042019. View Article : Google Scholar : PubMed/NCBI
|