Introduction
Previous research has demonstrated that 80–90% of
pancreatic cancer cases are pancreatic ductal adenocarcinoma
(PDAC). In the present study, pancreatic cancer refers to PDAC.
Pancreatic cancer mainly occurs in the head of the pancreas,
accounting for >70% of all cases. The most common clinical
symptom of pancreatic cancer is abdominal pain, often accompanied
by jaundice and new-onset diabetes mellitus (1). Currently, surgery is the only
available strategy used to eradicate pancreatic cancer. However, as
the pancreas is located at the back of the abdomen, the symptoms of
early-stage pancreatic cancer are not obvious and the majority of
patients have already developed near invasion and distant
metastasis at the time of diagnosis; thus, only a limited number of
patients have the opportunity to receive surgery (2). Although PDAC has a low incidence, it
constitutes the seventh leading cause of cancer-related mortality
worldwide and the fourth leading cause of cancer-related mortality
in the USA (3,4). Despite continuous improvements in
tumor diagnosis and treatment strategies in general over the past
several decades, and the notable progress in the 5-year survival
rate of patients with certain tumor types, such as leukemia, the
5-year survival rate of patients with pancreatic cancer remains low
(8.5%) in the USA (4).
Due to the poor efficacy of surgery for PDAC and the
limited efficacy of recent chemotherapeutic regimens, the
development of novel targeted drugs is of utmost importance. PDAC
involves a number of mutated genes, including KRAS, BRCA2, INK4A,
LκB1 and CDKN2A (5). Among these,
the KRAS gene mutation exists in the majority of cases of advanced
pancreatic cancer (6), which is
most commonly found in codon 12 (7). Previous studies have indicated that
the KRAS gene mutation is one of the earliest genetic events in the
progression of human pancreatic intraepithelial neoplasia (PanIN)
(8,9). At first, due to the universality and
importance of KRAS mutations in pancreatic cancer, a number of
small molecule inhibitor studies targeting mutant RAS protein have
been performed; however, to date, these have proven unsuccessful
(10). Researchers have tried to
identify the signal transduction pathways which play a vital role
downstream of RAS, in order to inhibit the occurrence and
development of PDAC by suppressing these pathways (11). NF-κB is constitutively activated in
the majority (67–70%) patients with PDAC (12,13),
which can promote cell proliferation, angiogenesis and invasion
(14). Thus, NF-κB is considered a
highly promising target for PDAC treatment. However, directly
targeting NF-κB proteins by small-molecule inhibitors has proven
unsuccessful for >30 years (15–17);
however, efforts against potential NF-κB inducers [such as tumor
necrosis factor (TNF)-α, interleukin (IL)-1α and Toll-like receptor
(TLR) family members] have achieved some effects (18–20).
TLR and IL-1 family receptors are key molecules for
human cells to recognize microorganisms or endogenous ligands and
inflammatory mediators, which play a vital role in the activation
of the NF-κB pathway (21). Ling
et al (22) revealed that
KrasG12D-activated AP-1 induced IL-1α, which activated NF-κB and
its target genes, IL-1α and p62, to initiate IL-1α/p62 feedforward
loops for inducing and sustaining NF-κB activity. Furthermore,
IL-1α overexpression was shown to be associated with Kras mutation,
NF-κB activity and the poor survival in of patients with PDAC.
Zhang et al (23)
demonstrated that the IL-1 receptor-associated kinase 4 (IRAK4),
the master kinase that relays signaling downstream of TLRs, was
activated in human PDAC samples and positively correlated with
activated NF-κB, which was associated with a high post-operative
relapse and a poor patient survival. Therefore, MyD88, as the
common intermediate messenger for NF-κB activation by these
inducers, may play a vital role in PDAC therapy.
Myeloid differentiation factor 88 (MyD88) MyD88 is a
member of the TLR/IL-1R family and the death domain family; apart
from TLR3, they all transmit signals through MyD88 (24). MyD88 is a soluble cytoplasmic
protein with three functional domains. The N-terminus is a domain
with 90 amino acid residues, mainly mediating interactions between
proteins containing dead sequences. The C-terminus Toll and
intermediate regions, which contain 130 amino acid residues, mainly
transmit signals by recruiting junction proteins. Previous studies
have demonstrated that an elevated MyD88 expression promotes tumor
growth and metastasis via TLR/IL-1R signaling in hepatocellular
carcinoma (HCC) and is related to the low survival rate of patients
with PDAC (25,26). Zhu et al (27) revealed that blocking MyD88 signaling
markedly attenuated the development of PDAC-associated cachexia.
MyD88-dependent inflammation is crucial in the pathophysiology of
pancreatic cancer progression and contributes to a high mortality
rate (27). Thus, it was
hypothesized that MyD88 inhibition, potentially via the specific
MyD88 small molecule inhibitor, ST2825 (28), which has been shown to inhibit HCC
cell proliferation and promote cell apoptosis (29), may serve as an effective target
strategy for PDAC.
In order to identify factors and strategies with
which to enhance the efficacy of PDAC chemotherapeutics, the
present study aimed to determine the role of MyD88 in PDAC and
whether MyD88 inhibition by ST2825 would suppress the progression
of PDAC. Furthermore, the present study investigated whether NF-κB
activation plays a vital role in the effects of ST2825, and also
aimed to identify the underlying signaling pathways.
Materials and methods
Tissues, cell lines and reagents
A tissue chip of PDAC and paracancerous tissues
(HPanAde170Sur01) was obtained from Shanghai Outdo Biotech Co.,
Ltd. The human PDAC cell lines, AsPC-1 (ATCC cat. no. CRL-1682,
RRID:CVCL_0152), BxPC-3 (ATCC cat. no. CRL-1687, RRID:CVCL_0186),
CFPAC-1 (ATCC cat. no. CRL-1918, RRID:CVCL_1119), PANC-1 (ATCC cat.
no. CRL-1469, RRID:CVCL_0480) and hTERT-HPNE (ATCC cat. no.
CRL-4023, RRID:CVCL_C466) were purchased from the American Type
Culture Collection. All cells were maintained in RPMI-1640 medium
containing 10% fetal bovine serum (Thermo Fisher Scientific, Inc.)
and cultured at 37°C in an atmosphere of 5% CO2. The
drugs, activators and inhibitors used included ST2825 (HY-50937)
and IL-1α (HY-P7027) from MedChem Express.
Cell interference and
transfection
The cells were cultured in six-ell plates with 3 µl
short interfering RNA (siRNA) and 7 µl Lipofectamine
3000® (Thermo Fisher Scientific, Inc.) for 6 h. The
siRNA, named p21(h)-si-1,2,3 (si-1, 5′-GCGATGGAACTTCGACTTTGT-3′;
si-2, 5′-GCTCTACATCTTCTGCCTTAG-3′; si-3,
5′-GCAGACCAGCATGACAGATTT-3′) was designed and synthesized by
GenePharma Co., Ltd. The AKT1 recombinant plasmids were designed
and synthesized by Ruibo Bio-Technology Co., Ltd. and were used
following the manufacturer's protocol. The AKT1 recombinant
lentivirus was designed and synthesized by Hanbio Biological
Technology Co. The PANC-1 cells were counted the day prior to viral
infection; 10,000 cells were inoculated in a well of a 12-well
plate and cultured overnight at 37°C. The virus was diluted with
serum-free medium, and added in the 12-well plate for infection.
The number of viruses was added according to the recommended
infection MOI for PANC-1 cells (MOI=10). Following 6 h of infection
using 3 µl Polyjet reagent (SignaGen Laboratories), the solution
containing the virus was removed and RPMI-1640 medium containing
10% fetal bovine serum was added for cell culture. Following 48 h
of culture at 37°C the fluorescence intensity of the cells was
observed under a fluorescence microscope (LEXT OLS4100, Olympus
Corporation). At the same time, 3 µg/ml puromycin (HY-B1743A,
MedChem Express) was added for cell screening, which lasted for ~2
weeks. After cell screening, reverse transcription-quantitative PCR
(RT-qPCR) and western blot analysis were performed to detect the
stable expression of the target gene.
Nuclear and cytoplasmic extraction and
western blot analysis
The nuclear and cytoplasmic extraction reagents were
utilized according to the manufacturer instructions (NE-PER™, cat.
no. 78833, Thermo Fisher Scientific, Inc.). Total proteins were
then extracted from the cells by incubating in RIPA cell lysis
buffer with 1% PMSF and phosphatase inhibitors (Wuhan Servicebio
Technology Co., Ltd.) on ice for 30 min. Following centrifugation
(1,000 × g, 4°C, 5 min), the supernatant was collected. Nuclear and
cytoplasm extracts were collected using Nuclear and Cytoplasmic
Extraction Reagents (NE-PER™, Thermo Fisher Scientific, Inc.). The
cells were pelleted and re-suspended in 400 ml cold Buffer A at
4°C. The cells were set on ice for 10 min and then vortexed for 10
sec. Following centrifugation (1,000 × g, 4°C, 5 min), the
supernatant fraction was saved as crude cytoplasm extract. The
pellet was re-suspended in 20 to 100 ml cold Buffer C basing on the
starting number of cells and incubated on ice for 20 min for
high-salt extraction. Nuclear and cytoplasm extracts were collected
and cleared by centrifugation. Subsequently, 30 µg
total/nuclear/cytoplasm protein were measured using a BCA protein
assay kit (Thermo Fisher Scientific, Inc.). The proteins were then
separated using 4–12% SurePAGE Bis-Tris gels [Gen Script (Nanjing)
Co., Ltd.] at consistent 120 V for 60 min. The separated proteins
were then transferred onto PVDF membranes at consistent 350 mA for
60 min. Following transfer, the membrane was blocked using 5% BSA
solution for 2 h at 4°C, followed by overnight incubation at 4°C
with the following primary antibodies. Antibodies against the
following proteins were used: GAPDH, Lamin B1, p21, p53, MyD88,
p100, p105 (all from Abcam); Bax, Bcl-2 (Proteintech, Rosemont, IL,
USA); Cdk1, phosphorylate-Cdk1, Cyclin B1, Chk1,
phosphorylated-Chk1, phosphorylated-AKT1, AKT1, phosphorylated-p65,
p65, Caspase-3, cleaved Caspase-3, PARP, cleaved PARP (Cell
Signaling Technology, Inc.); and p50, p52, IKKα and IKKβ (Santa
Cruz Biotechnology, Inc.). Target proteins were detected by
anti-mouse or anti-rabbit antibodies for 1 h at room temperature.
Detection was carried out using the ECL kit (Wuhan Servicebio
Technology Co., Ltd.). Visualization was performed using the
ChemiDoc MP system (Bio-Rad Laboratories, Inc.). The grayscale
detection of protein bands was completed using Image Lab 6.1
software (Bio-Rad Laboratories, Inc.). GAPDH was used to normalize
the protein expression. The detailed research resource identifiers
(RRIDs), catalogue numbers and dilutions of the antibodies are
presented in Table SI.
RT-qPCR
TRIzol® reagent (Thermo Fisher
Scientific, Inc.) was used for the extraction of total RNA from the
cells. NanoDrop ND-1000 UV/visible photometer (Thermo Fisher
Scientific, Inc.) was used to assess RNA purity. Reactions were
performed using the ChamQ universal SYBR master mix (Vazyme
Biotech) in the Bio-Rad CFX96 RealTime System. Pre-incubation at
95°C for 120 sec, amplification at 40 cycles of 95°C for 30 sec,
60°C for 30 sec and 72°C for 30 sec and then a final extension at
72°C for 5 min. mRNA expression was calculated using the
2−ΔΔCq method (30).
Shanghai Biological Engineering Technology Co. designed and
synthesized all primers used (5′-3′) (Table SII). All data were normalized
relative to GAPDH.
ST2825 and IL-1α treatment
As, to the best of our knowledge, ST2825 has never
been used in PDAC cell lines prior to the present study, the
concentration of ST2825 used herein was based on the overall
consideration of the manufacturer's certificate (MedChem Express)
and previous studies. In vitro, the working concentration of
ST2825 used in previous studies (28,29,31)
ranged from 2 to 30 µmol/l, and was mainly 5 and 10 µmol/l. In
order to determine the accurate IC50 value, a gradient of 0 to 80
µmol/l (0, 5, 10, 20, 40 and 80 µmol/l) was set. The PDAC cells was
treated with 5 or 10 µmol/l of ST2825 for 24 h prior to use in
further experiments, apart from the colony formation assay. In
vivo, previous study (28)
demonstrated that animals were orally administered ST2825 at doses
ranging from 50 to 200 mg/kg, or intraperitoneally at a dose of 25
mg/kg daily in 7 days. According to the information provided, in
the present study, the animals were intraperitoneally injected with
ST2825 on days 7, 10, 13, 16, 19 and 22 at a dose of 20 mg/kg. The
concentration of IL-1 α (10 ng/ml) used in PANC-1 cells were based
on a previous study (19).
Flow cytometry for apoptosis and cell
cycle analysis
Apoptosis and the cell cycle were detected using a
Digital BD LSR II flow cytometer (BD Biosciences). The apoptosis of
the cells was detected using an Annexin V-FITC/PI apoptosis
detection kit (BD Biosciences). The cells were collected and
suspended with binding buffer (BD Biosciences), then mixed with 5
µl fluorescein isothiocyanate-labeled Annexin V and 5 µl propidium
iodide to detect apoptosis. The cells in Q2 and Q4 were considered
as apoptotic cells. In the examination of cell cycle progression,
cells were collected and preserved in 75% alcohol for 24 h, and
then mixed with cell cycle liquid (BD Biosciences) to examine cell
cycle distribution.
3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide
(MTT) assay for measuring the IC50 values
The cells were seeded in 96-well plates
(4×103 cells per well). The attached cells were treated
with ST2825 (0, 5, 10, 20, 40 and 80 µmol/l) 24 h later. Following
48 h of incubation at 37°C, an MTT kit (C11019-2; Ruibo
Bio-Technology Co., Ltd.) was used to measure active cells, and the
absorbance was then detected at 570 nm using an iMark™ Microplate
Absorbance Reader (Bio-Rad Laboratories, Inc.).
Colony formation assay
The cells were cultured at 10,00 cells per 10 cm
dish. The cells were then treated with 1 µmol/l ST2825 for 2 weeks.
The colonies were fixed with formalin (MilliporeSigma) within 30
min and stained with Wright-Giemsa dye (Nanjing Jiancheng Yuehao
Technology Co., Ltd.) within 1 h at 37°C and counted using an
inverted phase contrast microscope (IX79, Olympus Corporation).
Transcriptome sequencing and
analysis
The negative control and 5 µmol/l ST2825-treated (48
h) PANC-1 cells were collected and prepared for sequencing. The
expression of mRNA between the two groups was compared and
analyzed, including genome mapping, differential expressed gene
screening and Kyoto Encyclopedia of Genes and Genomes (KEGG)
pathway enrichment analysis. The detailed procedures were performed
as previously described (32).
Histological and immunohistochemical
(IHC) analyses
The PANC-1 cell slides in six-well plates were fixed
in 4% formalin (MilliporeSigma) for 15 min and washed with PBS
twice. The cells were permeabilized with 0.5% Triton X-100 (Beijing
Solarbio Science & Technology Co., Ltd.) for 15 min and blocked
with 5% BSA solution (Beyotime Institute of Biotechnology) for 1 h.
The cells were then incubated with primary antibodies against
phosphorylated-p65 (1:200, ab86299, Abcam,) overnight at 4°C after
blocking non-specific binding. The cells were then washed with PBS
three times and stained with the anti-rabbit secondary antibody
(1:10,000, ab205718, Abcam) and counterstained with DAPI (1:1,000,
5 µg/ml, ab104139 Abcam) to visualize the nuclei. The slides were
observed using a fluorescence microscope (LEXT OLS4100, Olympus
Corporation) and imaged. The sections of tumors from the nude mice
used in the present study (as described below) were stained with
antibodies against MyD88 (ab133739, Abcam, 1:500, rabbit),
phosphorylated-p65 (ab86299, Abcam, 1:100, rabbit),
phosphorylated-AKT1 (4060, Cell Signaling Technology, Inc., 1:100,
rabbit) or p21 (ab54562, Abcam, 1:500, mouse). Briefly, the PDAC
tissues were excised 1 day after the collection of tumors. The
tissue sections were formalin-fixed, paraffin-embedded and
subjected to immune-staining using the streptavidin-peroxidase
technique. The sections were then subjected to heat with 0.01 mol/l
citrated buffer (Beyotime Institute of Biotechnology) and incubated
at 4°C overnight with the primary antibodies. The slides were
washed by Tris-buffered saline buffer (Beyotime Institute of
Biotechnology) and incubated for 30 min with the anti-rabbit
secondary antibody (1:10,000, ab205718, Abcam,) or anti-mouse
secondary antibody (1:10,000, ab205719, Abcam,) before being
counterstained with Meyer's hematoxylin for 5 min at 37°C (Beyotime
Institute of Biotechnology). The slides were observed using a
confocal microscope (FV3000, Olympus Corporation). The PDAC tissue
chip was stained with antibodies against myd88 from Outdo Biotech
Company, Shanghai, China (HPan-Ade170Sur-01). All procedures
performed in studies involving human participants were in
accordance with the ethical standards of the Shanghai Outdo Biotech
Company Ethics Committee and with the 1964 Declaration of Helsinki
and its later amendments or comparable ethical standards mentioned
in the product description. The tissues mentioned above were all
subjected to IHC staining using a previously described method
(19). Tissue sections were scanned
using Pannoramic MIDI (3D HISTECH). H-SCORE=∑ (PI xI)=(percentage
of cells of weak intensity ×1) + (percentage of cells of moderate
intensity ×2) + percentage of cells of strong intensity ×3). The
staining results were evaluated by two experienced pathologists in
a double-blinded manner.
ELISA and phosphorylation chip
assessment
PANC-1 cell nuclear fractions were isolated and the
binding activity of NF-κB with dsDNA was examined using an NF-κB
p65 Transcription Factor Assay kit (ab133112, Abcam) following the
manufacturer's protocol. The PANC-1 cells were treated with 0 and 5
µmol/l ST2825 for 48 h. The cell lysates were collected, added to
an NF-κB Phospho-Antibody Array (PCS248, Full Moon BioSystems), and
detected by Wayen Biotechnologies Inc. according to the
manufacturer's protocol. The analysis method was carried out as
previously described (33).
Chromatin immunoprecipitation (ChIP)
and co immunoprecipitation (Co-IP)
The PANC-1 cells were treated with 0 and 5 µmol/l
with ST2825 for 48 h. The MAGnify™ Chromatin Immunoprecipitation
System (cat. no. 492024, Thermo Fisher Scientific, Inc.) was used
to detect the p65-DNA interaction (p65-AKT1,
p65-CDKN1A and p65-CCND-1), as previously described
(34). Co-IP was performed using
the Pierce Crosslink Immunoprecipitation kit (cat. no. 26147,
Thermo Fisher Scientific, Inc.) as previously described (33). Flag-MyD88 and Myc-MyD88 plasmids
were designed and provided by Wuhan Genecreate Biological
Engineering Co. For transfection, the cells were inoculated in a
10-cm cell culture dish the day prior to transfection, with a cell
density of 70–80% at the time of transfection and a culture medium
of DMEM + 10% FBS. A total of 60 µl transfection reagent PEI
diluted to 800 µl medium, 20 µg (Flag-MyD88 and Myc-MyD88, per 10
µg) diluted to 800 µl medium was then added, and mixed and
incubated at room temperature for 20 min, and replenished to 3 ml.
The culture medium was removed from the dish, and 3 ml transfection
complex was prepared as in the previous step, and incubated at 37°C
for 5–6 h. The culture medium was removed and replaced with 2 ml
complete culture medium and incubated at 37°C for 48 h. Rat
anti-human Flag (cat. no. SAB4200071, MilliporeSigma, 2.5 µg/ml)
and mouse anti-human Myc (cat. no. M4439, MilliporeSigma, 2.5
µg/ml) antibodies were used to incubate with pre-treated proteins
at 4°CC overnight for immunoprecipitation.
Animal experiments
All mice were housed in accordance with the
guidelines of the Zhejiang Medical Experimental Animal Care
Commission. A total of 10 male (BALB/C, 5 weeks old) and 24 female
nude mice (BALB/C, 5 weeks old) were provided by the Key Laboratory
of Precision Diagnosis and Treatment for Hepatobiliary and
Pancreatic Tumor of Zhejiang Province. The mice were housed in
groups (5 or 6 mice per cage) in a specific-pathogen-free room with
filtered air and controlled temperature (24±2°C), relative humidity
(45–65%) and light/dark cycle (12/12 h), and water and food were
adequately provided. In the animal experiment with 10 male mice,
1×106 PANC-1 cells were injected into each mouse
subcutaneously in the axillary region on day 0, and the mice were
randomly divided into two groups (negative control and ST2825
group; 5 mice per group). ST2825 was intraperitoneally injected on
days 7, 10, 13, 16, 19 and 22 at a dose of 20 mg/kg. In the animal
experiment with 24 female mice, the animals were randomly divided
into four groups (negative control, ST2825, ST2825 after IL-1α and
ST2825 + AKT1 groups; 6 mice per group). A total of
1×106 PANC-1 cells were injected into each mouse
subcutaneously in the axillary region on day 0. The PANC-1 cells
used in the ST2825 after IL-1α group were pre-treated with 10 ng/ml
IL-1α for 1 week prior to injection; the PANC-1 cells used in the
ST2825 + AKT1 group were transfected with AKT1 recombinant plasmids
24 h prior to injection. ST2825 was intraperitoneally injected on
days 7, 10, 13, 16, 19 and 22 at a dose of 20 mg/kg. All mice were
observed for body weight and tumor volume within 4 weeks and were
then sacrificed by cervical dislocation; the condition of the
animals and their tumors were examined every 3 days, and the tumors
were weighed and collected for IHC analyses. All animal experiments
were approved by the Zhejiang Medical Experimental Animal Care
Commission. The humane endpoints were set as follows: i) A tumor
burden >10% of body weight, in an adult mouse; a tumor should
not exceed 20 mm in any dimension; ii) the tumor cannot not reach a
position that severely affects the normal functioning of the
animal, or the growth of the tumor causes animal pain; iii) the
weight loss of animals exceeded 20% of their normal body weight
(taking into account the proportion of tumors); iv) ulcers or
infections at tumor growth points; v) tumor metastasis to other
tissues and organs; vi) persistent spontaneous damage caused by
tumor growth; vii) tumor growth interfered with dietary activities.
In the present study, 1 mouse reached the fourth humane endpoint,
and the mouse was sacrificed immediately following the observation
of the ulcer and the data of the mice were excluded.
Gene expression profiling interactive
analysis (GEPIA)
GEPIA (http://gepia.cancer-pku.cn/) was used in the present
study to examine the expression of MyD88 in various types of
cancer. The correlation between the expression level of MyD88 with
that of RELA, RELB, REL, NFκB1, NFκB2, AKT1 and CDNK1A was
examined. In addition, the overall and disease-free survival plots
were analyzed depending on the expression level of MyD88 in
PDAC.
Statistical analysis
SPSS Statistics 19 software (SPSS, Inc.) was used
for all statistical analyses. Data are presented as the mean ± SD.
The t-test was used to analyze significant differences when only
two groups were being compared at the same time, and the Bonferroni
test was used for the multiple comparisons. For the Kaplan-Meier
analysis, the hazard ratio was calculated based on the Cox PH Model
with a 95% confidence interval, the log-rank test was used to
identify significant differences between the two groups. Pearson's
correlation analysis was used for the correlation analysis. The
Chi-squared test was used for categorical ordinal data in Table I. All statistical tests were
two-sided and P<0.05 was considered to indicate a statistically
significant difference.
 | Table I.Analysis of the clinical
characteristics of the 64 patients from the PDAC tissue chip. |
Table I.
Analysis of the clinical
characteristics of the 64 patients from the PDAC tissue chip.
| Characteristic | Low-MyD88 group
(n=32) | High-MyD88 group
(n=32) | P-value |
|---|
| Sex |
|
| 0.6107 |
|
Male | 20 | 18 |
|
|
Female | 12 | 14 |
|
| Age, years |
|
| 0.3513 |
|
>50 | 27 | 24 |
|
|
≤50 | 5 | 8 |
|
| Tumor size, cm |
|
| 0.0417a |
|
>4 | 9 | 15 |
|
| ≤4 | 23 | 17 |
|
| TMN stage |
|
| 0.1329 |
|
I–IIA | 20 | 14 |
|
|
IIB-IV | 12 | 18 |
|
| Tumor
differentiation |
|
| 0.6056 |
|
I–II | 21 | 19 |
|
|
III | 11 | 13 |
|
Results
MyD88 is highly expressed in PDAC and
indicates a worse clinical outcome; MyD88 inhibitor ST2825
suppresses tumor growth in vitro and in vivo
Based on GEPIA [and The Cancer Genome Atlas (TCGA)
database], the MyD88 gene expression profile across all tumor
samples and paired normal tissues revealed a significantly higher
MyD88 expression in PDAC (named as PAAD in Fig. S1) than in normal tissue (Figs. 1A and S1). To investigate the association
between MyD88 and the clinical outcomes of patients with PDAC, the
association between MyD88 expression and the survival rate in GEPIA
was analyzed. The overall survival rate was longer in the low MyD88
group, but did not significantly differ from that in the high MyD88
group (Fig. 1B, P=0.14), whereas
the disease-free survival rate in the low MyD88 group was
significantly longer (Fig. 1C,
P=0.0074). MyD88 protein expression was further detected using IHC
in a PDAC tissue chip containing 64 pairs of tumor and
paracancerous tissues with available clinical information for
patients, including survival time. MyD88 expression was
significantly higher in the tumor tissues compared with the normal
tissues (Fig. 1D and E). Depending
on the MyD88 IHC score in the tumor tissues, the 64 patients were
divided into two groups. Kaplan-Meier analysis demonstrated that
patients with a lower MyD88 expression (32 patients) exhibited a
significantly longer overall survival time than those with a higher
MyD88 expression (32 patients) (Fig.
1F, P=0.0168). Further analysis of the patient clinical
information revealed that the tumor size differed significantly
between the two groups, being larger in the group with a high MyD88
expression (Table I).
 | Figure 1.MyD88 is highly expressed in PDAC and
indicates a worse clinical outcome. The MyD88 inhibitor, ST2825,
suppressed tumor growth in vitro and in vivo. (A)
Gene expression of MyD88 in PDAC (n=179) and paracancerous tissues
(n=171) from TCGA database. (B and C) Overall survival time and
disease-free survival time of the patients in the high MyD88 group
(n=45) compared with those in the low MyD88 group (n=45) from TCGA
database. (B and C) Overall survive time and disease-free survive
time of the high MyD88 group (N=45) compared with the low MyD88
group (n=45) from the TCGA database. (D and E) Immunohistochemical
staining and H-score of MyD88 in PDAC and paracancerous tissues
from the tissue chip (n=64). (F) Overall survival time of the high
MyD88 group (n=32) compared with the low MyD88 group (n=32) from
the tissue chip. (G and H) Western blot analysis of the expression
of MyD88 in HPNE, CFPAC-1, PANC-1, AsPC-1 and BxPC-3 cell lines. (I
and J) MTT assay was used to determine the IC50 values and colony
formation assay was used to determine the growth of PANC-1 and
BxPC-3 cell lines treated with ST2825. (K) Image of each nude mouse
and its hypodermic tumor, body/tumor weight changes in the mice
treated with the negative control and ST2825. (L and M)
Immunohistochemical staining (×1 and ×20 magnification) and H-score
of MyD88 in tissues of the negative control and ST2825 groups. All
experiments were performed in triplicate (apart from the animal
experiments) and the data are presented as the mean ± SD. The
t-test was used for statistical analysis, *P<0.05, **P<0.01
and ***P<0.001; N.S., not significant. PDAC, pancreatic ductal
adenocarcinoma; MyD88, myeloid differentiation factor 88; IC50,
half maximal inhibitory concentration; PAAD, pancreatic
adenocarcinoma; NC, negative control; ST, ST2825. |
In a panel of four human PDAC cell lines and HPNE (a
type of immortalized human pancreatic epithelial cell line), the
PANC-1 and BxPC-3 cells expressed higher levels of MyD88 protein
than the HPNE cells, as determined using western blot analysis,
exhibiting a significant difference compared to the PANC-1 cells
(Fig. 1G and H). Therefore, the
PANC-1 and BxPC-3 cells were selected as the main experimental cell
line for use in further experiments. Following treatment with 5,
10, 20, 40 and 80 µmol/l ST2825, and MTT assay, the IC50 value of
ST2825 in the PANC-1 cells was found to be 12.96 µmol/l (Fig. 1I, top panel), and that in the BxPC-3
cells was found to be 18.39 µmol/l (Fig. 1J, top panel). To examine the effects
of ST2825 on cell proliferation, a colony formation assay were
performed following treatment of the PANC-1 and BxPC-3 cells with 1
µmol/l ST2825; the results revealed that ST2825 significantly
inhibited the growth of the PDAC cells (Fig. 1I and J, bottom panels). In addition,
to examine whether ST2825 inhibits PDAC tumor growth in
vivo, 10 BALB/C nude mice were injected hypodermically with
1×106 PANC-1 cells and randomly divided into two groups
(n=5 per group). The tumors were lobulated, hard in texture and
appeared grayish white or light yellowish white in color, with
unclear boundaries with their surrounding tissues. Tumors from the
mice in the ST2825 group treated with ST2825 were significantly
smaller than those in the negative control group, whereas no
significant difference was observed in mouse body weight between
the two groups (Fig. 1K).
Furthermore, IHC analysis of the tumor xenografts revealed that
MyD88 expression did not differ significantly between the two
groups (Fig. 1L and M).
The MyD88 inhibitor, ST2825, induces
the cell cycle arrest and apoptosis of PDAC cells
To examine the effects of ST2825 on PDAC cells,
alterations in the cell cycle and apoptosis of PANC-1 and BxPC-3
cells following treatment with 0, 5 and 10 µmol/l ST2825 were
examined using flow cytometry. The ratio of cells at the G2/M
phases in the three groups of PANC-1 cells (negative control, 5 and
10 µmol/l ST2825) was 12.99±1.45, 54.54±0.99 and 86.93±3.37%,
respectively (Fig. 2A and B), and
in the BxPC-3 cells this was 7.87±1.29, 12.10±3.51 and 54.63±6.07%,
respectively (Fig. 2C and D), which
revealed that ST2825 treatment significantly arrested the PANC-1
and BxPC-3 cells at the G2/M phase with the increasing ST2825
concentration. The ratio of apoptotic cells in the three groups of
PANC-1 cells (negative control, 5 and 10 µmol/l ST2825) was
4.97±1.20, 12.70±2.17 and 37.60±5.10%, respectively (Fig. 2E and F), and that in the BxPC-3
cells was 4.33±0.80, 7.37±0.51 and 11.47±0.76%, respectively
(Fig. 2G and H), which revealed
that ST2825 treatment induced significant PDAC cell death by
apoptosis in a concentration-dependent manner.
 | Figure 2.The MyD88 inhibitor, ST2825, induces
the cell cycle arrest and apoptosis of PDAC cells. (A and B) Flow
cytometric analysis of the cell cycle to determine the changes in
the ratio of PANC-1 cells in the G0/G1, S and G2/M phases following
treatment with ST2825. (C and D) Flow cytometric analysis of the
cell cycle to determine the changes of the ratio of BxPC-3 cells in
the G0/G1, S, and G2/M phases following treatment with ST2825. (E
and F) Flow cytometric analysis in the Annexin V-FITC/PI apoptosis
assay to determine the ratio of apoptotic PANC-1 cells following
treatment with ST2825. (G and H) Flow cytometric analysis in the
Annexin V-FITC/PI apoptosis assay to determine the ratio of
apoptotic BxPC-3 cells following treatment with ST2825. (I and J)
Western blot analysis of the three groups of PANC-1/BxPC-3 cells as
in panels A and C to determine the changes of expression of cell
cycle-related proteins. (K and L) Western blot analysis of the
three groups of PANC-1/BxPC-3 cells as in panels E and G to
determine the changes in the expression of apoptosis-related
proteins. All experiments were performed in triplicate and the data
are presented as the mean ± SD. The t-test with the Bonferroni test
was used for statistical analysis, *P<0.05, **P<0.01 and
***P<0.001; N.S., not significant. PDAC, pancreatic ductal
adenocarcinoma; MyD88, myeloid differentiation factor 88; NC,
negative control; ST, ST2825. |
To confirm the aforementioned results, the levels of
cell cycle arrest- and apoptosis-related proteins were examined
using western blot analysis. The levels of Cdk1 and Cyclin B1 and
other related protein levels (the Cdk1-Cyclin B1 complex), acting
as the key promoter of the G2-to-M phase progression, were
evaluated. Cdk1 expression was significantly decreased, while
Cyclin B1 expression was increased with the increasing ST2825
concentration. The expression of phosphorylated Chk1 was increased
while that of Chk1 was decreased, which phosphorylated cdk1 to
reduce the level of the Cdk1-Cyclin B1 complex (Figs. 2I and J, and S2). Furthermore, p21 expression was
significantly increased concomitant with a decrease in p53
expression, which may underlie the significant G2/M phase cell
cycle arrest upon Cdk1 decline (Fig. 2I
and J). Moreover, the levels of pro-apoptotic proteins (such as
cleaved PARP, cleaved caspase-3 and Bax) increased with the
increasing ST2825 concentration, as shown by western blot analysis,
whereas the level of Bcl-2 (a type of apoptosis inhibitory protein)
significantly decreased (Figs. 2K and
L, and S2).
ST2825 inhibits MyD88 dimerization to
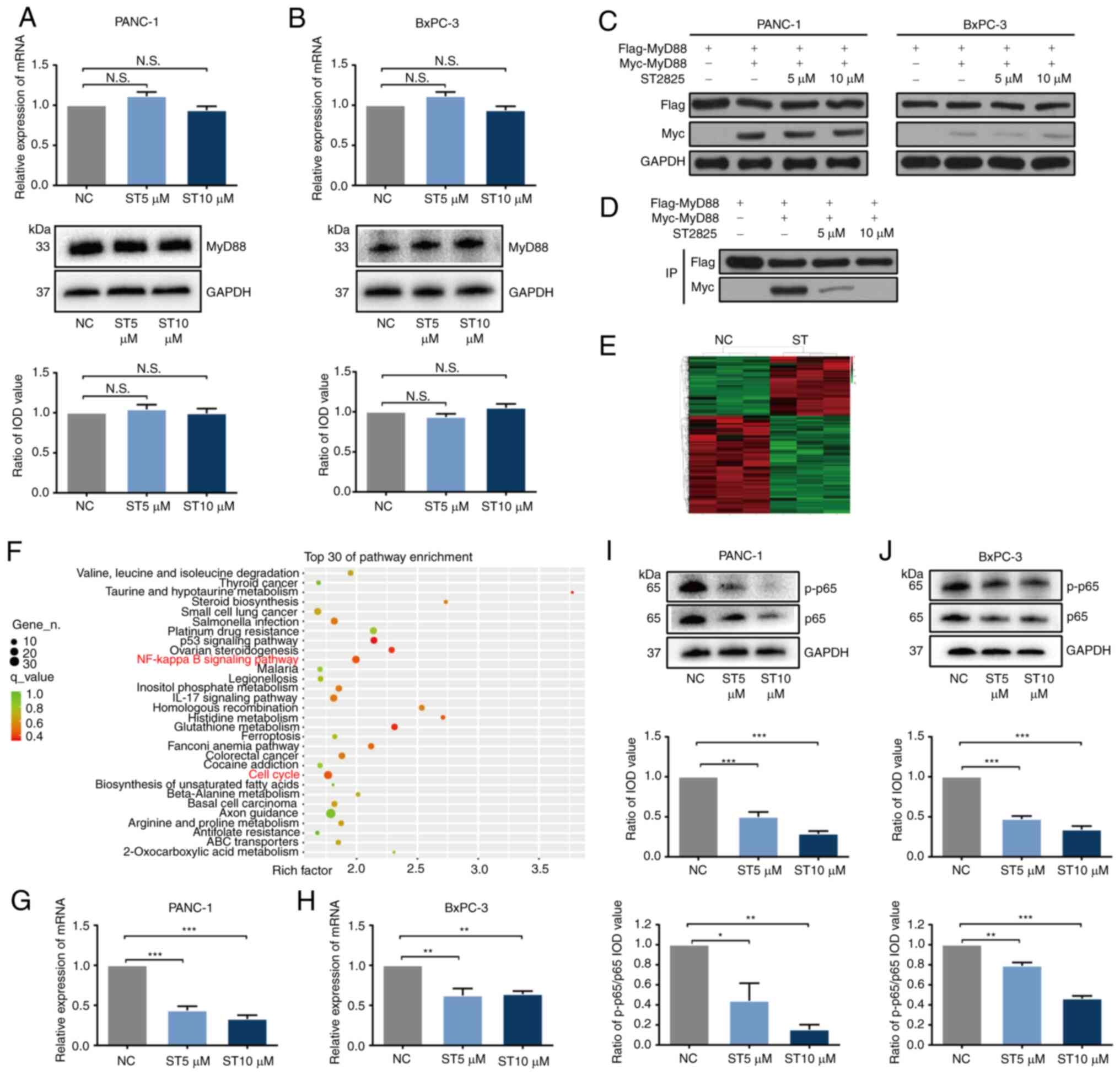
inactivate the NF-κB pathway in PDAC cells
To investigate the detailed underlying mechanisms of
the effects of ST2825 on MyD88, the PANC-1 and BxPC-3 cells were
treated with 0, 5 and 10 µmol/l ST2825. The expression of MyD88 was
not significantly altered, as shown by RT-qPCR and western blot
analysis (Fig. 3A and B); both the
mRNA and protein levels of MyD88 were not markedly altered
following treatment with ST2825. As MyD88 expression in PDAC cells
was unaltered by ST2825, to confirm whether ST2825 inhibits MyD88
by dedimerization, plasmids with Flag-MyD88 and Myc-MyD88 were
constructed, which were then transfected into PANC-1 and BxPC-3
cells. As the expression of Flag and Myc was higher in the PANC-1
cells than in the BxPC-3 cells (as determined using western blot
analysis), the PANC-1 cells were selected for use in further co-IP
experiments (Fig. 3C). The
Flag-complex was then collected by co-IP and the components were
detected by western blot analysis. ST2825 treatment decreased the
Myc-myd88 level in a concentration-dependent manner (Fig. 3D), indicating that ST2825 inhibited
MyD88 dimerization to inactivate MyD88 in PDAC cells, which was
consistent with the previously published mechanism in 293T cells
(28).
To reveal the molecular pathway involved in the
effects of ST2825 transcriptome sequencing of normal and
ST2825-treated PANC-1 cells was performed. The results of cluster
analysis revealed that ST2825 mRNA expression was altered in PANC-1
cells (Fig. 3E). KEGG enrichment
analysis indicated that the NF-κB signaling pathway was distinctly
inhibited in the ST2825-treated PDAC cells (Fig. 3F). In addition, the correlation
between MyD88 expression and members of the NF-κB signaling
pathway, including RELA (p65), RELB (RelB), REL (c-Rel), NFκB1
(p50), NFκB2 (p52), CHUK (IKKα) and IκBκB (IKKβ) was analyzed in
GEPIA; the MyD88 mRNA level significantly positively correlated
with all of these genes (Fig. S3).
To confirm these results, RELA (key gene of the NF-κB signaling
pathway) expression was examined using RT-qPCR and western blot
analysis in the PANC-1 and BxPC-3 cells. The RELA mRNA level was
significantly decreased (Fig. 3G and
H), and the p65 level was significantly decreased as was the
ratio of phosphorylated-p65/p65 proteins with the increasing ST2825
concentration (Fig. 3I and J),
indicating that ST2825 decreased p65 expression concomitantly with
p65 dephosphorylation.
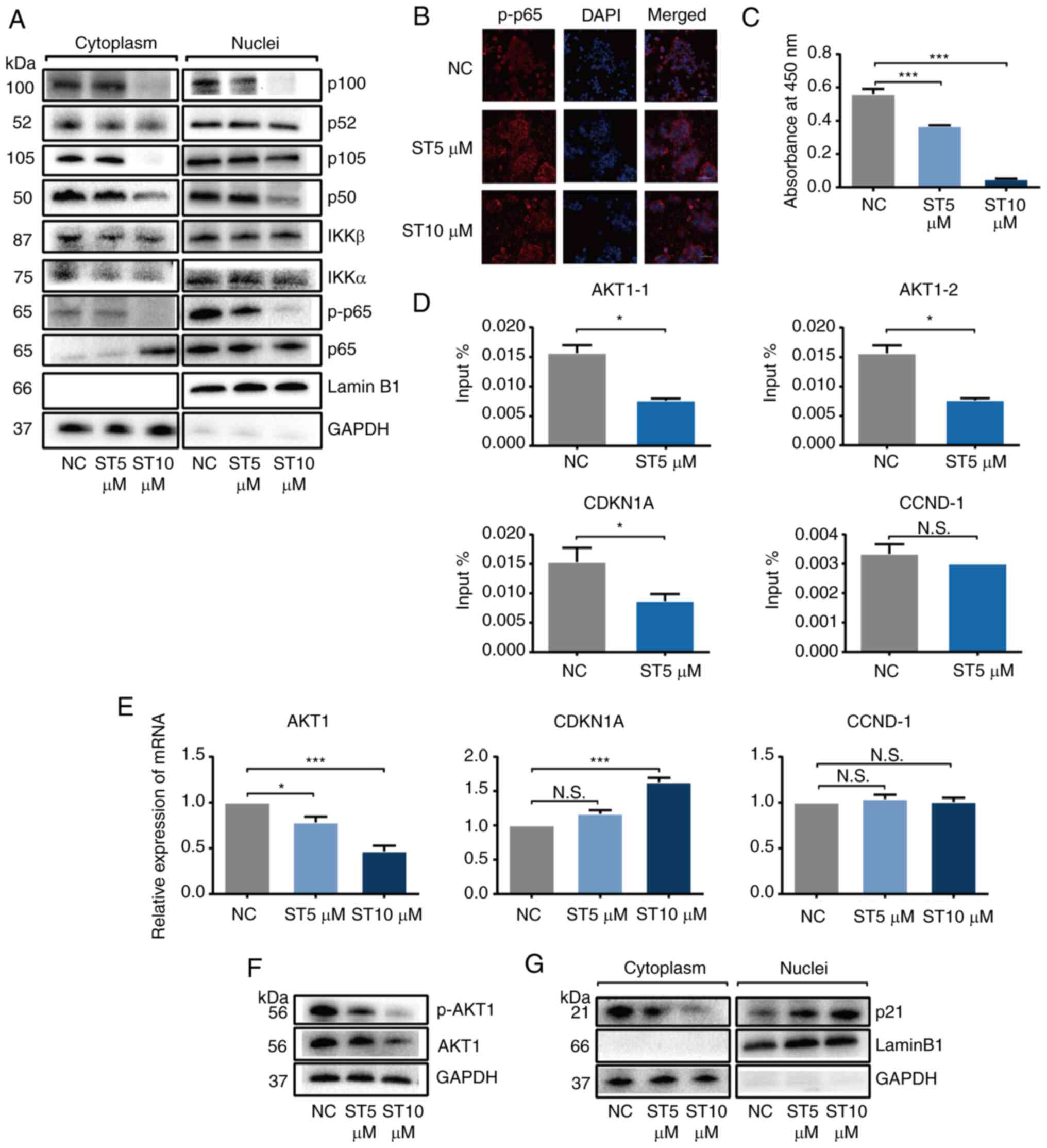
ST2825 inhibits AKT1 expression and
induces p21 overexpression by inhibiting NF-κB transcriptional
activity in PDAC cells
Based on the obtained data, it was suggested that
inhibition of the NF-κB pathway constituted the main mechanism of
action of ST2825. A series of experiments were then performed to
investigate whether ST2825 inhibits NF-κB (as a classic
transcription factor family) activity. To investigate whether
ST2825 inhibits the entry of activated NF-κB proteins into the
nucleus, the PANC-1 cells were treated with 0, 5 and 10 µmol/l
ST2825. Western blot analysis revealed that ST2825 decreased the
content of phosphorylated-p65 and other vital NF-κB proteins,
including p52, p50, IKKα and IKKβ in the nuclear extracts in a
concentration-dependent manner, indicating that ST2825 inhibited
NF-κB activation through both canonical and non-canonical pathways
(Fig. 4A). Confocal microscopy
imaging of the immunostaining results confirmed that ST2825
inhibited the intranuclear protein content of phosphorylated-p65 in
a concentration-dependent manner (Fig.
4B). Moreover, to clarify the binding ability of NF-κB protein
and its targeted DNA, the PANC-1 cells were treated with ST2825 and
the extracted nuclear protein was examined using an NF-κB/p65
Transcription Factor Assay kit. The results demonstrated that
ST2825 significantly decreased the binding ability of p65 and dsDNA
in a concentration-dependent manner (Fig. 4C).
Subsequently, the authors wished to determine the
vital target genes of NF-κB which underlie the effects of ST2825.
Based on the results of transcriptome sequencing mentioned above
(Table SIII) and a NF-κB
phosphorylation chip analysis (Table
SIV), AKT1 and CDKN1A (p21) were selected as key target gene
candidates for NF-κB. The ChIP analysis revealed that treatment
with 5 µmol/l ST2825 significantly inhibited p65 binding to AKT1
and CDKN1A, but did not alter the binding ability of p65 to CCND-1
(as a known p65 binding gene), suggesting that ST2825 selectively
affected downstream genes of NF-κB (Fig. 4D). Moreover in GEPIA, the MyD88 mRNA
level significantly and positively correlated with AKT1 and CDKN1A
(Fig. S3).
The present study then examined the expression of
candidate downstream NF-κB targeting genes using RT-qPCR and
western blot analysis following ST2825 treatment. The result of
RT-qPCR revealed a significantly decreased AKT1 and an increased
CDKN1A expression with the increasing ST2825 concentration;
however, the level of CCND-1 was not altered (Fig. 4E). The protein levels of AKT1 and
phosphorylated-AKT1 were decreased (Figs. 4F and S2) and that of p21 was increased as
aforementioned (Fig. 2I and J) with
the increasing ST2825 concentration. Furthermore, western blot
analysis revealed that the p21 content increased in the nuclear
extracts, whereas it decreased in the cytoplasmic extracts
following treatment with ST2825 in a concentration-dependent manner
(Fig. 4G). A previous study
reported that phosphorylated-AKT1 inhibited p21 from entering the
nucleus to induce cell cycle arrest (35). The results mentioned above indicated
that ST2825 promoted p21 entry into the nucleus to induce G2/M
phase cell cycle arrest followed by apoptosis by decreasing AKT1
expression concomitantly with p21 overexpression.
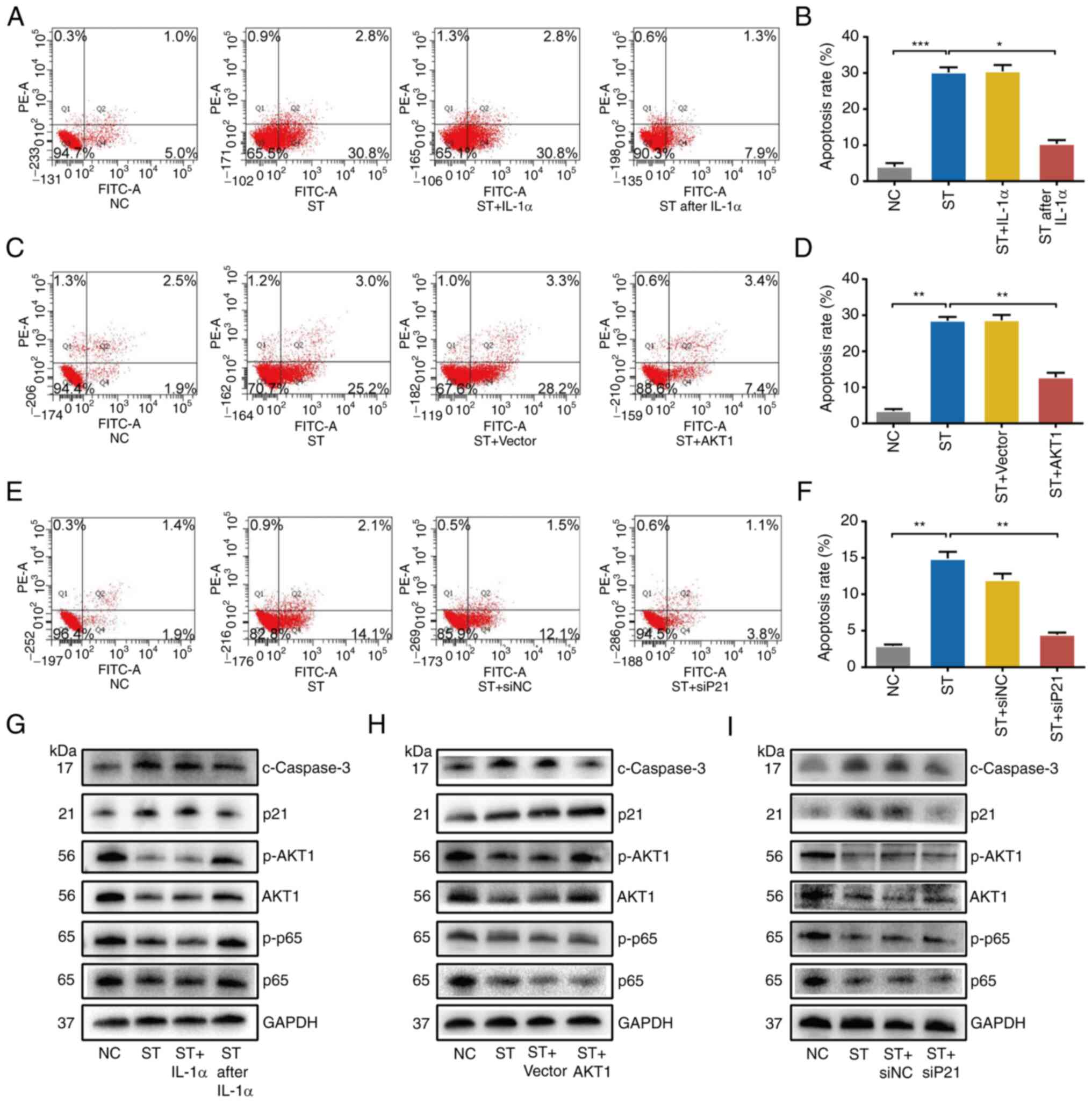
NF-κB activation, AKT1 overexpression
or p21 knockdown partially reverse the effects of ST2825 on PDAC
cells
To confirm the role of the NF-κB/AKT1/p21 pathway
for in the observed phenomena, a series of rescue experiments were
performed. The PANC-1 cells were treated with 10 µmol/l ST2825 with
a combination of IL-1α as a NF-κB activator. The results of Annexin
V-FITC/PI assay revealed that when the PDAC cells were concurrently
treated with a combination of IL-1α (10 ng/ml) and ST2825, cell
apoptosis was not lower than that following treatment with ST2825
alone. However, when IL-1α (10 ng/ml) was added 24 h prior to
ST2825 treatment, it significantly reversed apoptosis induced by
ST2825 (Fig. 5A and B). Western
blot analysis confirmed that the latter treatment reversed the
effects on p65, phosphorylated-p65, AKT1, phosphorylated-AKT1, p21
and cleaved Caspase-3 expression induced by ST2825 (Fig. 5G).
Furthermore, recombinant plasmid was used to
overexpress AKT1 (Fig. S4). This
was then transfected into PANC-1 cells with a combination of 10
µmol/l ST2825. The results of Annexin V-FITC/PI assay demonstrated
that the increasing ratio of apoptotic cells induced by ST2825 was
partially reversed by AKT1 overexpression (Fig. 5C and D). Western blot analysis
revealed that AKT1 overexpression also reversed the effects on
akt1, phosphorylated-akt1 and cleaved caspase-3, but not those on
p65, phosphorylated-p65 and p21 induced by ST2825 (Fig. 5H).
Subsequently, siRNA-p21-1, 2 and 3 were used to
knock down the expression of p21 in PANC-1 cells (Fig. S4). The mixture of siRNA-2 and 3 was
selected for use in further experiments. The expression p21 was
knocked down using siRNA with a combination of 5 µmol/l ST2825
treatment in PANC-1 cells. Flow cytometry revealed that the
ST2825-induced G2/M phase arrest and subsequent apoptosis were
partially reversed by p21 knockdown (Figs. 5E and F, and S4). Western blot analysis also revealed
that p21 knockdown reversed the effects of ST2825 on p21 and
cleaved Caspase-3, but not on p65, phosphorylated-p65, AKT1 and
phosphorylated-AKT1 (Fig. 5G).
Taken together, these data indicated that the NF-κB/AKT1/p21
pathway played a vital role in the effects of ST2825 on PDAC, and
AKT1 and p21 were the key downstream factors of NF-κB.
NF-κB activation or AKT1
overexpression antagonizes the inhibitory effects of ST2825 on PDAC
cells in vivo
To confirm the mechanisms of the ST2825-mediated
inhibition of PDAC cells in vivo, 24 mice were
hypodermically injected with 1×106 PANC-1 cells and
randomly assigned to four groups (negative control, ST2825, ST2825
after IL-1α, and ST2825 + AKT1; 6 mice per group) in an ectopic
xenograft nude mouse model. Recombinant lentivirus was used to
establish a stable akt1-overexpressing PANC-1 cell line (Fig. S4). Consistent with the results
obtained in vitro, the tumor weight was evidently smaller
following ST2825 treatment alone than following the combined
treatments (Fig. 6A). The weight of
the nude mouse did not differ significantly among all four groups
(Fig. 6B), whereas tumor weights in
the ST2825 group were significantly smaller than those in the other
three groups (Fig. 6C).
Furthermore, IHC analysis of the tumor xenografts
revealed that MyD88 expression did not differ significantly between
the four groups (Fig. 6D and E).
The decrease in the phosphorylated-p65 level in the ST2825 group
compared with the negative control groups was partially reversed in
the ST2825 after IL-1α group, but not in the ST2825 + AKT1 group
(Fig. 6F and G). The decrease in
the phosphorylated-AKT1 level in the ST2825 group compared with the
negative control groups was partially reversed in the other two
groups (Fig. 6H and I). The
increase in the p21 level in the ST2825 group compared with the
negative control groups was partially reversed in the ST2825 after
IL-1α group, but not in the ST2825 + AKT1 group (Fig. 6J and K). These results were all
consistent with those obtained in vitro.
Discussion
Previous studies have demonstrated that MyD88 plays
a divergent role in carcinogenesis, with previous studies reporting
that MyD88 contributes to the spontaneous tumorigenesis of skin and
colon cancer (36,37); however, MyD88 has also been shown to
be protective in virus-induced carcinogenesis (38). From the data obtained from TCGA, no
significant difference was observed in the expression of MyD88
between most tumors and their adjacent tissues, such as breast
cancer, lung cancer and prostate cancer; the expression of MyD88 in
a few tumors was significantly lower than that in adjacent tumors,
including diffuse large cell lymphoma, renal chromophobe cell tumor
and thymic carcinoma; there were also a few tumors in which the
expression of MyD88 was significantly higher than that near the
tumor, such as the topic of the present study, PDAC (Fig. S1). In the present study, it was
found that the inhibition of MyD88 by ST2825 induced the G2/M cell
cycle arrest and apoptosis of PDAC cells. The expression of MyD88
in a pancreatic cancer chip was detected in pancreatic cancer and
its adjacent tissues. The results revealed that the expression
level of MyD88 in PDAC tissues was significantly higher than that
in adjacent normal tissues, which was consistent with the
aforementioned research results. In addition, according to the
level of MyD88 expression in HCC, a previous study divided the
patients with HCC into a MyD88 high expression group and low
expression group (25). The results
revealed that the proportion of HCC in phase III–IV and the
recurrence rate following surgery in the MyD88 high expression
group was significantly higher; in addition, the overall survival
rate and disease-free survival rate at 1, 3 and 5 years after
surgery were much lower than that in the MyD88 low expression
group. This suggests that the expression of MyD88 may be a
potential prognostic indicator for patients with liver cancer
(25). The present study found that
in pancreatic cancer, patients with a high expression of MyD88 had
a larger tumor volume and a shorter survival rate, suggesting that
MyD88 may also be a potential prognostic indicator for patients
with pancreatic cancer.
Previous research has indicated that ST2825, a
selective inhibitor of MyD88, significantly inhibits the
proliferation of liver cancer cells and promotes the apoptosis of
HepG-2 in a concentration-dependent manner (29). Loiarro et al (28) found that ST2825 suppressed the
inflammatory reaction via the MyD88/IRAK/NF-κB signaling pathway.
However, the specific mechanisms of the effects of ST2825 on MyD88
in tumors were not verified. In the present study, it was found
that in pancreatic cancer, ST2825 inhibited the function of MyD88
by inhibiting the dimerization of MyD88, which is consistent with
previous extra-tumor research results (28).
The MyD88 signaling pathway, most often associated
with IL-1R and TLR, regulates the pro-inflammatory feedback
mechanism, participates in the tissue repair response and activates
oncogenes. In tumors, the effects of MyD88 on tumor progression are
mediated through TLR/IL-1R-dependent and -independent mechanisms.
As previously demonstrated, in a TLR/IL-1R-dependent manner, MyD88
is activated following stimulation by lipopolysaccharide and IL-1,
and plays a role in the activation of NF-κB, p38 or other pathways
through changes in downstream tumor regulatory factors, such as
MMP7, COX2, IL-6 and TNF-a (37,39).
It has also been shown that, in a TLR/IL-1R-independent manner, in
the process of RAS-mediated tumorigenesis, the MyD88 mutation
interacted with ERK rather than IRAK, leading to the loss of its
ability to regulate cell transformation (40).
Previous studies have indicated that MyD88, as a
common intermediate messenger between NF-κB and its inducers, may
constitute a novel therapeutic target for PDAC (18–20).
Consistent with this finding, the present study demonstrated that
ST2825 effectively inhibited PDAC development via the
NF-κB/AKT1/p21 pathway in a TLR/IL-1R-dependent manner. The results
indicated that the inhibition of the NF-κB pathway constituted the
main mechanism of ST2825, as the process of NF-κB acting as a
classic transcription factor family (including phosphorylation,
nuclear import, specific DNA binding and downstream gene
expression) was affected by ST2825.
As a classical transcription factor, NF-κB can play
a role in tumors by regulating a variety of downstream targeted
genes. A previous study found that ST2825 inhibited the
NF-κB-mediated transcription and translation of cyclin-D1 in HCC
(29). In the present study,
according to the results of transcriptome sequencing and a NF-κB
phosphorylation chip, AKT1 and cyclin-related genes were used as
the main potential target factors. The results revealed that ST2825
induced severe G2/M cell cycle arrest; however, its key factor, the
Cdk1-Cyclin B1 complex, did not exhibit any corresponding change,
although the protein level of Cdk1 was reduced and the ratio of
p-Cdk1/Cdk1 was slightly increased, which led to the decrease in
the Cdk1-Cyclin B1 complex; the decreased level of the Cdk1-Cyclin
B1 complex was not solely responsible however, for the range of
G2/M cell cycle arrest. Thus, it was hypothesized that other
factors were involved and played a decisive role. It was found that
following ST2825 treatment, p21, which was known as an inhibitor of
cell cycle progression that acting in the nucleus (41,42),
was increased significantly. It is known that p21 is a classic
downstream factor of p53, which can block the G2 to M phase
transition; however, in the present study, the elevation of p21 was
accompanied with a decrease in p53 expression, which indicated that
p21 expression was increased by ST2825 in a p53-independent manner.
According to the results of transcriptome sequencing, the mRNA
level of p53 was not altered following treatment with ST2825
(Table SIII); it was thus
speculated that ST2825 may affect the protein stability of p53,
although the expression of MDM2 (Table
SIII), which is known as a pro-degradation factor of p53 was
also not altered. Thus, the detailed mechanism responsible for the
decrease in p53 expression remains unclear, and this warrants
further investigation. In addition, p21 expression increased
following ST2825 treatment in the nuclear extracts, but decreased
in the cytoplasmic extracts with a decrease in AKT1 expression.
Together, these data indicated that ST2825 inhibited NF-κB
transcriptional activity to increase p21 expression and promote its
nuclear localization through the decreased expression of AKT1,
which resulted in Cdk1-Cyclin B1 complex inhibition, thereby
inducing G2/M cell cycle arrest and subsequent apoptosis.
The results of subsequent rescue experiments
revealed that the addition of IL-1α, the overexpression of AKT1 or
the knockdown of p21 partially inhibited the pro-apoptotic effects
of ST2825. As regards IL-1α treatment, we found that the addition
of 10 ng/ml IL-1α after ST2825 or simultaneously with ST2825 (5
µmol/l) did not reverse the effects of ST2825; however, when IL-1α
was added prior to ST2825 treatment, it partially reversed the
pro-apoptotic effects of ST2825, indicating that IL-1α was a
upstream factor of MyD88 in the NF-κB pathway (Fig. S5). From these results, the authors
also wished to determine whether the addition of IL-1α with ST2825
by intraperitoneally injection would also not be effective in in
vivo experiments. For this purpose, the cells in the ‘ST after
IL-1α’ group were incubated with 10 ng/ml IL-1α for 1 week before
the injection to simulate a NF-κB activated model. Before the in
vivo experiment, the in vitro preliminary experiment
revealed that NF-κB was effectively activated by treatment with 10
ng/ml IL-1α for 1 week (Fig. S5).
The model was successful. Furthermore, the results of western blot
analysis demonstrated that IL-1α increased the level of
phosphorylated p65 and phosphorylated akt1, and reduced the
expression of p21. However, the overexpression of AKT1 did not
affect the phosphorylation level of p65 and the expression of p21.
In addition, the knockdown of p21 did not affect the activation
level of p65 and akt1. These results clarified the upstream and
downstream relationship of these factors.
In summary, pro-inflammatory factors, such as IL-1α
activate the transcriptional activity of NF-κB through MyD88 in
PDAC, thus regulating downstream factors (AKT1 and p21) to play a
role in promoting tumor growth. ST2825 can block this process by
inhibiting the dimerization of MyD88.
However, the present study had some limitations. As
ST2825 is not an FDA-approved therapeutic drug, the effects of
MyD88 inhibitor could not be confirmed in clinical trials. The
detailed mechanisms responsible for the decrease in p53 expression
induced by ST2825 are unclear and further investigations are thus
required for clarifications. In addition, more practical animal
models are warranted to verify the experimental results in
vitro; thus, further studies are warranted.
In conclusion, the present study demonstrates that
ST2825 induces the G2/M cell cycle arrest and apoptosis of PDAC
cells via the MyD88/NF-κB/AKT1/p21 pathway. MyD88 may serve as a
potential therapeutic target in PDAC. ST2825 may serve as a novel
agent for the targeted therapy of PDAC in the future.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural Science
Foundation of Zhejiang Province (grant no. LY22H160017) and the
Medical Science and Technology Foundation of Zhejiang Province
(grant no. 2020KY751).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SL, TH, BZ, QZ and SY were involved in the
conception and design of the study. SL and YZ were involved in the
development of study methodology. SL TH and YZ were involved in the
acquisition of data. SL and YZ were involved in the analysis and
interpretation of data. SL, BZ and QZ were involved in the writing,
reviewing and/or revision of the manuscript. BZ, QZ and SY were
involved in the administrative, technical, or material support
aspects of the study. YS supervised the study. All authors have
read and approved the final manuscript. SL, TH and SY confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
All animal experiments were approved by the Zhejiang
Medical Experimental Animal Care Commission and the Ethics
Committee of the Second Affiliation Hospital of Zhejiang University
School of Medicine. All procedures performed of the tissue ship
involving human participants were approved by the Shanghai Qutdo
Biotech Company Ethics Committee.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
PDAC
|
pancreatic ductal adenocarcinoma
|
|
TLR
|
Toll-like receptor
|
|
PanIN
|
human pancreatic intraepithelial
neoplasia
|
|
RT-qPCR
|
reverse transcription-quantitative
PCR
|
|
IL
|
interleukin
|
|
MyD88
|
myeloid differentiation factor 88
|
|
ChIP
|
chromatin immunoprecipitation
|
|
Co-IP
|
co-immunoprecipitation
|
|
GEPIA
|
gene expression profiling interactive
analysis
|
|
TCGA
|
The Cancer Genome Atlas
|
|
IC50
|
half-maximal inhibitory
concentration
|
|
IHC
|
immunohistochemical
|
References
|
1
|
Saluja A and Maitra A: Pancreatitis and
pancreatic cancer. Gastroenterology. 156:1937–1940. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Neoptolemos JP, Kleeff J, Michl P,
Costello E, Greenhalf W and Palmer DH: Therapeutic developments in
pancreatic cancer: Current and future perspectives. Nat Rev
Gastroenterol Hepatol. 15:333–348. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mizrahi JD, Surana R, Valle JW and Shroff
RT: Pancreatic cancer. Lancet. 395:2008–2020. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Siegel RL, Miller KD, Fuchs HE and Jemal
A: Cancer statistics, 2022. CA Cancer J Clin. 72:7–33. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Matsuoka T and Yashiro M: Molecular
targets for the treatment of pancreatic cancer: Clinical and
experimental studies. World J Gastroenterol. 22:776–789. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rozenblum E, Schutte M, Goggins M, Hahn
SA, Panzer S, Zahurak M, Goodman SN, Sohn TA, Hruban RH, Yeoet CJ,
et al: Tumor-suppressive pathways in pancreatic carcinoma. Cancer
Res. 57:1731–1734. 1997.PubMed/NCBI
|
|
7
|
Yashiro M, Carethers JM, Laghi L, Saito K,
Slezak P, Jaramillo E, Rubio C, Koizumi K, Hirakawa K and Boland
CR: Genetic pathways in the evolution of morphologically distinct
colorectal neoplasms. Cancer Res. 61:2676–2683. 2001.PubMed/NCBI
|
|
8
|
Moskaluk CA, Hruban RH and Kern SE: p16
and K-ras gene mutations in the intraductal precursors of human
pancreatic adenocarcinoma. Cancer Res. 57:2140–2143.
1997.PubMed/NCBI
|
|
9
|
Hruban RH, van Mansfeld AD, Offerhaus GJ,
van Weering DH, Allison DC, Goodman SN, Kensler TW, Bose KK,
Cameron JL and Bos JL: K-ras oncogene activation in adenocarcinoma
of the human pancreas. A study of 82 carcinomas using a combination
of mutant-enriched polymerase chain reaction analysis and
allele-specific oligonucleotide hybridization. Am J Pathol.
143:545–554. 1993.PubMed/NCBI
|
|
10
|
Kosmidis C, Sapalidis K, Kotidis E,
Mixalopoulos N, Zarogoulidis P, Tsavlis D, Baka S, Man YG and
Kanellos J: Pancreatic cancer from bench to bedside: Molecular
pathways and treatment options. Ann Transl Med. 4:1652016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bryant KL, Mancias JD, Kimmelman AC and
Der CJ: KRAS: Feeding pancreatic cancer proliferation. Trends
Biochem Sci. 39:91–100. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Prabhu L, Mundade R, Korc M, Loehrer PJ
and Lu T: Critical role of NF-κB in pancreatic cancer. Oncotarget.
5:10969–10975. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang W, Abbruzzese JL, Evans DB, Larry L,
Cleary KR and Chiao PJ: The nuclear factor-kappa B RelA
transcription factor is constitutively activated in human
pancreatic adenocarcinoma cells. Clin Cancer Res. 5:119–127.
1999.PubMed/NCBI
|
|
14
|
Fujioka S, Sclabas GM, Schmidt C,
Frederick WA, Dong QG, Abbruzzese JL, Evans DB, Baker C and Chiao
PJ: Function of nuclear factor kappaB in pancreatic cancer
metastasis. Clin Cancer Res. 9:346–354. 2003.PubMed/NCBI
|
|
15
|
Mo W, Chen J, Patel A, Zhang L, Chau V, Li
YJ, Cho WS, Lim K, Xu J, Lazar AJ, et al: CXCR4/CXCL12 mediate
autocrine cell-cycle progression in NF1-associated malignant
peripheral nerve sheath tumors. Cell. 152:1077–1090. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Oeckinghaus A and Ghosh S: The NF-kappaB
family of transcription factors and its regulation. Cold Spring
Harb Perspect Biol. 4:a0000342009.PubMed/NCBI
|
|
17
|
Karandish F and Mallik S: Biomarkers and
targeted therapy in pancreatic cancer. Biomarkers Cancer. 8 (Suppl
1):S27–S35. 2016.PubMed/NCBI
|
|
18
|
Takahashi H, Funahashi H, Sawai H, Matsuo
Y, Yamamoto M, Okada Y, Takeyama H and Manabe T: Synthetic serine
protease inhibitor, gabexate mesilate, prevents nuclear
factor-kappaB activation and increases TNF-alpha-mediated apoptosis
in human pancreatic cancer cells. Dig Dis Sci. 52:2646–2652. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhuang Z, Ju HQ, Aguilar M, Gocho T, Li H,
Iida T, Lee H, Fan X, Zhou H, Ling J, et al: IL1 receptor
antagonist inhibits pancreatic cancer growth by abrogating NF-κB
activation. Clin Cancer Res. 22:1432–1444. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sun YL, Wu CS, Ma JX, Yang Y, Man XH, Wu
HY and Li SD: Toll-like receptor 4 promotes angiogenesis in
pancreatic cancer via PI3K/AKT signaling. Exp Cell Res.
347:274–282. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Niu J, Li Z, Peng B and Chiao PJ:
Identification of an autoregulatory feedback pathway involving
interleukin-1a in induction of constitutive NF-kappaB activation in
pancreatic cancer cells. J Biol Chem. 279:16452–16462. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ling JH, Kang Y, Zhao RY, Xia QH, Lee DF,
Chang Z, Li J, Peng BL, Fleming JB, Wang HM, et al:
KrasG12D-Induced IKK2/β/NF-κB activation by IL-1α and p62
feedforward loops is required for development of pancreatic ductal
adenocarcinoma. Cancer Cell. 21:105–120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang DX, Li L, Jiang HM, Knolhoff BL,
Lockhart AC, Wang-Gillam A, DeNardo DG, Ruzinova MB and Lim KH:
Constitutive IRAK4 activation underlies poor prognosis and
chemoresistance in pancreatic ductal adenocarcinoma. Clin Cancer
Res. 23:1748–1759. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Salcedo R, Cataisson C, Hasan U, Yuspa SH
and Trinchieri G: MyD88 and its divergent toll in carcinogenesis.
Trends Immunol. 34:232–238. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liang B, Chen R, Wang T, Cao L, Liu Y, Yin
F, Zhu M, Fan X, Liang Y, Zhang L, et al: Myeloid differentiation
Factor 88 promotes growth and metastasis of human hepatocellular
carcinoma. Clin Cancer Res. 19:2905–2915. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Song B, Zhang C, Li G, Jin G and Liu C:
MiR-940 inhibited pancreatic ductal adenocarcinoma growth by
targeting MyD88. Cell Physiol Biochem. 35:1167–1177. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhu XX, Burfeind KG, Michaelis KA, Braun
TP, Olson B, Pelz KR, Morgan TK and Marks DL: MyD88 signalling is
critical in the development of pancreatic cancer cachexia. J
Cachexia Sarcopeni. 10:378–390. 2019. View Article : Google Scholar
|
|
28
|
Loiarro M, Capolunghi F, Fantò N, Gallo G,
Campo S, Arseni B, Carsetti R, Carminati P, Santis RD, Ruggiero V,
et al: Pivotal advance: Inhibition of MyD88 dimerization and
recruitment of IRAK1 and IRAK4 by a novel peptidomimetic compound.
J Leukoc Biol. 82:801–810. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Deng Y, Sun J and Zhang LD: Effect of
ST2825 on the proliferation and apoptosis of human hepatocellular
carcinoma cells. Genet Mol Res. 15:150168262016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Van Tassell BW, Seropian IM, Toldo S,
Salloum FN, Smithson L, Varma A, Hoke NN, Gelwix C, Chau V and
Abbate A: Pharmacologic inhibition of myeloid differentiation
factor 88 (MyD88) prevents left ventricular dilation and
hypertrophy after experimental acute myocardial infarction in the
mouse. J Cardiovasc Pharmacol. 55:385–390. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jiang S, Li F, Li X, Wang L, Zhang L, Lu
C, Zheng L and Yan M: Transcriptome analysis of PK-15 cells in
innate immune response to porcine deltacoronavirus infection. PLoS
One. 14:e02231772019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rinaldi L, Delle DR, Catalanotti B,
Torres-Quesada O, Enzler F, Moraca F, Nisticò R, Chiuso F, Piccinin
S, Bachmann V, et al: Feedback inhibition of cAMP effector
signaling by a chaperone-assisted ubiquitin system. Nat Commun.
10:25722019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu D, Li L, Zhang XX, Wan DY, Xi BX, Hu
Z, Ding WC, Zhu D, Wang XL, Wang W, et al: SIX1 promotes tumor
lymphangiogenesis by coordinating TGFβ signals that increase
expression of VEGF-C. Cancer Res. 74:5597–5607. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ignacio RMC, Dong YL, Kabir SM, Choi H,
Lee E, Wilson AJ, Beeghly-Fadiel A, Whalen MM and Son D: CXCR2 is a
negative regulator of p21 in p53-dependent and independent manner
via Akt-mediated Mdm2 in ovarian cancer. Oncotarget. 9:9751–9765.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mittal D, Saccheri F, Vénéreau E, Pusterla
T, Bianchi ME and Rescigno M: TLR4-mediated skin carcinogenesis is
dependent on immune and radioresistant cells. EMBO J. 29:2242–2252.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rakoff-Nahoum S and Medzhitov R:
Regulation of spontaneous intestinal tumorigenesis through the
adaptor protein MyD88. Science. 317:124–127. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fathallah I, Parroche P, Gruffat H,
Zannetti C, Johansson H, Yue J, Manet E, Tommasino M, Sylla BS and
Hasan UA: EBV latent membrane protein 1 is a negative regulator of
TLR9. J Immunol. 185:6439–6447. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Naugler WE, Sakurai T, Kim S, Maeda S, Kim
K, Elsharkawy AM and Karin M: Gender disparity in liver cancer due
to sex differences in MyD88-dependent IL-6 production. Science.
317:121–124. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Coste I, Le Corf K, Kfoury A, Hmitou I,
Druillennec S, Hainaut P, Eychene A, Lebecque S and Renno T: Dual
function of MyD88 in RAS signaling and inflammation, leading to
mouse and human cell transformation. J Clin Invest. 120:3663–3667.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Watanabe S, Yamaguchi S, Fujii N, Eguchi
N, Katsuta H, Sugishima S, Iwasaka T and Kaku T: Nuclear
Co-expression of p21 and p27 induced effective cell-cycle arrest in
T24 cells treated with BCG. Cytotechnology. 71:219–229. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chang XT, Chai ZB, Zou JR, Wang HX, Wang
Y, Zheng YB, Wu H and Liu CY: PADI3 induces cell cycle arrest via
the Sirt2/AKT/p21 pathway and acts as a tumor suppressor gene in
colon cancer. Cancer Biol Med. 16:729–742. 2019. View Article : Google Scholar : PubMed/NCBI
|