Introduction
Endometrial cancer is the most common type of cancer
of the female genital tract in developed countries, including
Taiwan (1). In the United States,
endometrial cancer accounts for 6.9% of cancer diagnoses in women,
with a predicted 61,180 new cases and 12,160 fatalities in 2019
(2). Different endometrial cancer
histological subtypes and molecular traits have been reported. Type
I endometrial cancer is associated with unopposed estrogen
stimulation, consists of low-grade cells that are more prevalent
and has an improved prognosis than type II endometrial cancer,
which is not driven by estrogen and is comprised of high-grade
cells (3). Unopposed estrogen
therapy, early menarche, late menopause, tamoxifen therapy,
infertility and chronic anovulation are risk factors connected to
excessive unopposed exposure of the endometrium to estrogen
(4). In addition, the metabolic
combination of obesity, hypertension and diabetes, as well as other
metabolic illnesses, are strongly associated with endometrial
cancer (5). Endometrioid
endometrial cancer has a well-established link to obesity, with
relative risks of ~1.5 for overweight individuals, 2.5 for
individuals with class 1 obesity [body mass index (BMI), 30.0–34.9
kg/m2], 4.5 for individuals with class 2 obesity (BMI,
35.0–39.9 kg/m2) and 7.1 for individuals with class 3
obesity (BMI, ≥40.0 kg/m2) (3,6).
Fumarate hydratase (FH), also known as fumarase, is
a tricarboxylic acid (TCA) cycle enzyme that catalyzes the
reversible hydration of fumarate to malate (7). The TCA cycle is the ultimate
convergent pathway for the oxidation of lipids, carbohydrates and
amino acids, in a series of metabolic processes that take place
inside the mitochondria (8).
According to a previous report, genetic mutations in the TCA
cycle-related mitochondrial enzymes, including isocitrate
dehydrogenase, succinate dehydrogenase and FH all cause tumor
growth, suggesting that metabolic dysregulation can operate as a
cancer driver in addition to being a result of oncogenic
transformation (9). FH has dual
localization in the cytosol or mitochondria, depending on the
N-terminus peptide sequence (10).
A study has reported germline heterozygous mutations
of FH in patients with multiple cutaneous and uterine
leiomyomatosis (MCUL) and hereditary leiomyomatosis and renal cell
cancer with or without renal cancer (HLRCC), demonstrating the
allelic relationship between MCUL and HLRCC (11). In another study, 86% of FH-negative
tumors determined by immunohistochemistry had FH mutations, half of
which being germline mutations (12). Other than its mutational status in
uterine leiomyosarcoma and uterine fibroids, the role of FH in
endometrial cancer is mostly unclear.
In the present study, the role of FH in endometrial
cancer was explored. It was demonstrated that FH functions as a
tumor suppressor, with the potential to be developed as a
prognostic biomarker and therapeutic target.
Materials and methods
Patient samples
Endometrial cancer tissues were obtained from
patients (n=62, aged 26-82 years old) who had undergone surgical
treatment at the Department of Surgery, Kaohsiung Medical
University Hospital [Kaohsiung, Taiwan]. All participants in this
study were recruited between March 2025 and March 2017. Ethical
approval [IRB No.: KMUHIRB-E (1)-20150026] was obtained from the Ethics
Committee of Kaohsiung Medical University Hospital. Informed
patient consent was waived by the Institutional Review Board due to
the retrospective nature of the study.
Immunohistochemistry
All tissues were procured from formalin-fixed and
paraffin-embedded endometrial tissue blocks. Immunohistochemical
(IHC) staining for FH in endometrial tissues was performed using
the Bond-Max system (Leica Microsystems GmbH). Sections were
deparaffinized using Bond Dewax Solution (Leica Microsystems GmbH)
and rehydrated using graded alcohol. Heat-induced antigen retrieval
was achieved using Bond Epitope Retrieval Solution 1 (Leica
Microsystems GmbH) for 20 min at 98°C. After washing steps,
peroxidase blocking was carried out for 10 min using Bond Polymer
(Leica Microsystems GmbH). Tissues were again washed and then
incubated with the primary antibody, FH (cat. no. GTX110128;
1:100), for 30 min at room temperature. Post-primary IgG linker
reagent was applied for 8 min, and the slides were incubated with
polymeric horseradish peroxidase IgG reagent for 8 min to localize
the primary antibodies. Diaminobenzidine (DAB) was used as the
substrate to detect antigen-antibody binding. Then, hematoxylin was
used to counterstain nuclei for 5 min at room temperature. Images
of immunohistochemically stained sections were captured using Nikon
Eclipse E600 fluorescence microscope (Nikon Corporation). Relative
expression of FH in the endometrial cancer specimens was quantified
by two pathologists independently. For the endometrial cancer
samples, each specimen was assigned to one of four groups based on
the percentage of positively stained normal and tumor cells: 0
(0–4%), 1 (5–24%), 2 (25–49%), 3 (50–74%) or 4 (75–100%). In
addition, the immunostaining intensity was graded as: 0 (negative),
1 (weak), 2 (moderate) or 3 (strong), with the total score
calculated by multiplying the percentage of positively stained
cells by the graded intensity of staining for every sample.
Patients with a score <4.50 were categorized as the low FH
expression group and those with a score ≥4.50 were categorized as
the high FH expression group.
Cell culture
Ishikawa, RL95-2, HEC1A, AN3CA and KLE human
endometrial cancer cell lines were obtained from the Bioresource
Collection and Research Center (Hsinchu, Taiwan) and cultured in
RPMI 1640 (Gibco; Thermo Fisher Scientific, Inc.) and Dulbecco's
modified Eagle's medium (Gibco; Thermo Fisher Scientific, Inc.).
All cell lines were incubated in a humidified incubator at 37°C and
5% CO2. All culture media contained 10% fetal bovine
serum (FBS; Biological Industries; Sartorius AG), 1% penicillin G
and streptomycin.
Virus transfection for FH knockdown
and overexpression
To knockdown FH expression in endometrial cancer
cells, lentivirus carrying a pLKO.1_puro lentiviral vector (from
National RNAi Core Facility, Academia Sinica, Taipei, Taiwan) that
expressed double-stranded short hairpin (sh)RNA oligonucleotides
targeting the sequences of human FH (three clones) were used: i)
Clone 2: ID TRCN0000310398, target sequence: CAACGATCATGTTAATAAA,
shRNA sequence:
GCCCAACGATCATGTTAATAAACTCGAGTTTATTAACATGATCGTTGGGTTTTTG; ii) clone
5: ID TRCN0000052465, target sequence: CCCAACGATCATGTTAATAAA, shRNA
sequence:
CCGGCCCAACGATCATGTTAATAAACTCGAGTTTATTAACATGATCGTTGGGTTTTTG; and
iii) clone 6 ID: TRCN0000299140, target sequence:
GTGGTTATGTTCAACAAGTAA, shRNA sequence:
CCGGGTGGTTATGTTCAACAAGTAACTCGAGTTACTTGTTGAACATAACCACTTTTTG
(National RNAi Core Facility, Academia Sinica, Taipei, Taiwan).
A pLKO.1_puro lentiviral vector expressing shRNA
targeting firefly luciferase, unrelated to the human genome
sequence, was used as a negative control clone ID: TRCN0000052466,
target sequence: GTGGTTATGTTCAACAAGTAA, shRNA sequence:
GGGTGGTTATGTTCAACAAGTAACTCGAGTTACTTGTTGAACATAACCACTTTTTG (from
National RNAi Core Facility, Academia Sinica, Taipei, Taiwan).
A ready-to-use lentivirus particle containing the
pReceiver Lv105 lentiviral vector, which expressed the human FH
gene, was purchased from GeneCopoeia, Inc. for overexpression of FH
in endometrial cancer cells. Lentivirus particles containing an
empty pReceiver Lv105 lentiviral vector (GeneCopoeia, Inc.) were
used as a negative control.
Briefly, the cells were seeded at 5×105
cells/well in 6 cm plates (Corning, Inc.) and incubated overnight
at 37°C in 5% CO2 atmosphere. Lentiviral infection was
achieved by adding virus solution to cells in culture media
containing 8 g/ml polybrene (TR-1003; Sigma-Aldrich; Merck KGaA).
The number of viruses was added according to the recommended
infection MOI for Ishikawa, RL95-2, KLE, and AN3CA cells (MOI=5).
Following a 24 h incubation at 37°C in 5% CO2
atmosphere, 2 g/ml puromycin (cat. no. A11138-03; Gibco; Thermo
Fisher Scientific, Inc.) was added for selection. Selected cells
were cultured in 2 g/ml puromycin for 2 weeks to establish cells
with stable overexpression or knockdown of FH.
2,3-Bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide
(XTT) colorimetric assay
The cell proliferation rate was determined using a
XTT colorimetric assay (Roche Diagnostics GmbH) following the
detailed procedure in accordance with a previous report (13).
Phosphokinase array
A human phosphokinase antibody array (ARY003C;
R&D systems, Inc.) was applied to explore the kinases that are
affected by FH in endometrial cancer cells. The site-specific
phosphorylation of 43 kinases was determined in a single sample.
Total cell lysates of RL95-2 cells with or without FH knockdown
were harvested for phosphokinase array analysis according to the
manufacturer's instruction.
Bioinformatic database analysis
FH and twist RNA Seq datasets of uterine corpus
endometrial carcinoma (UCEC) in The Cancer Genome Atlas (TCGA)
database were retrieved from TCGA website (Project ID: TCGA-UCEC;
http://xena.ucsc.edu). The Pearson's correlation
between interested proteins was analyzed using the data of patients
from TCGA-UCEC.
FH, VIM and CLDN1 datasets of uterus in the
Genotype-Tissue Expression (GTex) database were retrieved from the
GTex website (https://gtexportal.org/home/). The correlation between
FH and VIM and FH and CLDN1 was analyzed using the data of patients
from GTex-Uterus.
Transwell migration and invasion
assays
Cell migration assays were performed in 24-well
plates with Transwell (Corning Inc.) membrane filter inserts (6.5
mm diameter, 8 µm pore size). Endometrial cancer cells, after FH
overexpression or knockdown, were trypsinized, suspended in
serum-free medium and seeded (1×105 cells) in the upper
chamber of the Transwell filters. Medium containing 10% FBS was
added to the lower chamber and the plates were incubated for 24 h
at 37°C. Following incubation, cells were stained with crystal
violet for 2 h at room temperature. Non-migrating cells were
removed by wiping the upper surface of the filter. Migrated cells
were imaged using an Olympus SZX10 stereo light microscope (Olympus
Corporation) and analyzed using ImageJ software (ij153-win-java8;
National Institutes of Health).
For invasion assays, BioCoat Matrigel (BD
Biosciences) invasion chambers were rehydrated according to the
manufacturer's instructions and subsequent steps were identical to
the migration assay.
To study the effect of EGFR phosphorylation on
endometrial cancer cell metastasis, Gefitinib, an EGFR
phosphorylation inhibitor, was purchased from Sigma-Aldrich (Merck
KGaA; cat. no. SML1657). RL95-2 endometrial cancer cells were
treated with Gefitinib for 24 h before performing the migration and
invasion assays.
Western blotting
Western blotting was performed to assess the
knockdown efficiency following lentivirus infection and to assess
the protein expression of other proteins. The detailed procedure
according to a previous study was followed (13). In brief, the cells were lysed with
RIPA buffer (20 mM Tris-HCL pH 7. 4, 150 mM NaCl, 1 mM EDTA, 1%
Triton-X100, 1% sodium deoxycholate, 0.1% SDS) and cell lysates
were collected. The BCA protein assay (cat. no. 23225;
Sigma-Aldrich; Merck KGaA) was used to quantify total protein. The
samples were electrophoresed on a SDS-PAGE gel (8–12%; 20 µg/lane).
After protein transfer, the polyvinylidene fluoride (PVDF) membrane
was blocked with 2% BSA in 1X TBST solution for 1 h at room
temperature. The membranes were incubated overnight with primary
antibodies at 4°C. Antibodies against FH (cat. no. GTX110128;
1:2,000), vimentin (cat. no. GTX100619; 1:1,000), twist (cat. no.
GTX127310; 1:1,000), EGFR (cat. no. GTX100448; 1:1,000) and
phosphorylated (p-)EGFR (cat. no. GTX133599; 1:1,000) were
purchased from GeneTex Inc. Antibodies against JNK1/2/3 (cat. no.
ab179461; 1:1,000) and p-JNK1/2/3 (cat. no. ab124956; 1:1,000) were
purchased from Abcam. An antibody against β-actin (cat. no. A5441;
1:5,000; Sigma-Aldrich; Merck KGaA) was used as the internal
control. After the incubation with primary antibodies and
subsequent washing, PVDF membranes were incubated with rabbit (HRP
conjugate; cat. no. GTX2131101; 1:5,000; GeneTex, Inc.) or mouse
(HRP conjugate; cat. no. GTX213111; 1:5,000; GeneTex, Inc.)
secondary antibodies for 1 h at room temperature. The protein bands
on the PVDF membrane were visualized using enhanced
chemiluminescence reagent (PerkinElmer, Inc.) and Image Lab
software 6.0.1 (Bio-Rad Laboratories, Inc.).
Statistical analysis
All statistical analyses were performed using the
SPSS 14.0 statistical package for PC (SPSS, Inc.). Comparisons
between FH expression with various variables, including stage,
tumor size, grade lymph node metastasis and myometrium invasion,
were investigated by χ2 test. Student's t-test
(unpaired) was used to compare the difference between two groups.
One-way analysis of variance with post-hoc Tukey's test was used
for multiple group comparisons. P<0.05 was considered to
indicate a statistically significant difference.
Results
FH expression in endometrial cancer
tissues is negatively associated with tumor size and lymph node
metastasis
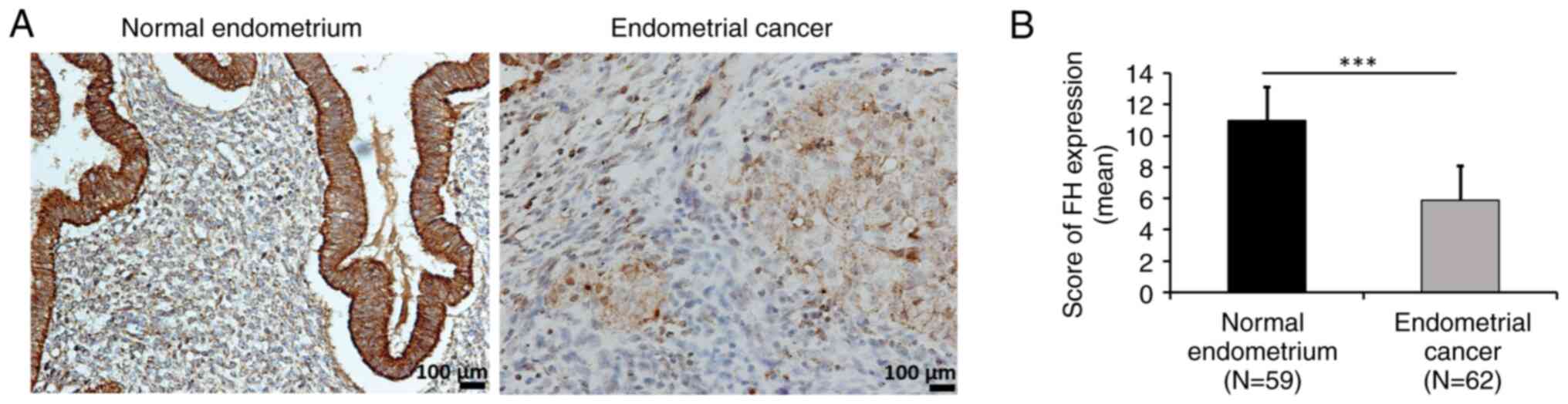
To evaluate FH expression in normal endometrium and
endometrial cancer tissues, IHC staining of 59 normal endometrial
and 62 endometrial cancer tissue samples was performed. It was
demonstrated that endometrial cancer tissue had low FH expression
compared with normal endometrial tissue (Fig. 1A and B). FH expression was further
correlated with the clinicopathological characteristics of patients
with endometrial cancer and it was found that FH had a negative
correlation with tumor size (P=0.028) and lymph node metastasis
(P=0.044; Table I). Moreover, the
patients were classified into an FH-low group (<4.50%) and an
FH-high group (≥4.50%; Table I)
using receiver operating characteristic (ROC) curve.
 | Table I.Correlation of FH with
clinicopathological characteristics in endometrial cancer
patients. |
Table I.
Correlation of FH with
clinicopathological characteristics in endometrial cancer
patients.
|
| FH |
|
|---|
|
|
|
|
|---|
| Variable | Low (score
<4.50) n (%) | High (score ≥4.50)
n (%) |
P-valuea |
|---|
| Pathologic
stage |
|
| 0.580 |
| I | 7 (11.3) | 10 (16.1) |
|
|
II/III/IV | 22 (35.5) | 23 (37.1) |
|
| Tumor size |
|
| 0.028a |
| <2
cm | 23 (37.1) | 32 (51.6) |
|
| ≥2
cm | 6 (9.7) | 1 (1.6) |
|
| Gradeb |
|
| 0.053 |
| G1 | 17 (31.0) | 24 (43.6) |
|
|
G2/G3 | 10 (18.1) | 4 (7.3) |
|
| Lymph node
metastasis |
|
| 0.044a |
|
Negative | 22 (35.5) | 31 (50.0) |
|
|
Positive | 7 (11.3) | 2 (3.2) |
|
| Myometrium
invasionc |
|
| 0.363 |
|
<1/2d | 16 (27.1) | 13 (22.0) |
|
|
≥1/2e | 13 (22.0) | 17 (28.9) |
|
Downregulation of FH expression
enhances, while overexpression of FH reduces, endometrial cancer
cell migration and invasion abilities
The endogenous expression of FH was further assessed
in five endometrial cancer cell lines. The result demonstrated that
the HEC1A, Ishikawa and RL95-2 cell lines (which are relatively
more invasive cell lines) had low FH protein expression, while the
AN3CA and KLE cell lines (which are relatively less invasive cell
lines) had high FH protein expressions (Fig. 2A). FH was subsequently recombinantly
overexpressed in Ishikawa cells (which had low endogenous FH
expression) and knocked down in RL95-2 cells (which had high
endogenous FH expression), to evaluate the effect of FH expression
on the proliferation, migration and invasion abilities of
endometrial cancer cells. A total of three shRNAs for FH expression
knockdown were assessed. It was discovered that KD6 had the best
knockdown efficiency and KD6 was therefore used in further
experiments.
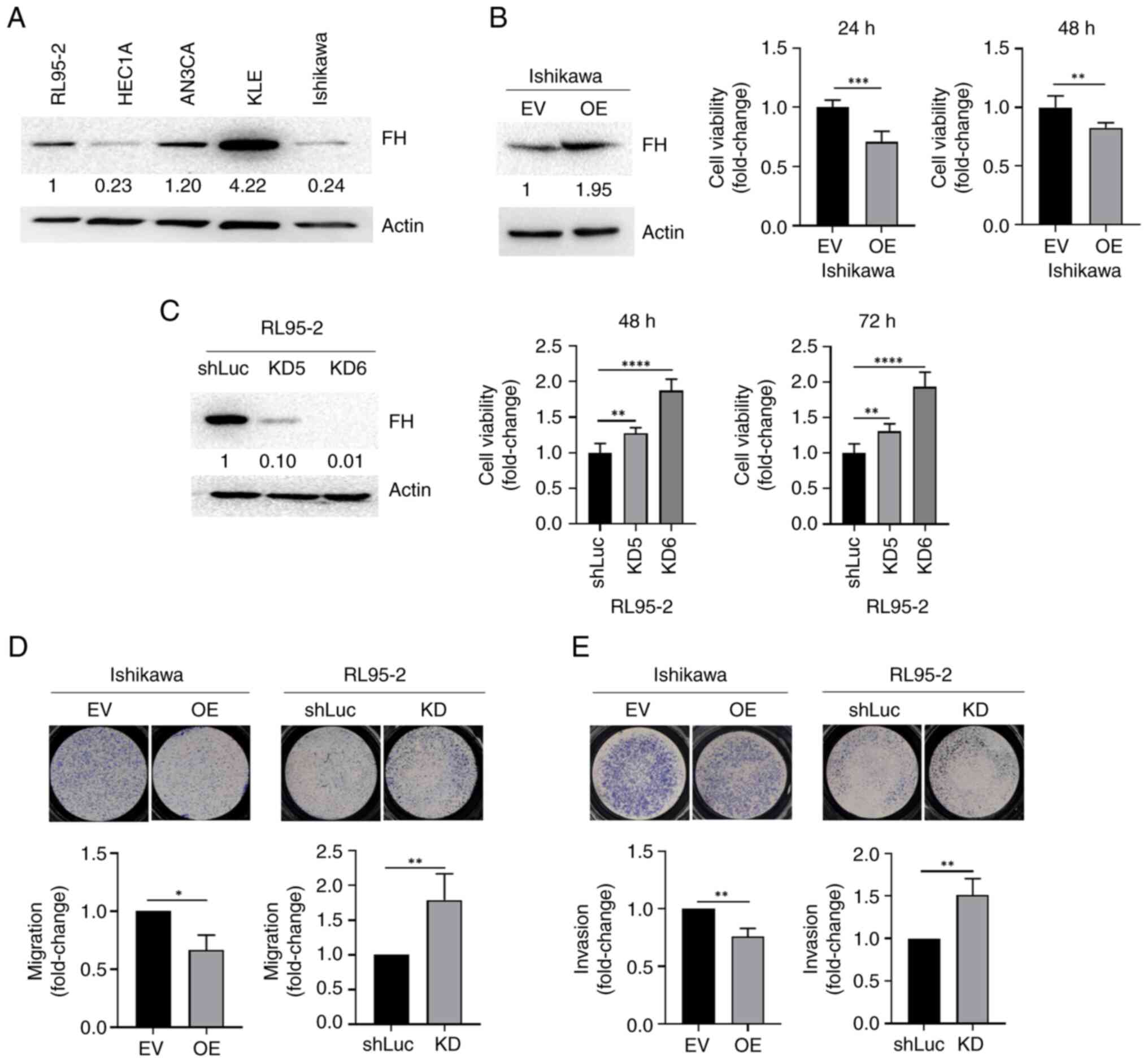 | Figure 2.KD of FH increases, while OE of FH
decreases, the migration ability of endometrial cancer cells. (A)
Western blot analysis of endogenous FH levels in human endometrial
cancer cell lines. (B) Left, western blot showing FH OE efficiency;
right, cell proliferation in Ishikawa cells infected with EV
controls or FH OE lentivirus. Student's t-test was used to compare
the difference between two groups. (C) Left, western blot showing
FH KD efficiency of two clones; right, cell proliferation in RL95-2
cells infected with shluc or FH shRNAs. One-way analysis of
variance with post-hoc Tukey's test was used for multiple group
comparisons. (D) Migration ability of FH-overexpressing Ishikawa
cells and FH-KD RL95-2 cells. (E) Cancer invasion ability of
FH-overexpressing Ishikawa cells and FH-KD RL95-2 cells. The data
are presented as the mean ± SD. *P<0.05, **P<0.01,
***P<0.001, ****P<0.0001. FH, fumarate hydratase; KD,
knockdown; FH, fumarate hydratase; OE, overexpression; EV, empty
vector; sh, short hairpin; shLuc, firefly luciferase-specific
shRNA. |
The cell proliferation assay results revealed that
the proliferation of FH-overexpressing Ishikawa cells was
decreased, while the proliferation of FH-knockdown RL95-2 cells was
increased (Fig. 2B and C).
Furthermore, the migration and invasion abilities of
FH-overexpressing Ishikawa cells were also decreased. By contrast,
the migration and invasion abilities of FH-knockdown RL95-2 cells
were increased. The migration ability of cells transfected with KD2
and KD5 clones was also assessed and it was found that cell
migration was also increased significantly compared with the
control group (Fig. S1B).
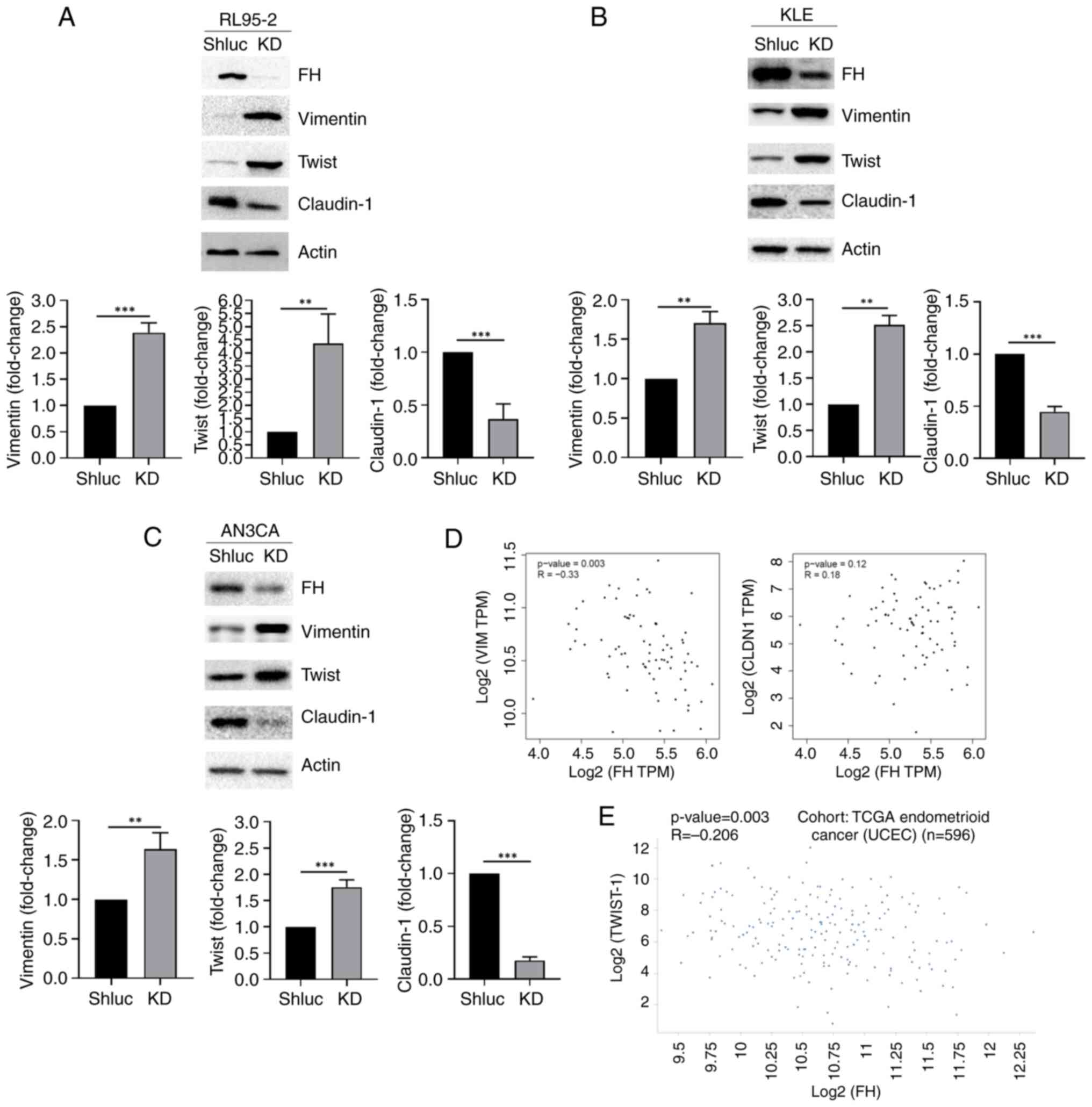
Expression of mesenchymal markers,
vimentin and twist, are upregulated in FH-knockdown endometrial
cancer cells
It is shown in Table
I that FH expression was negatively correlated with lymph node
metastasis. Therefore, the protein expression of various
epithelial-mesenchymal transition (EMT) markers, which play
critical role in cancer cell metastasis (14), in FH-knockdown RL95-2, KLE and AN3CA
cells was further evaluated. It was demonstrated that expression of
the mesenchymal markers, vimentin and twist, was upregulated in
FH-knockdown cells; by contrast, expression of the epithelial
marker, claudin 1, was downregulated significantly compared with
the control group (Fig. 3A-C). To
access whether FH mRNA expression is correlated with the mRNA
expression of EMT markers, the expression levels in the TCGA
(https://xena.ucsc.edu/) and GTex (https://gtexportal.org/home/datasets)
datasets were analyzed. A negative correlation was observed between
FH and two mesenchymal markers, vimentin (r=−0.33; P=0.003) and
twist (r=−0.206; P=0.003), whereas a positive, but not
statistically significant, correlation was observed between FH and
the epithelial marker, claudin-1 (r=0.18, P=0.12), as shown in
Fig. 3D and E.
EGFR phosphorylation is upregulated in
FH-knockdown endometrial cancer cells
Next, a human phosphokinase array was used to
identify the possible kinases regulating FH-mediated endometrial
cancer cell behavior. The results demonstrated that the levels of
p-JNK1/2/3 and p-EGFR were increased in FH-knockdown RL95-2 cells
compared with control cells (Figs.
4A and S2A), which was further
validated by western blotting (Fig.
4B-D). The p-EGFR protein level was significantly increased in
FH-knockdown endometrial cancer cell lines including RL95-2
(Fig. 4B), KLE (Fig. 4C) and AN3CA (Fig. 4D), while p-JNK1/2/3 expression did
not change in FH-knockdown RL95-2 cells (Fig. S2B). In addition, a connection
between FH and EGFR, mediated by TP53, was observed using the
STRING online database (https://string-db.org/) (Fig. S3).
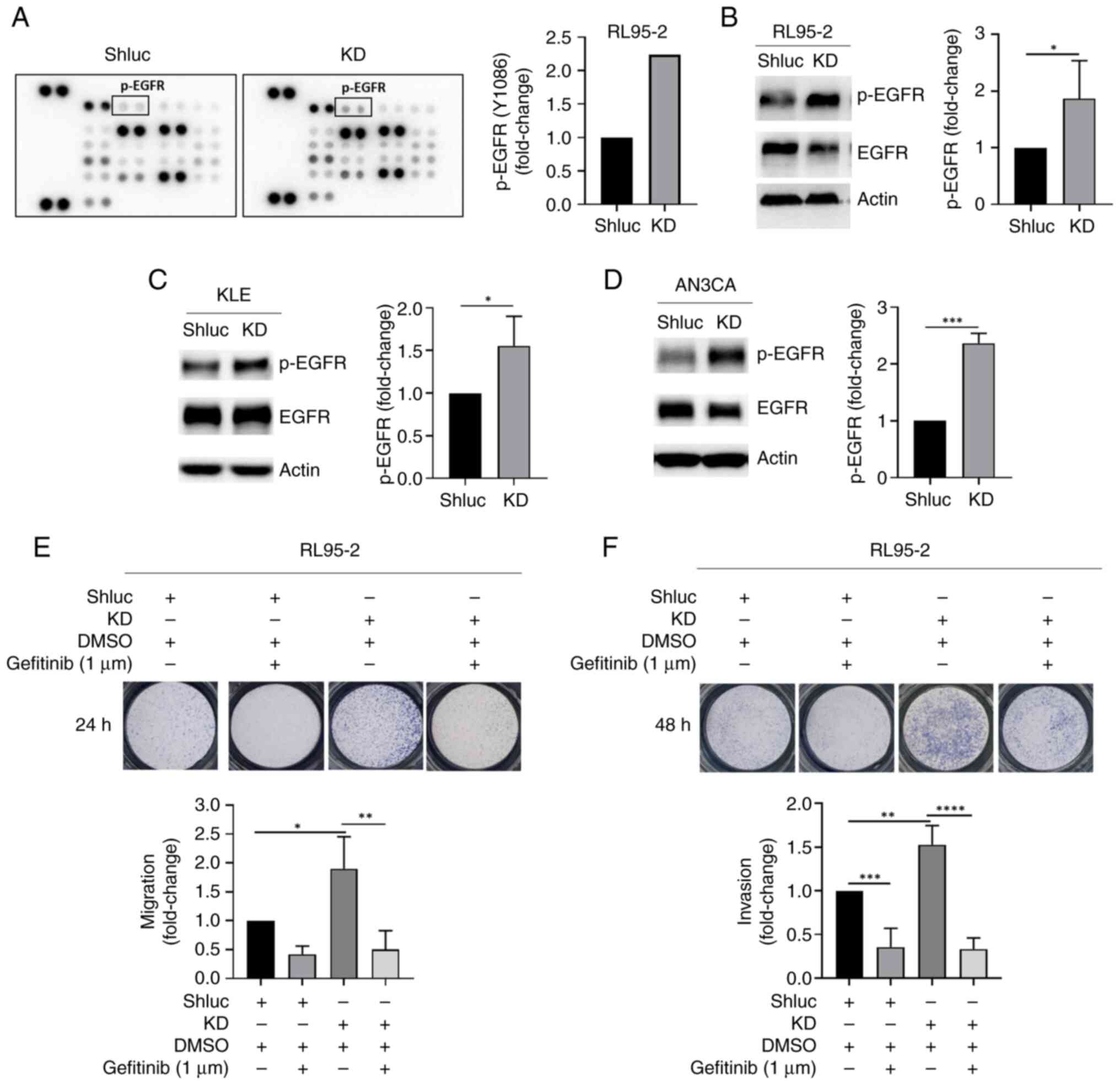 | Figure 4.EGFR phosphorylation is upregulated
in FH-KD RL95-2 cells. (A) Left, phosphokinase array showing
expression of various kinases in shluc and FH-KD RL95-2 cells;
right, fold change in p-EGFR kinase levels from the phosphokinase
array. (B) Left, western blot showing the protein expression levels
of p-EGFR and EGFR protein in RL95-2 shluc control and KD cells;
right, quantification of the p-EGFR protein expression levels. (C)
Left, western blot showing the protein expression levels of p-EGFR
and EGFR protein in KLE shluc control and KD cells; right,
quantification of the p-EGFR protein expression levels. (D) Left,
western blot showing the protein expression levels of p-EGFR and
EGFR protein in AN3CA shluc control and KD cells; right,
quantification of the p-EGFR protein expression levels. (E) Top,
migration and invasion abilities of RL95-2 following gefitinib
treatment (1 µM; 24 h); bottom, quantification of the results. (F)
Top, migration and invasion abilities of RL95-2 following gefitinib
treatment (1 µM, 24 h); bottom, quantification of the results. All
western blots were performed three times independently. Student's
t-test was used to compare the difference between two groups.
One-way analysis of variance with post-hoc Tukey's test was used
for multiple group comparisons. Data are presented as the mean ±
SD. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. FH,
fumarate hydratase; KD, knockdown; p-, phosphorylated; sh, short
hairpin; shLuc, firefly luciferase-specific shRNA. |
Further analysis was performed using the p-EGFR
inhibitor, gefitinib (1 µM), to treat FH-knockdown RL95-2 cells.
The results demonstrated that gefitinib inhibited p-EGFR protein
expression (Fig. S2C), while the
migration and invasion abilities of FH-knockdown cells were
significantly decreased after gefitinib treatment, compared with
the vehicle treatment group (Fig. 4C
and D). These results suggested that FH knockdown promoted EGFR
phosphorylation and hence upregulated the migration and invasion of
endometrial cancer cells.
Discussion
The role of FH in endometrial cancer is mostly
unclear. The present study, to the best of the authors' knowledge,
is the first study reporting that FH acts as a tumor suppressor in
endometrial cancer, as demonstrated by the negative correlation
observed between FH and tumor size or metastasis using clinical
data. In addition, the results of the present study demonstrated
that FH knockdown led to an increase in endometrial cancer cell
proliferation and metastasis and, since FH catalyzes the reversible
hydration of fumarate to malate (7), fumarate may have a role in this
process. Consistent with these findings, Sciacovelli et al
(15) reported that fumarate, which
may accumulate when FH is inactivated, promotes EMT through
activation of the transcription factors, snail1 and zeb1/2, by
silencing miR200 cluster expression. A negative correlation of FH
and mesenchymal marker vimentin, determined by
immunohistochemistry, has also been reported. In chromophobe renal
cell carcinoma and low-grade oncocytic renal tumor, there is a
positive staining for FH but a negative staining for vimentin
(16,17), while in FH-negative renal cell
carcinoma, 6 out of 8 cases are positive for vimentin expression
(18).
The present study demonstrated the upregulation of
the mesenchymal markers, vimentin and twist and the downregulation
of the epithelial marker, claudin-1, in FH-knockdown endometrial
cancer cells. In agreement with these findings, vimentin and twist
have been reported to be upregulated in endometrial cancer
(19,20) and associated with the poor survival
of patients with endometrial cancer (21). In addition, the high expression of
claudin-1 protein in endometrial cancer has been reported in a
previous study (22). Notably, the
subcellular localization of claudin-1 may determine its role as a
tumor suppressor or a promoter (23), with nuclear or cytoplasmic-localized
claudin-1 acting as an oncogene and cell membrane-localized
claudin-1 acting as a tumor suppressor (24). Whether regulation of FH definitively
influences subcellular localization of claudin-1 remains to be
determined.
FH mutations have been reported in renal cancer and
malignant paraganglioma (25,26).
Loss-of-function FH mutations cause an increase in fumarate and a
decrease in malate and citrate (27). Furthermore, FH deficiency promotes
renal tumor growth by inducing glucose uptake and angiogenesis
(28,29) and FH exerts oncogenic effects in
renal cell carcinoma through its ability to activate
hypoxia-inducible factor (HIF) by directly inhibiting prolyl
hydroxylases (30). Crosstalk
between HIF and EGFR has been described as a tumor-promoting
mechanism and EGFR signaling enhanced HIF activity through the
PI3K/AKT pathway (31). Moreover,
accumulation of fumarate, caused by FH mutations, promotes EMT and
increases cell migration (32). In
agreement with the aforementioned reports, especially for renal
cell carcinoma, FH knockdown resulted in an increase in endometrial
cancer cell proliferation and metastasis in the present study.
A previous study demonstrated that FH deficiency
resulted in diminished p53 levels in kidney cancer (33). p53 exerts tumor suppressor function
by upregulating tumor suppressor genes, the products of which
display an array of tumor suppression activities (33). Once activated, p53 enhances the
conversion of pyruvate to acetyl-CoA, allowing acetyl-CoA to enter
the TCA cycle and enhance mitochondrial respiration (34). However, the activity of p53 is
inhibited in the majority of cancer types (35) and a recent study demonstrated that
EGFR knockdown increased wild type p53 transcriptional activity
(36), which highlighted the role
of TP53 mutations in influencing prognosis and responsiveness to
EGFR-targeted therapy in non-small-cell lung cancer (37).
The results of the human phosphokinase array
analysis conducted in the present study suggested that p-EGFR
expression was upregulated in FH-knockdown cells and might mediate
malignant endometrial cancer cell behavior. EGFR is a receptor
tyrosine kinase that regulates cellular processes, including
proliferation, migration and survival and upregulation of EGFR has
been found to promote cancer cell metastasis in a variety of types
of cancer, including breast, pancreatic (38), gastric (39) and head and neck (40) cancer. Tamoxifen treatment activates
EGFR to promote endometrial cancer cell proliferation (41). In addition, another study found that
EGFR functions as a downstream effector of MUC20 to promote
endometrial cancer cell metastasis (42). In the present study, a novel
discovery linking EGFR activation to malignant endometrial cancer
cell behaviors when FH expression is suppressed was reported.
EGFR-tyrosine kinase inhibitors (EGFR-TKIs) are widely used for the
treatment of non-small cell lung cancer harboring EGFR-activating
mutations (43). Gefitinib arrests
PC-9 non-small cell lung cancer cells at the
G0/G1 phase (44) and induced apoptosis and autophagy in
A431 skin epidermoid carcinoma cells (45). The use of EGFR-TKIs is therefore
promising for the targeted treatment of EGFR-activated cancer
types, including endometrial cancer. Although overexpression of FH
is also a potential approach for the treatment of endometrial
cancer cells with downregulated FH expression, it is more practical
to target EGFR activation since several EGFR-TKIs are already
clinically available.
The clinical and translational significance of the
present study is two-fold. First, FH can be considered as a new
diagnostic and prognostic marker for endometrial cancer. Second,
novel therapeutic strategies targeting FH are worthy of further
investigation for personalized treatment management. However, the
present study has the limitation of not including an in vivo
animal study for validation. To provide a more complete picture for
the role of FH in endometrial cancer initiation and progression, an
in vivo model should be established to validate the in
vitro and clinical findings of the present study and to explore
the therapeutic potential of targeting FH for endometrial cancer
treatment.
In conclusion, the present study suggested that FH
functions as a tumor suppressor in endometrial cancer and presents
the potential of FH to be developed as a prognostic marker and
therapeutic target, after more extensive and multi-center clinical
studies.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the Ministry of
Science and Technology (grant nos. MOST 111-2314-B-037-011-, MOST
111-2314-B-037-020- and MOST 111-2314-B-037-046-) and the Center
for Intelligent Drug Systems and Smart Bio-devices (IDS2B) from the
Featured Areas Research Center Program within the framework of the
Higher Education Sprout Project by the Ministry of Education,
Taiwan. The present study was also supported by grants from
Kaohsiung Medical University Hospital (grant nos. KMUH110-0R43,
KMUH111-1R37 and KMUH-DK(A)112001) and Kaohsiung Medical University
(grant nos. KMU-DK(A)111005, NYCUKMU-111-P17 and
NSYSU-KMU-112-P04), Taiwan.
Availability of data and materials
The data that support the findings of this study are
available from the corresponding author upon reasonable
request.
Authors' contributions
Concept and design of the experiments was by Y-Y
Wang, A Vadhan, C-H Wu, C-Y Hsu, Y-C Chen, Y-K Chen, P-Y Chen, Y-C
Chang and S-S Yuan. The experiments were performed by Y-Y Wang, A
Vadhan, C-H Wu, C-Y Hsu, Y-C Chen and S-S Yuan. Y-Y Wang and S-S
Yuan confirm the authenticity of all the raw data. Data analysis
and discussion was performed by Y-Y Wang, A Vadhan, Y-K Chen, P-Y
Chen, H.D.H.N. and S-S Yuan. Y-Y Wang, Y-C Chang and S-S Yuan
contributed reagents, materials and analysis tools. Y-Y Wang, A
Vadhan, C-H Wu, C-Y Hsu, Y-C Chen, Y-K Chen, P-Y Chen, H.D.H.N.,
Y-C Chang and S-S Yuan prepared the manuscript. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Ethical approval was obtained from the Ethics
Committee of Kaohsiung Medical University Hospital.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that there are no conflicts of
interest.
References
|
1
|
Plataniotis G and Castiglione M; ESMO
Guidelines Working Group, : Endometrial cancer: ESMO clinical
practice guidelines for diagnosis, treatment and follow-up. Ann
Oncol. 21 (Suppl 5):v41–v45. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2019. CA Cancer J Clin. 69:7–34. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Setiawan VW, Yang HP, Pike MC, McCann SE,
Yu H, Xiang YB, Wolk A, Wentzensen N, Weiss NS, Webb PM, et al:
Type I and II endometrial cancers: Have they different risk
factors? J Clin Oncol. 31:2607–2618. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Braun MM, Overbeek-Wager EA and Grumbo RJ:
Diagnosis and management of endometrial cancer. Am Fam Physician.
93:468–474. 2016.PubMed/NCBI
|
|
5
|
Kyo S and Nakayama K: Endometrial cancer
as a metabolic disease with dysregulated PI3K signaling: Shedding
light on novel therapeutic strategies. Int J Mol Sci. 21:60732020.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lauby-Secretan B, Scoccianti C, Loomis D,
Grosse Y, Bianchini F and Straif K; International Agency for
Research on Cancer Handbook Working Group, : Body fatness and
cancer-viewpoint of the IARC Working Group. N Engl J Med.
375:794–798. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Frezza C: Mitochondrial metabolites:
Undercover signalling molecules. Interface Focus. 7:201601002017.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Akram M: Citric acid cycle and role of its
intermediates in metabolism. Cell Biochem Biophys. 68:475–478.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Raimundo N, Baysal BE and Shadel GS:
Revisiting the TCA cycle: Signaling to tumor formation. Trends Mol
Med. 17:641–649. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Picaud S, Kavanagh KL, Yue WW, Lee WH,
Muller-Knapp S, Gileadi O, Sacchettini J and Oppermann U:
Structural basis of fumarate hydratase deficiency. J Inherit Metab
Dis. 34:671–676. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tomlinson IPM, Alam NA, Rowan AJ, Barclay
E, Jaeger EE, Kelsell D, Leigh I, Gorman P, Lamlum H, Rahman S, et
al: Germline mutations in FH predispose to dominantly inherited
uterine fibroids, skin leiomyomata and papillary renal cell cancer.
Nat Genet. 30:406–610. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu C, Dillon J, Beavis AL, Liu Y,
Lombardo K, Fader AN, Hung CF, Wu TC, Vang R, Garcia JE and Xing D:
Prevalence of somatic and germline mutations of fumarate hydratase
in uterine leiomyomas from young patients. Histopathology.
76:354–365. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yuan SSF, Hou MF, Hsieh YC, Huang CY, Lee
YC, Chen YJ and Lo S: Role of MRE11 in cell proliferation, tumor
invasion, and DNA repair in breast cancer. J Natl Cancer Inst.
104:1485–1502. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yeung KT and Yang J:
Epithelial-mesenchymal transition in tumor metastasis. Mol Oncol.
11:28–39. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sciacovelli M, Gonçalves E, Johnson TI,
Zecchini VR, da Costa AS, Gaude E, Drubbel AV, Theobald SJ, Abbo
SR, Tran MG, et al: Fumarate is an epigenetic modifier that elicits
epithelial-to-mesenchymal transition. Nature. 537:544–547. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Michalova K, Tretiakova M, Pivovarcikova
K, Alaghehbandan R, Perez Montiel D, Ulamec M, Osunkoya A, Trpkov
K, Yuan G, Grossmann P, et al: Expanding the morphologic spectrum
of chromophobe renal cell carcinoma: A study of 8 cases with
papillary architecture. Ann Diagn Pathol. 44:1514482020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen T, Peng Y, Lei T, Wu C, Wang H and
Shi Y: Low-grade oncocytic tumour (LOT) of the kidney is
characterised by GATA3 positivity, FOXI1 negativity and mTOR
pathway mutations. Pathol Oncol Res. 29:16108522023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang W, Chu J, Zou YW, Jiang YX, Wei ZM,
Zhong DC, Liu Y, Li YJ and Yu WJ: Clinicopathological
characteristics of fumarate hydratase-deficient renal cell
carcinoma. Zhonghua Bing Li Xue Za Zhi. 48:120–126. 2019.(In
Chinese). PubMed/NCBI
|
|
19
|
Kyo S, Sakaguchi J, Ohno S, Mizumoto Y,
Maida Y, Hashimoto M, Nakamura M, Takakura M, Nakajima M, Masutomi
K and Inoue M: High twist expression is involved in infiltrative
endometrial cancer and affects patient survival. Hum Pathol.
37:431–438. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ihira K, Dong P, Xiong Y, Watari H, Konno
Y, Hanley SJ, Noguchi M, Hirata N, Suizu F, Yamada T, et al: EZH2
inhibition suppresses endometrial cancer progression via
miR-361/Twist axis. Oncotarget. 8:13509–13520. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wu HM, Huang HY, Schally AV, Chao A, Chou
HH, Leung PC and Wang HS: Growth hormone-releasing hormone
antagonist inhibits the invasiveness of human endometrial cancer
cells by down-regulating twist and N-cadherin expression.
Oncotarget. 8:4410–4421. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sobel G, Németh J, Kiss A, Lotz G, Szabó
I, Udvarhelyi N, Schaff Z and Páska C: Claudin 1 differentiates
endometrioid and serous papillary endometrial adenocarcinoma.
Gynecol Oncol. 103:591–598. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dhawan P, Singh AB, Deane NG, No Y, Shiou
SR, Schmidt C, Neff J, Washington MK and Beauchamp RD: Claudin-1
regulates cellular transformation and metastatic behavior in colon
cancer. J Clin Invest. 115:1765–1776. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bhat AA, Syed N, Therachiyil L, Nisar S,
Hashem S, Macha MA, Yadav SK, Krishnankutty R, Muralitharan S,
Al-Naemi H, et al: Claudin-1, a double-edged sword in cancer. Int J
Mol Sci. 21:5692020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hol JA, Jongmans MCJ, Littooij AS, de
Krijger RR, Kuiper RP, van Harssel JJT, Mensenkamp A, Simons M,
Tytgat GAM, van den Heuvel-Eibrink MM and van Grotel M: Renal cell
carcinoma in young FH mutation carriers: Case series and review of
the literature. Fam Cancer. 19:55–63. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Castro-Vega LJ, Buffet A, De Cubas AA,
Cascón A, Menara M, Khalifa E, Amar L, Azriel S, Bourdeau I, Chabre
O, et al: Germline mutations in FH confer predisposition to
malignant pheochromocytomas and paragangliomas. Hum Mol Genet.
23:2440–2446. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hvinden IC, Cadoux-Hudson T, Schofield CJ
and McCullagh JSO: Metabolic adaptations in cancers expressing
isocitrate dehydrogenase mutations. Cell Rep Med. 2:10046922021.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Isaacs JS, Jung YJ, Mole DR, Lee S,
Torres-Cabala C, Chung YL, Merino M, Trepel J, Zbar B, Toro J, et
al: HIF overexpression correlates with biallelic loss of fumarate
hydratase in renal cancer: Novel role of fumarate in regulation of
HIF stability. Cancer Cell. 8:143–153. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pollard PJ, Brière JJ, Alam NA, Barwell J,
Barclay E, Wortham NC, Hunt T, Mitchell M, Olpin S, Moat SJ, et al:
Accumulation of Krebs cycle intermediates and over-expression of
HIF1alpha in tumours which result from germline FH and SDH
mutations. Hum Mol Genet. 14:2231–2239. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lindner AK, Tulchiner G, Seeber A, Siska
PJ, Thurnher M and Pichler R: Targeting strategies in the treatment
of fumarate hydratase deficient renal cell carcinoma. Front Oncol.
12:9060142022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Peng XH, Karna P, Cao Z, Jiang B-H, Zhou M
and Yang L: Cross-talk between epidermal growth factor receptor and
hypoxia-inducible factor-1alpha signal pathways increases
resistance to apoptosis by up-regulating survivin gene expression.
J Biol Chem. 281:25903–25914. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bhattacharya D and Scimè A: Metabolic
regulation of epithelial to mesenchymal transition: Implications
for endocrine cancer. Front Endocrinol (Lausanne). 10:7732019.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kruiswijk F, Labuschagne CF and Vousden
KH: p53 in survival, death and metabolic health: A lifeguard with a
licence to kill. Nat Rev Mol Cell Biol. 16:393–405. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Contractor T and Harris CR: p53 negatively
regulates transcription of the pyruvate dehydrogenase kinase Pdk2.
Cancer Res. 72:560–567. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Muller PA and Vousden KH: p53 mutations in
cancer. Nat Cell Biol. 15:2–8. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ding J, Li X, Khan S, Zhang C, Gao F, Sen
S, Wasylishen AR, Zhao Y, Lozano G, Koul D and Alfred Yung WK: EGFR
suppresses p53 function by promoting p53 binding to DNA-PKcs: A
noncanonical regulatory axis between EGFR and wild-type p53 in
glioblastoma. Neuro Oncol. 24:1712–1725. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Canale M, Andrikou K, Priano I, Cravero P,
Pasini L, Urbini M, Delmonte A, Crinò L, Bronte G and Ulivi P: The
role of TP53 mutations in EGFR-mutated non-small-cell lung cancer:
Clinical significance and implications for therapy. Cancers
(Basel). 14:11432022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee J and Kim JH: Kaempferol inhibits
pancreatic cancer cell growth and migration through the blockade of
EGFR-related pathway in vitro. PLoS One. 11:e01552642016.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jiang L, Lan T, Chen Y, Sang J, Li Y, Wu
M, Tao Y, Wang Y, Qian H and Gu L: PKG II inhibits EGF/EGFR-induced
migration of gastric cancer cells. PLoS One. 8:e616742013.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Holz C, Niehr F, Boyko M, Hristozova T,
Distel L, Budach V and Tinhofer I:
Epithelial-mesenchymal-transition induced by EGFR activation
interferes with cell migration and response to irradiation and
cetuximab in head and neck cancer cells. Radiother Oncol.
101:158–164. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang L, Li Y, Lan L, Liu R, Wu Y, Qu Q
and Wen K: Tamoxifen has a proliferative effect in endometrial
carcinoma mediated via the GPER/EGFR/ERK/cyclin D1 pathway: A
retrospective study and an in vitro study. Mol Cell Endocrinol.
437:51–61. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen CH, Wang SW, Chen CW, Huang MR, Hung
JS, Huang HC, Lin HH, Chen RJ, Shyu MK and Huang MC: MUC20
overexpression predicts poor prognosis and enhances EGF-induced
malignant phenotypes via activation of the EGFR-STAT3 pathway in
endometrial cancer. Gynecol Oncol. 128:560–567. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Singh M and Jadhav HR: Targeting non-small
cell lung cancer with small-molecule EGFR tyrosine kinase
inhibitors. Drug Discov Today. 23:745–753. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang L, Qi Y, Xing K, Qian S, Zhang P and
Wu X: A novel strategy of EGFR-TKI combined chemotherapy in the
treatment of human lung cancer with EGFR-sensitive mutation. Oncol
Rep. 40:1046–1054. 2018.PubMed/NCBI
|
|
45
|
Wang J, Wang C, Hu X, Yu C, Zhou L, Ding Z
and Zhou M: Gefitinib-mediated apoptosis is enhanced via inhibition
of autophagy by chloroquine diphosphate in cutaneous squamous cell
carcinoma cells. Oncol Lett. 18:368–374. 2019.PubMed/NCBI
|