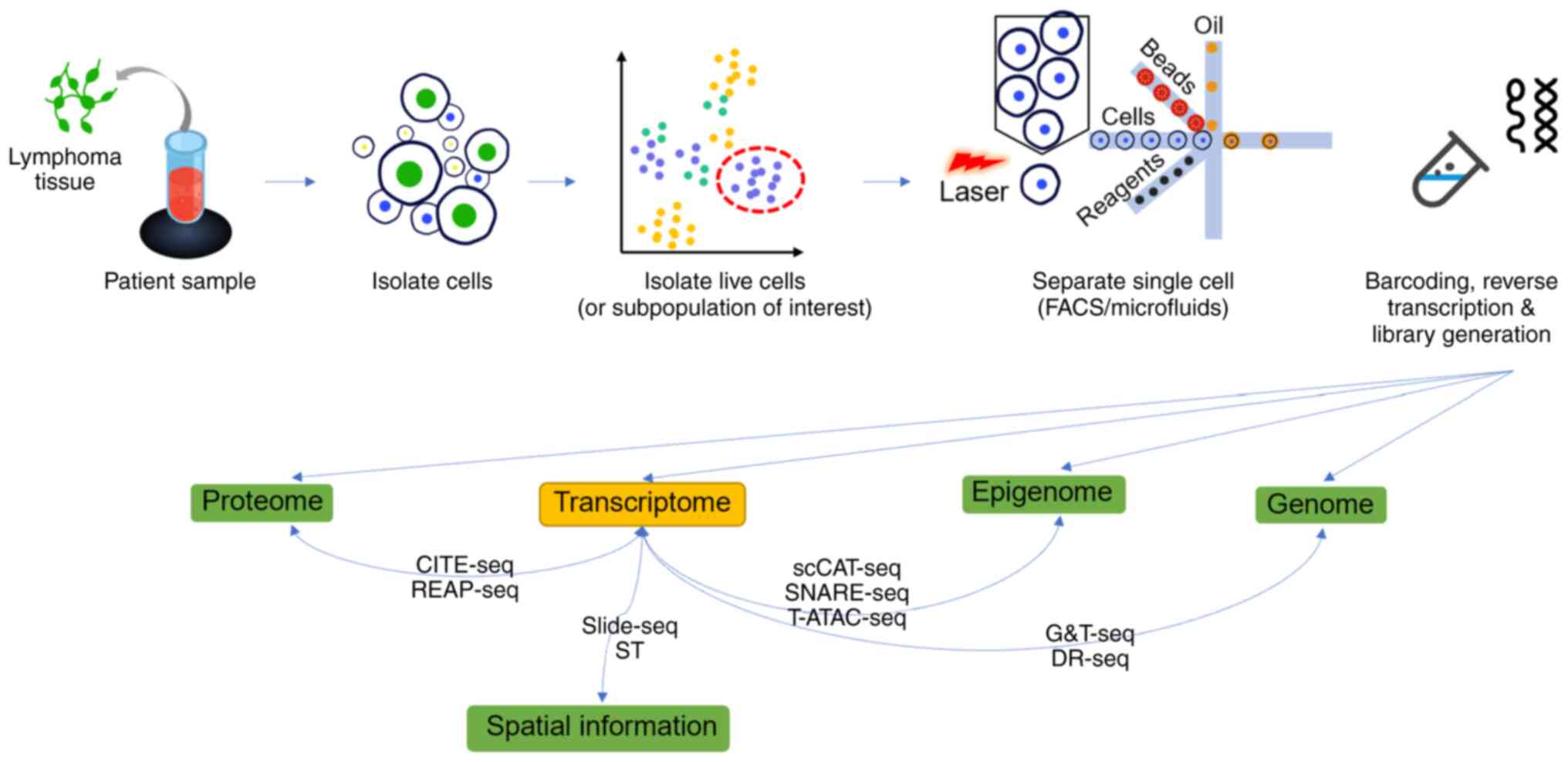
Lymphoma is a sizable collection of
immunocyte-derived malignancies that exhibit molecular aberrations
in various aspects (1). Variations
in genotype, phenotype and metabolic events lead to changes in
lymphoma cell morphology, progression rate and metastatic potential
(2). However, conventional
high-volume detection techniques, such as chromatin
immunoprecipitation, real-time quantitative polymerase chain
reaction, and western blotting, do not provide comprehensive
analyses of the tumor cell landscape (3). Previously, novel techniques and
analytic tools have been developed to promote research and
understand lymphoma. These techniques and tools include capturing
mRNAs from cell lysates using arrays of thousands of
oligonucleotide probes (4),
identifying differentially expressed genes and signaling pathways
between two samples using Gene Set Enrichment Analysis (5), and inferring sample cell composition
by deconvolution of the bulk transcriptome (6,7).
Nevertheless, these techniques and tools fundamentally reflect the
functions of lymphoma cells as a whole and do not describe the
molecular mechanisms at the level of a single lymphoma cell.
Presently, single-cell sequencing technologies involving genomics,
transcriptomics, proteomics and epigenomics employ high-resolution
characterization, which allows researchers to understand precisely
tumor heterogeneity, the tumor microenvironment (TME), and tumor
progression (8). Single-cell
spatial transcriptomics is also used to understand these tumor
events (9) (Fig. 1).
By utilizing the rapidly-evolving single-cell
multi-omics techniques to identify the different types of lymphoma,
it is possible to reveal heterogeneity (1,10,11)
and drug resistance (12), identify
presumptive precancerous groups (13) and evolutionary processes (14,15),
identify new biomarkers (16,17),
and explore TMEs (18,19). Lymphoma research has advanced to a
more sophisticated level with the existence of single-cell
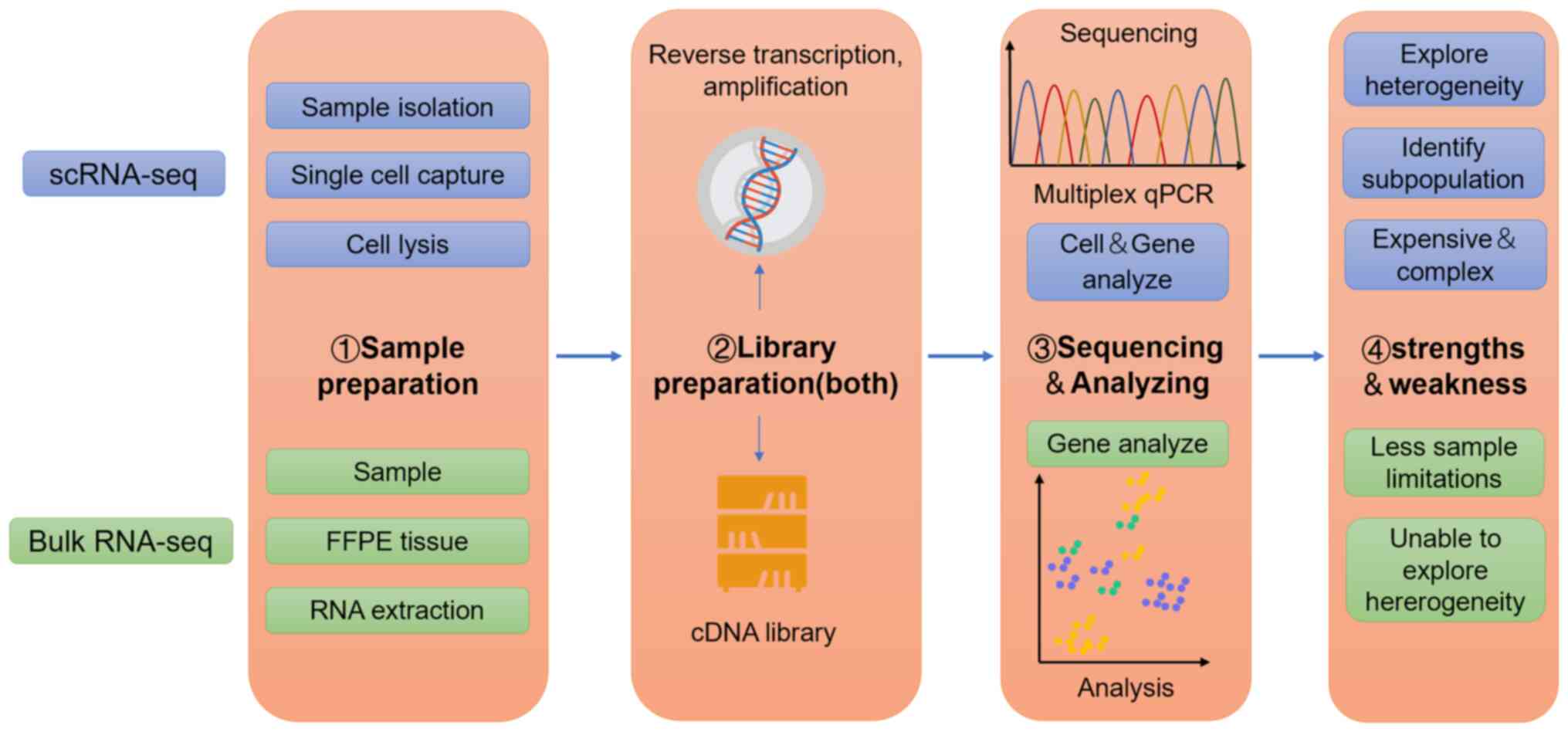
multi-omics approaches. Using single-cell RNA sequencing
(scRNA-seq) as an example, in Fig.
2 the differences and similarities between the workflow of
scRNA-seq and bulk RNA seq are illustrated.
Presently, the generally acknowledged platforms of
high-throughput single-cell sequencing are Drop-Seq (20), inDrop (21), and 10X Genomics (22). The scRNA-seq technologies commonly
used in lymphoma analysis are listed in Table I.
The multiparametric classification approach from the
World Health Organization and the revised European and American
Lymphoma classification (36) have
tremendously aided lymphoma clinical and translational research,
which serves as a foundation for discovering the causes of
molecular changes in these tumors (37). Although a diagnosis of Hodgkin's
lymphoma according to the existence of Reed-Sternberg (R-S) cells
has been proposed for a long time, the lymphomagenesis of Hodgkin's
lymphoma remains unclear (38).
Given that malignant R-S cells derived from germinal center (GC) B
cells (39,40) represent a minority of lymphoma
cells, an inadequate biopsy may fail to identify these cells and
make an accurate diagnosis (41).
Non-Hodgkin's lymphoma accounts for ~3% of all cancer occurrences
and deaths worldwide, making it the most common malignant tumor in
the hematological field (42). Each
of its >40 subtypes has specific driver genetic mutations and
distinct risk factors (43). Thus,
it is the extensive and remarkable heterogeneity of lymphoma cells
that requires to finely characterize lymphoma cell molecules.
Currently, there is rapid progress in the accurate
characterization of single-cell immune cells, which allows for
comprehensive detection of lymphoma and precise clinical diagnosis.
For instance, phenotyping CD molecules (CD3+,
CD4+ and CD8+) on T cells can help identify
the specific cellular status and advance the development of
immunotherapy (44). It has been
shown that using just 6 biomarkers (CD95, CD73, RB, CD39, CD38 and
CD27) can define tonsillar (and also lymph node) B cells and
characterize B cells (45).
Furthermore, single-cell sequencing analysis of NK cells from
patients with acute myeloid leukemia (AML) revealed similar
patient-specific patterns of NKp30 and CD27 expression, and
downregulation of CD160 transcription on NK cells is related to the
reduced survival of patients with AML, suggesting that CD160 is
promising precision medicine biomarker (46). A single-cell histological assay of a
subpopulation of T cells associated with Hodgkin's lymphoma
revealed prominent expression of the immunosuppressor receptor
LAG3, which offers a new approach targeting immune checkpoints in
Hodgkin's lymphoma, second only to PD-1 (47). High-resolution clustering and
combinatorial gene characterization using large scRNA-seq datasets
of human γδ T cells allow in-depth characterization of these cells,
which can specifically identify T cell receptor (TCR) Vδ1 and TCR
Vδ2 subpopulations (48).
Repertoire and Gene Expression by Sequencing (RAGE-seq), which
combines short-read transcript analysis based on single-cell
libraries of barcodes with long-read sequences and targeted capture
of B cell receptor (BCR) or the TCR mRNA transcriptome, can
identify the full-length antigen receptor profiles of B or T cells
(49).
These single-cell multi-omics techniques have been
applied to study various cells in the human immune cell population,
and they have great potential in identifying new subpopulations of
lymphoma cells or in exploring new biomarkers.
In the field of tumor research, single-cell
multi-omics techniques have transformed the existing knowledge of
the biological features of lymphoma lesions (3). Different technologies, such as
scRNA-seq, scDNA-seq, single-cell chromatin
immunoprecipitation-seq, and methylation sequencing, have been
widely used in lymphoma research, and their specific features have
been well characterized.
At the genetic level, the rather mature single-cell
combinatorial indexing RNA sequencing can provide information on
lymphoma copy number variation (50). In addition, genomic mutations can
also be identified in different types of lymphoma with single-cell
precision. Stratified clustering of NK cells showed that a total of
56 genomic mutations based on the JAK-STAT pathway and TP53 were
present in 102 EBV patient samples (51), which is in line with previous bulk
analysis. Single-cell exome sequencing and protein profiling (SNV
and small insertions) of GI-DLBCL and non-GI-DLBCL exhibited
changes in gene mutation frequency, among which ID3, CCDN3 and TP53
were increased, while BCL2, CREBBP and MYD88 were significantly
decreased (52). In an exploration
of lymphoma molecular drivers, researchers identified signature
mutations in CD79B and MYD88 L265P as well as a CDKN2A deletion in
BCR and NF-kB pathways (53). As
for the clinical application, based on gene mutation data from the
NF-κB pathway, CCND1, as well as ATM according to scRNA-seq,
researchers classified mantle cell lymphoma into four genetic
subgroups as C1-C4 for the first time (54). In another study, single-cell genome
and exome sequencing showed that CDKN1B, SMARCB1 and DAZAP1
expression has a negative impact on patient prognosis, suggesting
that they could be used as biomarkers for patient prognosis
(55). Lymphoma heterogeneity has
been investigated among primary BM specimens by applying
single-cell targeted DNA sequencing through the Fluidigm C1 system
(56). As for the molecular
mechanism, single-cell gene transcription analysis of IG-non MYC
translocations in Burkitt's lymphoma, the most common type of
childhood lymphoma, revealed MYC oncogene dysregulation and
demonstrated that at least one BL subgroup precursor could be
expressed in each patient (57).
Furthermore, KIR2DL4 was reported to be overexpressed in malignant
NK cells by scRNA-seq, and KIR2DL4 promotes lymphoma pathogenesis
by mediating NK cell proliferation and apoptosis, which are
associated with certain signaling pathways such as AKT and NF-κB
(58). After analyzing the
transcriptome of tens of thousands single tumor cells from 6
primary FL patients, the results revealed that malignant B cells
showed more pronounced expression of the BCL2 gene and absence of
expression of MHC-II and CD52 genes. Moreover, the results also
identified the co-expression of B2M and CEBPA genes in Treg cells
with immune checkpoint molecules, which helps further map the
network of genes included within the immunoregulatory mechanism
(59). Thus, single-cell analysis
could help expand tumor genomic studies in different classes of
lymphoma and accelerate the transition from genetic analysis to
pharmacodynamic therapy in the future.
Targeted and functional proteomic analysis enables
bulk and thorough studies of proteins, particularly for
high-throughput screening of promising biomarkers from complex
tumor biological microenvironments. Currently, proteomics has
become one of the key fields of lymphoma research, and it is
considered as the most suitable approach to discovering new
biomarkers and personalized therapies, as well as it being a
high-throughput technique for revealing genotype-phenotype
uncoupling in lymphoma and tracking proteomic dynamics in relapsed
patients (60). For example,
through the E1A-binding protein p300, mice with knockdown BCL6
expression on the surface of lymphomas can block oncogenic
transformation by inhibiting the acetylation of Lys132 in the p53
gene, which upregulates cystein-1 to reduce BCL6 stability
(61).
Single-cell epigenomic analysis of lymphoma has also
greatly aided the analysis lymphoma diagnostic and prognostic
processes. The epigenome of lymphoma is defined by the regulation
of normal gene expression in cells, modified by histone proteins
and DNA, plus the action of non-coding RNA (62). Previous studies have demonstrated
strong associations between the differentiating expression of
miRNAs and the pathogenic progression of NKTCL tumors by
single-cell techniques targeting P53, cell-cycle related genes and
MAPK pathways (63,64). The reduced functions of miR-26 and
miR-101 cause EZH2 overexpression, whereas the over-activation of
miR-223 contributes to PRDM1 suppression (65,66).
Hypermethylation in the promoter region was studied by whole
methylation assay and methylation site-specific validation, and the
results demonstrated that the functions of TET2, PTPRK, SOCS6 and
PTPN6 can increase the expression of methylated genes (67,68).
Functionally, TET2 inactivation may lead to hypermethylation of
lymphoma promoters, and the negative regulation of JAK-STAT by
PTPRK, SOCS6 and PTPN6 suggests an alternative mechanism associated
with the activation of the JAK-STAT signaling pathway (67).
Besides gene transcription, protein expression and
epigenetic inheritance, single-cell technology also has roles in
other areas. For example, scATAC-seq can identify specific
chromatin motifs (69), whereas
scNGS can provide information about somatic mutations and cellular
heterogeneity (70). It is even
possible to reveal the mutation profiles of B cells in the entire
life cycle of a human by single-cell whole genome sequencing, thus
identifying potential cancer-causing mutations (71)
Taken together, single-cell multi-omics techniques
provide the productive characterization of the biological features
and intrinsic dynamics of lymphoma, which may widen our knowledge
of lymphoma and accelerate the pace of clinical diagnosis and
treatment.
A comprehensive understanding of tumorigenesis and
clonal evolution can help to unravel tumor heterogeneity as well as
targeted therapies for tumors. Although protein-altering lesions
have always been detected in lymphoma diagnosis, the tumor's clonal
structures and the initial progress remain unclear. Quantitative
single-cell genomics based on mass spectrometry is used
specifically to understand the pathogenesis of lymphomas, and its
incremental value in deciphering the complexity of lymphoma
entities to regulate the heterogeneity of molecular mechanisms is
currently a popular diagnostic approach and therapeutic strategy
(72). Single-cell multi-omics
techniques can reveal the clonal specificity of lymphoma and
establish the order of genetic events in lymphoma.
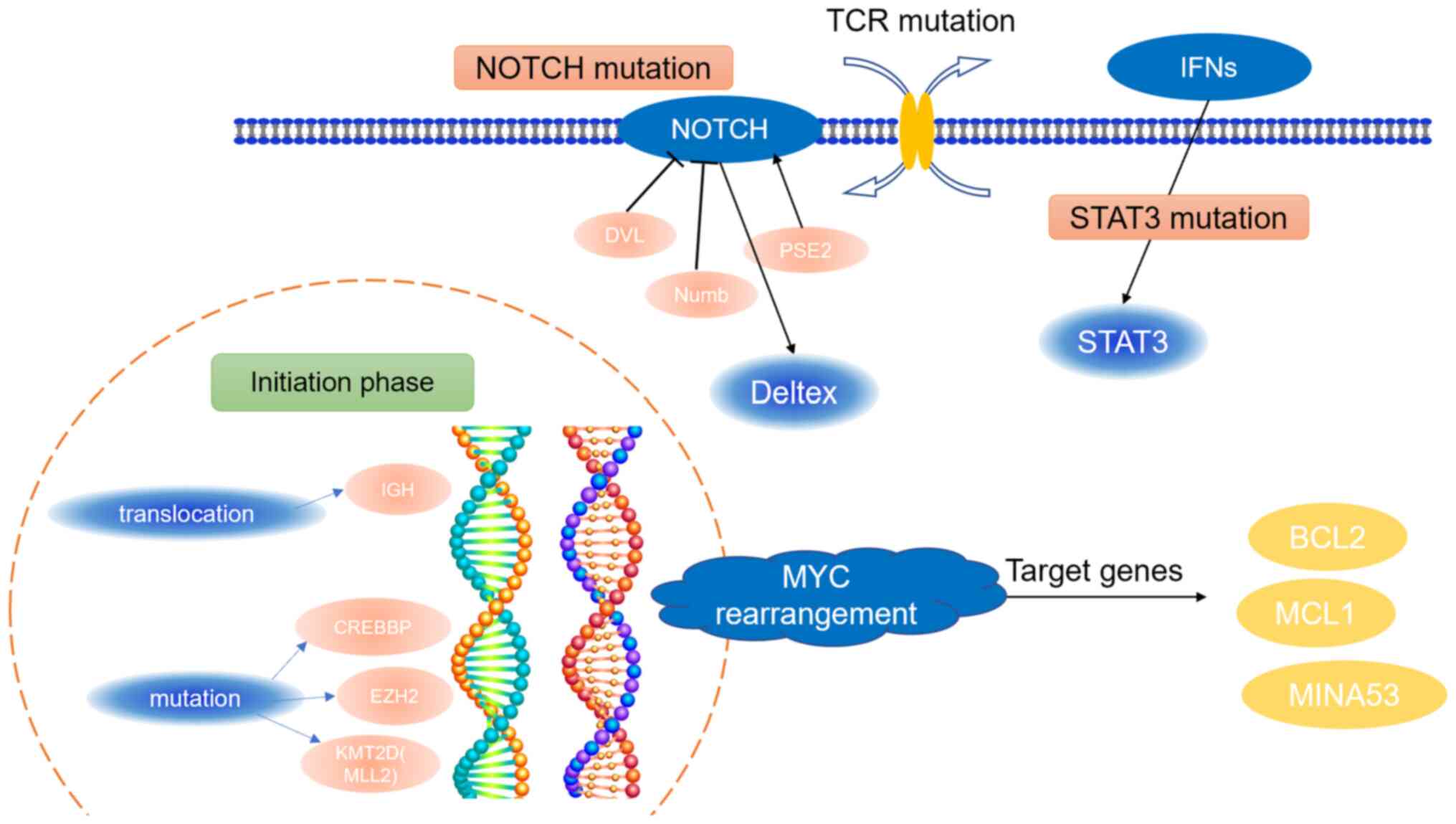
Lymphoma cells may have translocation mutations in
gene bases, although few types of lymphoma can result from
recurrent translocations involving specific genes. For example, the
IGH translocation on 14q32 is responsible for a certain proportion
of mature B-cell tumors (73).
Meanwhile, regulatory chromatin genes (CREBBP, EZH2 and KMT2D) have
also been shown to mutate in the early drivers in lymphoma
(74). These mutations indicate the
initiation phase of lymphoma development.
The clonal evolution of lymphoma is inevitably
accompanied by changes in the transcriptome of genes, and to
clarify genotyping, numerous experiments have been performed using
single-cell sequencing techniques to describe the clonal
progression of lymphoma. Fluorescence in situ hybridization
results identified that lymphoma cells had MYC rearrangements that
were not BCL2 or BCL6, but involved the immunoglobulin light or
heavy chain gene and the 8q24 region, which are markers of lymphoma
lesions (75). This finding is
remarkable because the prognosis of lymphoma with MYC translocation
is often worse than that of lymphoma alone (76). However, this is a limited ordering
study of lymphoma using bulk samples, and it did not explore the
tumor evolution. Single-cell capture and WGA of microfluidic
specimens have proved RAG-mediated structural vibrations to precede
lymphoma (77). The acquisition of
fusion genes and the loss of 9p21 (CDKN2A/B) accounts for the
intermediated clonal evolution of lymphoma (56). Gene rearrangements and chromosomal
translocations such as these play a non-negligible role in
promoting lymphoma development.
Multi-step mutations in NOTCH, the STAT3 signaling
pathway, and TCR are always late events in the evolution of
lymphoma and can further accelerate the proliferative potential of
tumor cells, leading to the development of highly malignant clones
that can lead to disease onset and progression (9).
Single-cell sequencing techniques allow the complete
clonal evolutionary structure of tumor cells to be reconstructed,
thus providing insights into the evolution of lymphoma and
revealing synergistic combinations that promote clonal expansion
and dominant mutations, which offers information for therapeutic
decisions (Fig. 3). For example,
according to 2017 World Health Organization criteria, lymphomas
with mutations in MYC and BCL2 or BCL6 during clonal evolution, as
mentioned above, and with concurrent DLBCL histological features
are no longer classified as DLBCL, but are referred to as advanced
B cell lymphomas with MYC and BCL2 or BCL6 rearrangements (41). These sub-entities tend to have a
worse prognosis for common DLBCL and often require the application
of different treatments (78–80).
To further dissect the heterogeneity of the origin
of lymphomas, the studies on the origin of lymphoma at the cellular
level were also summarized, as detailed below (Single-cell
multi-omics for modeling B-cell GCs).
Drug resistance in lymphoma can be broadly divided
into the development of resistance at genetic and transcriptional
levels within cancer cells and the development of resistance at
tissue and cellular levels. According to the widely accepted theory
of clonal evolution, the pre-emptive generation of heterogeneous
drug-resistant clonal mutations through therapeutic selection is
the most critical biological process (81). The ability to detect populations of
cells that survive under a period of anticancer treatment is
essential for drug resistance prediction or even reversal. It is
difficult for conventional genetic studies of bulk samples to
identify small numbers of drug-resistant cells. Today, through gene
expression analysis under single-cell resolution, these cells and
their mechanisms of drug resistance can be characterized (82).
Changes in gene levels are responsible for drug
resistance in certain cancers, including lymphoma. Transcriptome
sequencing analysis targeting the BTK gene [encodes a signaling
protein vital for B cell development, differentiation and signaling
(83)] found that after the
inhibition of BTK, the FC receptors (FCGR2A, FCGR2B and FCGR3A) on
the surface of B cells exhibited a higher level of expression
(84), and the FC receptors
promoted the internalization of the CD20 monoclonal antibody
rituximab [core drug against malignant B lymphocyte proliferation
(85)], thus reducing its clinical
efficacy (86). In addition,
through mutation or deletion, TP53, ATM, CDKN2A, KMT2D, and other
key lymphoma suppressor genes can be inactivated, leading to
genomic instability and promoting secondary mutations and drug
resistance (87).
At the cellular level, lymphoma resistance in the
macroscopic sense is mostly manifested by the presence of inherent
molecular mechanisms of genotoxic drug resistance (88), hypoxia (89) and DNA damage (90). Previous studies have used
single-cell transcriptomics techniques to deeply explore the
mechanisms of acquired drug resistance and the differential
alterations in the lymphoma microenvironment. Early experimental
manipulations of whole-genome sequencing and DNA microarray of
murine lymphoma have been performed to analyze CpG methylation,
mRNA expression and DNA sequences that are related to increased
drug resistance (91). Another
transcriptional analysis revealed that lymphomas with low GAPDH
expression predominantly adopt bovine/phosphorus metabolism and are
dependent on mTORC1 signaling and glutaminyl solubilization to
generate ATP (92), which indicates
a large metabolic reprogramming response and reflects the
acquisition mechanism of drug resistance (93). In another pan-cancer analysis of
lymphoma, the results revealed differences in mRNA expression using
data from The Cancer Genome Atlas and concluded that targeting the
high expression of TRPV channel-associated genes can enhance drug
resistance, and they may be promising biomarkers of patient
prognosis (94). In addition to
lymphoma, there is a single-cell muti-omics study showing that the
levels of MSH2, MSH6 and MLH1, together with homologous
recombination effectors such as BRCA2 and RAD51, are reduced in
rectal cancer, suggesting that just like single-cell organisms,
tumor cells can also promote drug-resistant persistent cells
through continuous mutation derivation (95).
Although lymphoma has multiple mechanisms of drug
resistance development, targeted single-cell studies have been used
for molecular sequencing and outcome assessment of drug resistance
in lymphoma. The single-cell multi-omics techniques not only
explore and detect, but also show therapeutic potential to drug
resistance in lymphoma. According to a single-cell analysis, KP772
can induce apoptosis of BCL-2-independent cells and upregulate the
Harakri gene, making KP772 a promising candidate for anti-multidrug
resistance of malignant lymphoma (96). Meanwhile, splicing modulators have
been clinically tested for the treatment of drug resistance in
lymphoma given their ability to interfere with drug metabolism or
absorption of gene expression such as FPGS, dCK and SLC29A1
(97).
In addition to changes at the genetic and cellular
levels, the mechanisms by which lymphomas develop drug resistance
are complex and also include changes in the TME.
Transcriptomic analyses from multiple independent
lymphoma cohorts were performed to describe four microenvironment
subtypes based on clinical behaviors and biological aberrations,
and this approach identified the ECM proteins DCN and BGN as novel
potential therapeutic targets for the TME (100). In another assessment of gene
expression profiles, it was revealed that SPARC expression in the
TME has fair inter-observer reproducibility and is a strong
prognostic reference (101). In
terms of treatment, the increased expression of CD8+
co-receptors (PTRPC and FYB) was found to enhance LCK and FYN
expression, thereby activating the signaling of T cells and greatly
contributing to immunocidal effects (84). A comprehensive transcriptomic atlas
from over 100,000 lymphoma non-hematopoietic cells (NHCs;
mesenchymal stromal and endothelial cells) at single-cell
resolution demonstrated that NHC heterogeneity in LNs can be
detected even in aggressive lymphomas, indicating the powerful
practicability of single-cell analysis of the NHC profile in
characterizing various TME subtypes (102).
It is well established that angiogenesis contributes
to the inevitable progression of lymphoma through signaling
transducers, cytokines and other components in the TME (103,104). Studies have shown that STAT3 is
overexpressed in primary central nervous system lymphoma tissues
vs. normal brain cells and vessels (105), and chromatin modifications occur
when PCNSL forms (106).
Single-cell histological analysis have shown that ROBO1, KAT2B and
KMT2D regulate the lymphoma microenvironment by disrupting the
functions of keratinocytes and stromal cells via non-histone
acetylation (107), including
histone deacetylase and DNA methyltransferase inhibitors are under
investigation, suggesting they may be a new therapeutic option for
lymphoma (108). Lysine
acetyltransferase inhibitors enable the activation of KAT2B
transcription to affect lymphoma angiogenesis (109). Recently, the study of gene impact
associated with epigenetic modifications and angiogenetic events to
fully profile the lymphoma microenvironment has gained research
momentum.
The TME is also closely related to immune escape and
drug resistance development in lymphoma, and studies at single-cell
resolution reveal several novel mechanisms, suggesting that the
microenvironment can suppress the host's antitumor immune activity
(110). The immune escape of
tumors is also a topic of research and an approach of clinical
care.
Taken together, single-cell multi-omics techniques
allow for the comprehensive characterization of the lymphoma
microenvironment and the application of the gained information to a
wider range of research.
GCs formed in secondary lymphatic organs undertake
the binding of antigens to mature primitive B cells. GC B cells can
produce numerous types of non-Hodgkin's lymphoma, such as DLBCL and
Burkitt's lymphoma, which develop through different pathogenic
mechanisms due to their origin in different stages of GC B cells
(111). Transcriptional profiling
revealed that DZ and LZ cells express RNA and the surface proteins
CXCR4 and CD83, respectively, reflecting a heterogeneity that
distinguishes them from other types of cells (112).
Single-cell multi-omics analysis revealed that
reduced TNFRSF14 expression and STAT6 activation improves the
inhibitory effect on GC B-cell signaling (113,114). The results showed that the
epigenetic modifiers EZH2 and CREBBP can directly impact on GC
B-cell's function. For example, upon CREBBP inactivation, MHC-II
and CD40 are suppressed, which disturbs the presentation of
antigens from lymphoma cells to CD4+ T cells (115).
Utilizing single-cell multi-omics techniques to
model GCs can improved the characterization of the origin of
lymphoma and provide more constructive insight into
lymphomagenesis.
The extensive heterogeneity of lymphoma is a major
obstacle to the clinical management and the pharmacological
treatment of this disease. As aforementioned in the sections
pertaining to the development of novel biomarkers and the
investigation of drug resistance and immune mechanisms in lymphoma,
single-cell technology is rapidly evolving and may be of great
value in the diagnosis and treatment of lymphoma. For example,
scRNA-seq can identify several suitable entities for targeted
therapy. Combined with capture technology, scRNA-seq can identify
transcriptomic profiles related to the efficacy and toxicity of
certain drugs, which has also been used to study the biomarkers of
toxicity (116). Similarly,
another study used scRNA-seq data to identify CD19 expression in
brain wall cells and reveal the mechanism of neurotoxicity in
CD19-targeted therapy (117). And
it is promising that combining single-cell multi-omics techniques
would be beneficial in the diagnosis of lymphoma, especially if the
pathology is poorly characterized, as scRNA-seq identified immune
cell biomarkers and determined essential biomarkers such as CD31,
CD84 and CD226, which were hugely over-regulated and are promising
to be the diagnostic biomarkers for lymphoma (118).
At the same time, numerous new assays based on
single-cell sequencing are emerging, and investigators worldwide
are working toward an easier and a more accurate application of
single-cell technologies for lymphoma detection, which may offer
constructive options for lymphoma treatment. For example, transient
transfection of short barcode oligonucleotides allows the
estimation of drug targets by transcriptional patterns of multiple
scRNA-sequences to assess the efficacy and cytotoxicity of
different drugs (119). Currently,
a study is underway on the identification of targeted neoantigens
by applying scRNA-seq combined with TCR sequencing for single-cell
genomics (120). In the sequencing
of paired single-cell RNA and TCR in patient tumors before and
after PD-1 immunotherapy, the researchers identified the
co-expression of biomarkers of chronic T cell apoptosis (121). The profiles of various T cells and
TCR populations from normal adjacent tissues of tumors and
peripheral blood was investigated by in-depth scRNA-seq and TCR
sequencing from common types of tumors (122). RNA velocity can predict the future
changes of each cell on a timescale of hours through the direct
estimation of unsprayed and spliced mRNA from common scRNA-seq
protocols, and it will greatly assist in the analysis of
developmental lineages and cell dynamics in the future, which has
been shown experimentally to be also applicable to human lymphoma
(123). Mostly natural
sequencing-by-synthesis for scRNA-seq is a novel approach
applicable to the Ultima genomics platform, which has been
benchmarked against scRNA-seq technologies and has a broader
application potential (124).
In addition to the aforementioned novel single-cell
technologies that have been applied to lymphoma, numerous other
technologies in the field of oncology may have application
potential in lymphoma, including CITE-seq, which enables
simultaneous comparison of protein abundance and mRNA expression in
specimens (34). In addition,
SUPeR-seq synchronously detects linear and circ-rRNA expression
levels in the tumor cell and the other control cell (125), whereas G&T-seq describes the
whole genome and transcriptome of any sample cell (126). scTrio-seq simultaneously profiles
the genomic, transcriptomic and epigenomic status of a single tumor
cell (127), whereas INS-seq, a
synthesis technique that enables large-scale parallel collection of
scRNA-seq data and detection of intracellular protein activity,
holds promise in exploring new immune subpopulations by analyzing
different intracellular immune signaling signatures as well as
metabolic activity and transcription factor binding (128). Currently, SCT provides a favorable
platform for cancer diagnosis through the development of specific
tumor biomarkers and individualized tumor therapy, and this
technology will revolutionize the prognosis and treatment of all
types of cancer on a global scale (129,130). MULTI-seq involves the multiplexed
analysis of scRNA-sequences and mononuclear RNA sequences via
lipid-labelled indexing, which can barcode all cells and nuclei
worldwide through accessible plasma membranes. This approach is
currently being used in triple-negative breast cancer (131).
The future of single-cell technology is unlimited,
as evidenced by the increasing number of new techniques and
improvements that are emerging (132). For example, RAGE-seq reconstructs
highly diverse sequences (49). To
advance lymphoma research and address lymphoma heterogeneity,
additional integrated single-cell multi-omics technologies are
essential to understand the cellular (tumor, stromal and immune)
information about transcriptomes, proteomes and epigenomes, and
even predict the state of each cell via pseudo-time detection of
abundant patient data. In the near future, it may be possible to
routinely analyze millions of cells; as a case in point, a pilot
study to characterize a human cell atlas with 35 trillion human
cells has reportedly begun (133).
Through multiple fusion approaches and technological
innovations, single-cell transcriptomics techniques can assess how
individual genetic drivers differently contribute to the fitness of
lymphoma cells and investigate the mechanisms of cellular
mutational symbiosis, multiple lesion interactions and clonal
evolution, as revealed by spatial transcriptomics and spatial
proteomics (134). Single-cell
spatial transcription technologies have confirmed the upregulated
expression of the major ligands CCR4, CCL17 and CCL22, and the
downregulated expression of certain biomarkers in NK cells and CTL
cells (135). Single-cell spatial
analysis was applied to the DZ/LZ region of GCs in the tested lymph
nodes, showing that MHC I/II expression and EZH2 mutation frequency
were different within this microenvironment, which could assist in
the diagnosis of GC lymphoma (136). And to reveal deeper analyses of
tumor-environment interactions, the spatial analysis identified
CXCR3 as biomarkers suggesting immune desert region, and also
indicated therapeutic targets such as CCR4 and TIM-3, which are
associated with combination treatment assays of lymphoma cellular
therapies (137). Although
knowledge of lymphoma spatial histology is currently in a state of
infancy, there already exists a glimpse of the great potential of
the combined application of single-cell multi-omics. Convergence
and cross-analysis of multi-omics on top of different histologic
assays may be a more important direction of development, and it is
expected that multi-omics research will change the treatment
paradigm of lymphoma in the future, and improve guiding the choice
of clinical treatments. With the help of single-cell technology,
this revolution is expanding throughout the field of immunology,
not just lymphoma (34,138).
Feedback on the developed single-cell multi-omics
platforms will be provided clinically through advanced cancer risk
stratification, which is likely to deliver accurate lymphoma
prognosis and detailed individual treatment.
Not applicable.
The present study was supported by the Natural Science
Foundation of Jilin (grant no. YDZJ202201ZYTS117).
Not applicable.
CJ and DZ authored or reviewed drafts of the paper,
and approved the final draft. JL provided figures and helped with
proofreading of draft. LB and LL prepared tables, and approved the
final draft. All authors read and approved the final version of the
manuscript. Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Ysebaert L, Quillet-Mary A, Tosolini M,
Pont F, Laurent C and Fournié JJ: Lymphoma heterogeneity unraveled
by single-cell transcriptomics. Front Immunol. 12:5976512021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bocci F, Gearhart-Serna L, Boareto M,
Ribeiro M, Ben-Jacob E, Devi GR, Levine H, Onuchic JN and Jolly MK:
Toward understanding cancer stem cell heterogeneity in the tumor
microenvironment. Proc Natl Acad Sci USA. 116:148–157. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lei Y, Tang R, Xu J, Wang W, Zhang B, Liu
J, Yu X and Shi S: Applications of single-cell sequencing in cancer
research: Progress and perspectives. J Hematol Oncol. 14:912021.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Alizadeh AA, Eisen MB, Davis RE, Ma C,
Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al:
Distinct types of diffuse large B-cell lymphoma identified by gene
expression profiling. Nature. 403:503–511. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Newman AM, Liu CL, Green MR, Gentles AJ,
Feng W, Xu Y, Hoang CD, Diehn M and Alizadeh AA: Robust enumeration
of cell subsets from tissue expression profiles. Nat Methods.
12:453–457. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gentles AJ, Newman AM, Liu CL, Bratman SV,
Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, et al: The
prognostic landscape of genes and infiltrating immune cells across
human cancers. Nat Med. 21:938–945. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kothalawala WJ, Barták BK, Nagy ZB,
Zsigrai S, Szigeti KA, Valcz G, Takács I, Kalmár A and Molnár B: A
detailed overview about the single-cell analyses of solid tumors
focusing on colorectal cancer. Pathol Oncol Res. 28:16103422022.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bingham GC, Lee F, Naba A and Barker TH:
Spatial-omics: Novel approaches to probe cell heterogeneity and
extracellular matrix biology. Matrix Biol. 91–92. 152–166.
2020.
|
|
10
|
Borcherding N, Voigt AP, Liu V, Link BK,
Zhang W and Jabbari A: Single-Cell profiling of cutaneous T-Cell
lymphoma reveals underlying heterogeneity associated with disease
progression. Clin Cancer Res. 25:2996–3005. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gaydosik AM, Tabib T, Geskin LJ, Bayan CA,
Conway JF, Lafyatis R and Fuschiotti P: Single-Cell lymphocyte
heterogeneity in advanced cutaneous T-cell lymphoma skin tumors.
Clin Cancer Res. 25:4443–4454. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang W, Yang B, Weng L, Li J, Bai J, Wang
T, Wang J, Ye J, Jing H, Jiao Y, et al: Single cell sequencing
reveals cell populations that predict primary resistance to
imatinib in chronic myeloid leukemia. Aging (Albany NY).
12:25337–25355. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ren J, Qu R, Rahman NT, Lewis JM, King
ALO, Liao X, Mirza FN, Carlson KR, Huang Y, Gigante S, et al:
Integrated transcriptome and trajectory analysis of cutaneous
T-cell lymphoma identifies putative precancer populations. Blood
Adv. 7:445–457. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yamagishi M, Kubokawa M, Kuze Y, Suzuki A,
Yokomizo A, Kobayashi S, Nakashima M, Makiyama J, Iwanaga M, Fukuda
T, et al: Chronological genome and single-cell transcriptome
integration characterizes the evolutionary process of adult T cell
leukemia-lymphoma. Nat Commun. 12:48212021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Haebe S, Shree T, Sathe A, Day G,
Czerwinski DK, Grimes SM, Lee H, Binkley MS, Long SR, Martin B, et
al: Single-cell analysis can define distinct evolution of tumor
sites in follicular lymphoma. Blood. 137:2869–2880. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Borcherding N, Severson KJ, Henderson N,
Ortolan LS, Rosenthal AC, Bellizzi AM, Liu V, Link BK, Mangold AR
and Jabbari A: Single-cell analysis of Sézary syndrome reveals
novel markers and shifting gene profiles associated with treatment.
Blood Adv. 7:321–335. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Valentin Hansen S, Høy Hansen M, Cédile O,
Møller MB, Haaber J, Abildgaard N and Guldborg Nyvold C: Detailed
characterization of the transcriptome of single B cells in mantle
cell lymphoma suggesting a potential use for SOX4. Sci Rep.
11:190922021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pritchett JC, Yang ZZ, Kim HJ, Villasboas
JC, Tang X, Jalali S, Cerhan JR, Feldman AL and Ansell SM:
High-dimensional and single-cell transcriptome analysis of the
tumor microenvironment in angioimmunoblastic T cell lymphoma
(AITL). Leukemia. 36:165–176. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wei B, Liu Z, Fan Y, Wang S, Dong C, Rao
W, Yang F, Cheng G and Zhang J: Analysis of cellular heterogeneity
in immune microenvironment of primary central nervous system
lymphoma by single-cell sequencing. Front Oncol. 11:6830072021.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Macosko EZ, Basu A, Satija R, Nemesh J,
Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck
EM, et al: Highly parallel genome-wide expression profiling of
individual cells using nanoliter droplets. Cell. 161:1202–1214.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Klein AM, Mazutis L, Akartuna I,
Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA and Kirschner
MW: Droplet barcoding for single-cell transcriptomics applied to
embryonic stem cells. Cell. 161:1187–1201. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zheng GX, Terry JM, Belgrader P, Ryvkin P,
Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et
al: Massively parallel digital transcriptional profiling of single
cells. Nat Commun. 8:140492017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gong H, Do D and Ramakrishnan R:
Single-Cell mRNA-Seq using the fluidigm C1 system and integrated
fluidics circuits. Methods Mol Biol. 1783:193–207. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Han X, Chen H, Huang D, Chen H, Fei L,
Cheng C, Huang H, Yuan GC and Guo G: Mapping human pluripotent stem
cell differentiation pathways using high throughput single-cell
RNA-sequencing. Genome Biol. 19:472018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ziegenhain C, Vieth B, Parekh S, Reinius
B, Guillaumet-Adkins A, Smets M, Leonhardt H, Heyn H, Hellmann I
and Enard W: Comparative analysis of single-cell RNA sequencing
methods. Mol Cell. 65:631–643.e4. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xin Y, Kim J, Ni M, Wei Y, Okamoto H, Lee
J, Adler C, Cavino K, Murphy AJ, Yancopoulos GD, et al: Use of the
Fluidigm C1 platform for RNA sequencing of single mouse pancreatic
islet cells. Proc Natl Acad Sci USA. 113:3293–3298. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gierahn TM, Wadsworth MH II, Hughes TK,
Bryson BD, Butler A, Satija R, Fortune S, Love JC and Shalek AK:
Seq-Well: Portable, low-cost RNA sequencing of single cells at high
throughput. Nat Methods. 14:395–398. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Aicher TP, Carroll S, Raddi G, Gierahn T,
Wadsworth MH II, Hughes TK, Love C and Shalek AK: Seq-Well: A
sample-efficient, portable picowell platform for massively parallel
single-cell RNA sequencing. Methods Mol Biol. 1979:111–132. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Han X, Wang R, Zhou Y, Fei L, Sun H, Lai
S, Saadatpour A, Zhou Z, Chen H, Ye F, et al: Mapping the mouse
cell atlas by microwell-seq. Cell. 173:13072018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lai S, Huang W, Xu Y, Jiang M, Chen H,
Cheng C, Lu Y, Huang H, Guo G and Han X: Comparative transcriptomic
analysis of hematopoietic system between human and mouse by
Microwell-seq. Cell Discov. 4:342018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Briggs JA, Weinreb C, Wagner DE, Megason
S, Peshkin L, Kirschner MW and Klein AM: The dynamics of gene
expression in vertebrate embryogenesis at single-cell resolution.
Science. 360:eaar57802018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tosches MA, Yamawaki TM, Naumann RK,
Jacobi AA, Tushev G and Laurent G: Evolution of pallium,
hippocampus, and cortical cell types revealed by single-cell
transcriptomics in reptiles. Science. 360:881–888. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jaitin DA, Kenigsberg E, Keren-Shaul H,
Elefant N, Paul F, Zaretsky I, Mildner A, Cohen N, Jung S, Tanay A
and Amit I: Massively parallel single-cell RNA-seq for marker-free
decomposition of tissues into cell types. Science. 343:776–779.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Stoeckius M, Hafemeister C, Stephenson W,
Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R and Smibert
P: Simultaneous epitope and transcriptome measurement in single
cells. Nat Methods. 14:865–868. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mimitou EP, Cheng A, Montalbano A, Hao S,
Stoeckius M, Legut M, Roush T, Herrera A, Papalexi E, Ouyang Z, et
al: Multiplexed detection of proteins, transcriptomes, clonotypes
and CRISPR perturbations in single cells. Nat Methods. 16:409–412.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Harris NL, Jaffe ES, Stein H, Banks PM,
Chan JK, Cleary ML, Delsol G, De Wolf-Peeters C, Falini B, Gatter
KC, et al: A revised European-American classification of lymphoid
neoplasms: A proposal from the International Lymphoma Study Group.
Blood. 84:1361–1392. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Barbui T, Thiele J, Gisslinger H,
Kvasnicka HM, Vannucchi AM, Guglielmelli P, Orazi A and Tefferi A:
The 2016 WHO classification and diagnostic criteria for
myeloproliferative neoplasms: Document summary and in-depth
discussion. Blood Cancer J. 8:152018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Glaser SL and Jarrett RF: The epidemiology
of Hodgkin's disease. Baillieres Clin Haematol. 9:401–416. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Marafioti T, Hummel M, Foss HD, Laumen H,
Korbjuhn P, Anagnostopoulos I, Lammert H, Demel G, Theil J, Wirth T
and Stein H: Hodgkin and reed-sternberg cells represent an
expansion of a single clone originating from a germinal center
B-cell with functional immunoglobulin gene rearrangements but
defective immunoglobulin transcription. Blood. 95:1443–1450. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kanzler H, Küppers R, Hansmann ML and
Rajewsky K: Hodgkin and Reed-Sternberg cells in Hodgkin's disease
represent the outgrowth of a dominant tumor clone derived from
(crippled) germinal center B cells. J Exp Med. 184:1495–1505. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Grimm KE and O'Malley DP: Aggressive B
cell lymphomas in the 2017 revised WHO classification of tumors of
hematopoietic and lymphoid tissues. Ann Diagn Pathol. 38:6–10.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Thandra KC, Barsouk A, Saginala K, Padala
SA, Barsouk A and Rawla P: Epidemiology of Non-Hodgkin's Lymphoma.
Med Sci (Basel). 9:52021.PubMed/NCBI
|
|
43
|
de Leval L and Jaffe ES: Lymphoma
Classification. Cancer J. 26:176–185. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
García-Sanz R and Jiménez C: Time to move
to the single-cell level: Applications of single-cell multi-omics
to hematological malignancies and Waldenström's Macroglobulinemia-A
particularly heterogeneous lymphoma. Cancers (Basel). 13:15412021.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Glass DR, Tsai AG, Oliveria JP, Hartmann
FJ, Kimmey SC, Calderon AA, Borges L, Glass MC, Wagar LE, Davis MM
and Bendall SC: An Integrated Multi-omic Single-cell atlas of human
B cell identity. Immunity. 53:217–232.e5. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Crinier A, Dumas PY, Escalière B,
Piperoglou C, Gil L, Villacreces A, Vély F, Ivanovic Z, Milpied P,
Narni-Mancinelli É and Vivier É: Single-cell profiling reveals the
trajectories of natural killer cell differentiation in bone marrow
and a stress signature induced by acute myeloid leukemia. Cell Mol
Immunol. 18:1290–1304. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Aoki T, Chong LC, Takata K, Milne K, Hav
M, Colombo A, Chavez EA, Nissen M, Wang X, Miyata-Takata T, et al:
Single-Cell transcriptome analysis reveals disease-defining T-cell
subsets in the tumor microenvironment of classic Hodgkin Lymphoma.
Cancer Discov. 10:406–421. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Pizzolato G, Kaminski H, Tosolini M,
Franchini DM, Pont F, Martins F, Valle C, Labourdette D, Cadot S,
Quillet-Mary A, et al: Single-cell RNA sequencing unveils the
shared and the distinct cytotoxic hallmarks of human TCRVδ1 and
TCRVδ2 γδ T lymphocytes. Proc Natl Acad Sci USA. 116:11906–11915.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Singh M, Al-Eryani G, Carswell S, Ferguson
JM, Blackburn J, Barton K, Roden D, Luciani F, Giang Phan T,
Junankar S, et al: High-throughput targeted long-read single cell
sequencing reveals the clonal and transcriptional landscape of
lymphocytes. Nat Commun. 10:31202019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Vitak SA, Torkenczy KA, Rosenkrantz JL,
Fields AJ, Christiansen L, Wong MH, Carbone L, Steemers FJ and Adey
A: Sequencing thousands of single-cell genomes with combinatorial
indexing. Nat Methods. 14:302–328. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Xiong J, Cui BW, Wang N, Dai YT, Zhang H,
Wang CF, Zhong HJ, Cheng S, Ou-Yang BS, Hu Y, et al: Genomic and
transcriptomic characterization of natural killer T cell lymphoma.
Cancer Cell. 37:403–419.e6. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Li P, Chai J, Chen Z, Liu Y, Wei J, Liu Y,
Zhao D, Ma J, Wang K, Li X, et al: Genomic mutation profile of
primary gastrointestinal diffuse large B-Cell Lymphoma. Front
Oncol. 11:6226482021. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Radke J, Ishaque N, Koll R, Gu Z, Schumann
E, Sieverling L, Uhrig S, Hübschmann D, Toprak UH, López C, et al:
The genomic and transcriptional landscape of primary central
nervous system lymphoma. Nat Commun. 13:25582022. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yi S, Yan Y, Jin M, Bhattacharya S, Wang
Y, Wu Y, Yang L, Gine E, Clot G, Chen L, et al: Genomic and
transcriptomic profiling reveals distinct molecular subsets
associated with outcomes in mantle cell lymphoma. J Clin Invest.
132:e1532832022. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Nadeu F, Martin-Garcia D, Clot G,
Díaz-Navarro A, Duran-Ferrer M, Navarro A, Vilarrasa-Blasi R, Kulis
M, Royo R, Gutiérrez-Abril J, et al: Genomic and epigenomic
insights into the origin, pathogenesis, and clinical behavior of
mantle cell lymphoma subtypes. Blood. 136:1419–1432. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
De Bie J, Demeyer S, Alberti-Servera L,
Geerdens E, Segers H, Broux M, De Keersmaecker K, Michaux L,
Vandenberghe P, Voet T, et al: Single-cell sequencing reveals the
origin and the order of mutation acquisition in T-cell acute
lymphoblastic leukemia. Leukemia. 32:1358–1369. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
López C, Kleinheinz K, Aukema SM, Rohde M,
Bernhart SH, Hübschmann D, Wagener R, Toprak UH, Raimondi F, Kreuz
M, et al: Genomic and transcriptomic changes complement each other
in the pathogenesis of sporadic Burkitt lymphoma. Nat Commun.
10:14592019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Küçük C, Hu X, Gong Q, Jiang B, Cornish A,
Gaulard P, McKeithan T and Chan WC: Diagnostic and biological
significance of KIR EXPRESSION PROFILE DETErmined by RNA-Seq in
Natural Killer/T-Cell Lymphoma. Am J Pathol. 186:1435–1441. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Andor N, Simonds EF, Czerwinski DK, Chen
J, Grimes SM, Wood-Bouwens C, Zheng GXY, Kubit MA, Greer S, Weiss
WA, et al: Single-cell RNA-Seq of follicular lymphoma reveals
malignant B-cell types and coexpression of T-cell immune
checkpoints. Blood. 133:1119–1129. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Huang Z, Ma L, Huang C, Li Q and Nice EC:
Proteomic profiling of human plasma for cancer biomarker discovery.
Proteomics. 17((6))2017.
|
|
61
|
Kim MK, Song JY, Koh DI, Kim JY, Hatano M,
Jeon BN, Kim MY, Cho SY, Kim KS and Hur MW: Reciprocal negative
regulation between the tumor suppressor protein p53 and B cell
CLL/lymphoma 6 (BCL6) via control of caspase-1 expression. J Biol
Chem. 294:299–313. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Daniunaite K, Jarmalaite S and Kriukiene
E: Epigenomic technologies for deciphering circulating tumor DNA.
Curr Opin Biotechnol. 55:23–29. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ng SB, Yan J, Huang G, Selvarajan V, Tay
JL, Lin B, Bi C, Tan J, Kwong YL, Shimizu N, et al: Dysregulated
microRNAs affect pathways and targets of biologic relevance in
nasal-type natural killer/T-cell lymphoma. Blood. 118:4919–4929.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Zhang X, Ji W, Huang R, Li L, Wang X, Li
L, Fu X, Sun Z, Li Z, Chen Q and Zhang M: MicroRNA-155 is a
potential molecular marker of natural killer/T-cell lymphoma.
Oncotarget. 7:53808–53819. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Yan J, Ng SB, Tay JL, Lin B, Koh TL, Tan
J, Selvarajan V, Liu SC, Bi C, Wang S, et al: EZH2 overexpression
in natural killer/T-cell lymphoma confers growth advantage
independently of histone methyltransferase activity. Blood.
121:4512–4520. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Liang L, Nong L, Zhang S, Zhao J, Ti H,
Dong Y, Zhang B and Li T: The downregulation of PRDM1/Blimp-1 is
associated with aberrant expression of miR-223 in extranodal
NK/T-cell lymphoma, nasal type. J Exp Clin Cancer Res. 33:72014.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Küçük C, Hu X, Jiang B, Klinkebiel D, Geng
H, Gong Q, Bouska A, Iqbal J, Gaulard P, McKeithan TW and Chan WC:
Global promoter methylation analysis reveals novel candidate tumor
suppressor genes in natural killer cell lymphoma. Clin Cancer Res.
21:1699–1711. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Chen YW, Guo T, Shen L, Wong KY, Tao Q,
Choi WW, Au-Yeung RK, Chan YP, Wong ML, Tang JC, et al:
Receptor-type tyrosine-protein phosphatase κ directly targets STAT3
activation for tumor suppression in nasal NK/T-cell lymphoma.
Blood. 125:1589–1600. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Ranzoni AM, Tangherloni A, Berest I, Riva
SG, Myers B, Strzelecka PM, Xu J, Panada E, Mohorianu I, Zaugg JB
and Cvejic A: Integrative Single-Cell RNA-Seq and ATAC-seq analysis
of human developmental hematopoiesis. Cell Stem Cell.
28:472–487.e7. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Yu J, Gemenetzis G, Kinny-Köster B, Habib
JR, Groot VP, Teinor J, Yin L, Pu N, Hasanain A, van Oosten F, et
al: Pancreatic circulating tumor cell detection by targeted
single-cell next-generation sequencing. Cancer Lett. 493:245–253.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Zhang L, Dong X, Lee M, Maslov AY, Wang T
and Vijg J: Single-cell whole-genome sequencing reveals the
functional landscape of somatic mutations in B lymphocytes across
the human lifespan. Proc Natl Acad Sci USA. 116:9014–9019. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Psatha K, Kollipara L, Voutyraki C,
Divanach P, Sickmann A, Rassidakis GZ, Drakos E and Aivaliotis M:
Deciphering lymphoma pathogenesis via state-of-the-art mass
spectrometry-based quantitative proteomics. J Chromatogr B Analyt
Technol Biomed Life Sci. 1047:2–14. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Bacher U, Haferlach T, Alpermann T, Kern
W, Schnittger S and Haferlach C: Several lymphoma-specific genetic
events in parallel can be found in mature B-cell neoplasms. Genes
Chromosomes Cancer. 50:43–50. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Okosun J, Bödör C, Wang J, Araf S, Yang
CY, Pan C, Boller S, Cittaro D, Bozek M, Iqbal S, et al: Integrated
genomic analysis identifies recurrent mutations and evolution
patterns driving the initiation and progression of follicular
lymphoma. Nat Genet. 46:176–181. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Sewastianik T, Prochorec-Sobieszek M,
Chapuy B and Juszczyński P: MYC deregulation in lymphoid tumors:
Molecular mechanisms, clinical consequences and therapeutic
implications. Biochim Biophys Acta. 1846:457–467. 2014.PubMed/NCBI
|
|
76
|
Rosenthal A and Rimsza L: Genomics of
aggressive B-cell lymphoma. Hematology Am Soc Hematol Educ Program.
2018:69–74. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Gawad C, Koh W and Quake SR: Dissecting
the clonal origins of childhood acute lymphoblastic leukemia by
single-cell genomics. Proc Natl Acad Sci USA. 111:17947–17952.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Snuderl M, Kolman OK, Chen YB, Hsu JJ,
Ackerman AM, Dal Cin P, Ferry JA, Harris NL, Hasserjian RP,
Zukerberg LR, et al: B-cell lymphomas with concurrent IGH-BCL2 and
MYC rearrangements are aggressive neoplasms with clinical and
pathologic features distinct from Burkitt lymphoma and diffuse
large B-cell lymphoma. Am J Surg Pathol. 34:327–340. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Huang W, Medeiros LJ, Lin P, Wang W, Tang
G, Khoury J, Konoplev S, Yin CC, Xu J, Oki Y and Li S:
MYC/BCL2/BCL6 triple hit lymphoma: A study of 40 patients with a
comparison to MYC/BCL2 and MYC/BCL6 double hit lymphomas. Mod
Pathol. 31:1470–1478. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Moore EM, Aggarwal N, Surti U and Swerdlow
SH: Further exploration of the complexities of large B-Cell
Lymphomas With MYC abnormalities and the importance of a blastoid
morphology. Am J Surg Pathol. 41:1155–1166. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Jiang Y, Redmond D, Nie K, Eng KW, Clozel
T, Martin P, Tan LH, Melnick AM, Tam W and Elemento O: Deep
sequencing reveals clonal evolution patterns and mutation events
associated with relapse in B-cell lymphomas. Genome Biol.
15:4322014. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Ding S, Chen X and Shen K: Single-cell RNA
sequencing in breast cancer: Understanding tumor heterogeneity and
paving roads to individualized therapy. Cancer Commun (Lond).
40:329–344. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Kim E, Hurtz C, Koehrer S, Wang Z,
Balasubramanian S, Chang BY, Müschen M, Davis RE and Burger JA:
Ibrutinib inhibits pre-BCR(+) B-cell acute lymphoblastic leukemia
progression by targeting BTK and BLK. Blood. 129:1155–1165. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Wang L, Mo S, Li X, He Y and Yang J:
Single-cell RNA-seq reveals the immune escape and drug resistance
mechanisms of mantle cell lymphoma. Cancer Biol Med. 17:726–739.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Marcus R, Imrie K, Solal-Celigny P,
Catalano JV, Dmoszynska A, Raposo JC, Offner FC, Gomez-Codina J,
Belch A, Cunningham D, et al: Phase III study of R-CVP compared
with cyclophosphamide, vincristine, and prednisone alone in
patients with previously untreated advanced follicular lymphoma. J
Clin Oncol. 26:4579–4586. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Lim SH, Vaughan AT, Ashton-Key M, Williams
EL, Dixon SV, Chan HT, Beers SA, French RR, Cox KL, Davies AJ, et
al: Fc gamma receptor IIb on target B cells promotes rituximab
internalization and reduces clinical efficacy. Blood.
118:2530–2540. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Zhang J, Dominguez-Sola D, Hussein S, Lee
JE, Holmes AB, Bansal M, Vlasevska S, Mo T, Tang H, Basso K, et al:
Disruption of KMT2D perturbs germinal center B cell development and
promotes lymphomagenesis. Nat Med. 21:1190–1198. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Arber DA, Orazi A, Hasserjian R, Thiele J,
Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M and Vardiman JW:
The 2016 revision to the World Health Organization classification
of myeloid neoplasms and acute leukemia. Blood. 127:2391–2405.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Swerdlow SH and Cook JR: As the world
turns, evolving lymphoma classifications-past, present and future.
Hum Pathol. 95:55–77. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Takagi M: DNA damage response and
hematological malignancy. Int J Hematol. 106:345–356. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Flinders C, Lam L, Rubbi L, Ferrari R,
Fitz-Gibbon S, Chen PY, Thompson M, Christofk H, B Agus D, Ruderman
D, et al: Epigenetic changes mediated by polycomb repressive
complex 2 and E2a are associated with drug resistance in a mouse
model of lymphoma. Genome Med. 8:542016. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Chiche J, Reverso-Meinietti J, Mouchotte
A, Rubio-Patiño C, Mhaidly R, Villa E, Bossowski JP, Proics E,
Grima-Reyes M, Paquet A, et al: GAPDH expression predicts the
response to R-CHOP, the tumor metabolic status, and the response of
DLBCL patients to metabolic inhibitors. Cell Metab.
29:1243–1257.e10. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Klener P and Klanova M: Drug Resistance in
Non-Hodgkin Lymphomas. Int J Mol Sci. 21:20812020. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Wang X, Li G, Zhang Y, Li L, Qiu L, Qian
Z, Zhou S, Wang X, Li Q and Zhang H: Pan-Cancer analysis reveals
genomic and clinical characteristics of TRPV Channel-related genes.
Front Oncol. 12:8131002022. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Russo M, Crisafulli G, Sogari A, Reilly
NM, Arena S, Lamba S, Bartolini A, Amodio V, Magrì A, Novara L, et
al: Adaptive mutability of colorectal cancers in response to
targeted therapies. Science. 366:1473–1480. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Kater L, Kater B, Jakupec MA, Keppler BK
and Prokop A: KP772 overcomes multiple drug resistance in malignant
lymphoma and leukemia cells in vitro by inducing Bcl-2-independent
apoptosis and upregulation of Harakiri. J Biol Inorg Chem.
26:897–907. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Sciarrillo R, Wojtuszkiewicz A, Assaraf
YG, Jansen G, Kaspers GJL, Giovannetti E and Cloos J: The role of
alternative splicing in cancer: From oncogenesis to drug
resistance. Drug Resist Updat. 53:1007282020. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Roider T, Seufert J, Uvarovskii A,
Frauhammer F, Bordas M, Abedpour N, Stolarczyk M, Mallm JP, Herbst
SA, Bruch PM, et al: Dissecting intratumour heterogeneity of nodal
B-cell lymphomas at the transcriptional, genetic and drug-response
levels. Nat Cell Biol. 22:896–906. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Wills QF, Livak KJ, Tipping AJ, Enver T,
Goldson AJ, Sexton DW and Holmes C: Single-cell gene expression
analysis reveals genetic associations masked in whole-tissue
experiments. Nat Biotechnol. 31:748–752. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Kotlov N, Bagaev A, Revuelta MV, Phillip
JM, Cacciapuoti MT, Antysheva Z, Svekolkin V, Tikhonova E,
Miheecheva N, Kuzkina N, et al: Clinical and Biological Subtypes of
B-cell lymphoma revealed by microenvironmental signatures. Cancer
Discov. 11:1468–1489. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Croci GA, Au-Yeung RKH, Reinke S, Staiger
AM, Koch K, Oschlies I, Richter J, Poeschel V, Held G, Loeffler M,
et al: SPARC-positive macrophages are the superior prognostic
factor in the microenvironment of diffuse large B-cell lymphoma and
independent of MYC rearrangement and double-/triple-hit status. Ann
Oncol. 32:1400–1409. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Abe Y, Sakata-Yanagimoto M, Fujisawa M,
Miyoshi H, Suehara Y, Hattori K, Kusakabe M, Sakamoto T, Nishikii
H, Nguyen TB, et al: A single-cell atlas of non-haematopoietic
cells in human lymph nodes and lymphoma reveals a landscape of
stromal remodelling. Nat Cell Biol. 24:565–578. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Ferreri AJ, Cwynarski K, Pulczynski E,
Ponzoni M, Deckert M, Politi LS, Torri V, Fox CP, Rosée PL, Schorb
E, et al: Chemoimmunotherapy with methotrexate, cytarabine,
thiotepa, and rituximab (MATRix regimen) in patients with primary
CNS lymphoma: Results of the first randomisation of the
International Extranodal Lymphoma Study Group-32 (IELSG32) phase 2
trial. Lancet Haematol. 3:e217–e227. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Bromberg JEC, Issa S, Bakunina K, Minnema
MC, Seute T, Durian M, Cull G, Schouten HC, Stevens WBC, Zijlstra
JM, et al: Rituximab in patients with primary CNS lymphoma (HOVON
105/ALLG NHL 24): A randomised, open-label, phase 3 intergroup
study. Lancet Oncol. 20:216–228. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Ruggieri S, Tamma R, Resta N, Albano F,
Coccaro N, Loconte D, Annese T, Errede M, Specchia G, Senetta R, et
al: Stat3-positive tumor cells contribute to vessels neoformation
in primary central nervous system lymphoma. Oncotarget.
8:31254–31269. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Zhou Y, Liu W, Xu Z, Zhu H, Xiao D, Su W,
Zeng R, Feng Y, Duan Y, Zhou J and Zhong M: Analysis of genomic
alteration in primary central nervous system lymphoma and the
expression of some related genes. Neoplasia. 20:1059–1069. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Ribatti D, Nico B, Ranieri G, Specchia G
and Vacca A: The role of angiogenesis in human non-Hodgkin
lymphomas. Neoplasia. 15:231–238. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Clozel T, Yang S, Elstrom RL, Tam W,
Martin P, Kormaksson M, Banerjee S, Vasanthakumar A, Culjkovic B,
Scott DW, et al: Mechanism-based epigenetic chemosensitization
therapy of diffuse large B-cell lymphoma. Cancer Discov.
3:1002–1019. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Hazar B, Paydas S, Zorludemir S, Sahin B
and Tuncer I: Prognostic significance of microvessel density and
vascular endothelial growth factor (VEGF) expression in
non-Hodgkin's lymphoma. Leuk Lymphoma. 44:2089–2093. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Carlo-Stella C and Santoro A:
Microenvironment-related biomarkers and novel targets in classical
Hodgkin's lymphoma. Biomark Med. 9:807–817. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Holmes AB, Corinaldesi C, Shen Q, Kumar R,
Compagno N, Wang Z, Nitzan M, Grunstein E, Pasqualucci L,
Dalla-Favera R and Basso K: Single-cell analysis of germinal-center
B cells informs on lymphoma cell of origin and outcome. J Exp Med.
217:e202004832020. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Mintz MA and Cyster JG: T follicular
helper cells in germinal center B cell selection and
lymphomagenesis. Immunol Rev. 296:48–61. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Turqueti-Neves A, Otte M, Prazeres da
Costa O, Höpken UE, Lipp M, Buch T and Voehringer D:
B-cell-intrinsic STAT6 signaling controls germinal center
formation. Eur J Immunol. 44:2130–2138. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Mintz MA, Felce JH, Chou MY, Mayya V, Xu
Y, Shui JW, An J, Li Z, Marson A, Okada T, et al: The HVEM-BTLA
Axis Restrains T cell help to germinal center B cells and functions
as a cell-extrinsic suppressor in lymphomagenesis. Immunity.
51:310–323.e7. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Hashwah H, Schmid CA, Kasser S, Bertram K,
Stelling A, Manz MG and Müller A: Inactivation of CREBBP expands
the germinal center B cell compartment, down-regulates MHCII
expression and promotes DLBCL growth. Proc Natl Acad Sci USA.
114:9701–9706. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Deng Q, Han G, Puebla-Osorio N, Ma MCJ,
Strati P, Chasen B, Dai E, Dang M, Jain N, Yang H, et al:
Characteristics of anti-CD19 CAR T cell infusion products
associated with efficacy and toxicity in patients with large B cell
lymphomas. Nat Med. 26:1878–1887. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Parker KR, Migliorini D, Perkey E, Yost
KE, Bhaduri A, Bagga P, Haris M, Wilson NE, Liu F, Gabunia K, et
al: Single-Cell analyses identify brain mural cells expressing CD19
as potential off-tumor targets for CAR-T immunotherapies. Cell.
183:126–142.e17. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Shi Y, Ding W, Gu W, Shen Y, Li H, Zheng
Z, Zheng X, Liu Y and Ling Y: Single-cell phenotypic profiling to
identify a set of immune cell protein biomarkers for relapsed and
refractory diffuse large B cell lymphoma: A single-center study. J
Leukoc Biol. 112:1633–1648. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Shin D, Lee W, Lee JH and Bang D:
Multiplexed single-cell RNA-seq via transient barcoding for
simultaneous expression profiling of various drug perturbations.
Sci Adv. 5:eaav22492019. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Krieg C, Nowicka M, Guglietta S, Schindler
S, Hartmann FJ, Weber LM, Dummer R, Robinson MD, Levesque MP and
Becher B: High-dimensional single-cell analysis predicts response
to anti-PD-1 immunotherapy. Nat Med. 24:144–153. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Yost KE, Satpathy AT, Wells DK, Qi Y, Wang
C, Kageyama R, McNamara KL, Granja JM, Sarin KY, Brown RA, et al:
Clonal replacement of tumor-specific T cells following PD-1
blockade. Nat Med. 25:1251–1259. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Wu TD, Madireddi S, de Almeida PE,
Banchereau R, Chen YJ, Chitre AS, Chiang EY, Iftikhar H, O'Gorman
WE, Au-Yeung A, et al: Peripheral T cell expansion predicts tumour
infiltration and clinical response. Nature. 579:274–278. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
La Manno G, Soldatov R, Zeisel A, Braun E,
Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lönnerberg
P, Furlan A, et al: RNA velocity of single cells. Nature.
560:494–498. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Simmons SK, Lithwick-Yanai G, Adiconis X,
Oberstrass F, Iremadze N, Geiger-Schuller K, Thakore PI, Frangieh
CJ, Barad O, Almogy G, et al: Mostly natural
sequencing-by-synthesis for scRNA-seq using Ultima sequencing. Nat
Biotechnol. 41:204–211. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Fan X, Zhang X, Wu X, Guo H, Hu Y, Tang F
and Huang Y: Single-cell RNA-seq transcriptome analysis of linear
and circular RNAs in mouse preimplantation embryos. Genome Biol.
16:1482015. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Macaulay IC, Haerty W, Kumar P, Li YI, Hu
TX, Teng MJ, Goolam M, Saurat N, Coupland P, Shirley LM, et al:
G&T-seq: Parallel sequencing of single-cell genomes and
transcriptomes. Nat Methods. 12:519–522. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Hou Y, Guo H, Cao C, Li X, Hu B, Zhu P, Wu
X, Wen L, Tang F, Huang Y and Peng J: Single-cell triple omics
sequencing reveals genetic, epigenetic, and transcriptomic
heterogeneity in hepatocellular carcinomas. Cell Res. 26:304–319.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Katzenelenbogen Y, Sheban F, Yalin A, Yofe
I, Svetlichnyy D, Jaitin DA, Bornstein C, Moshe A, Keren-Shaul H,
Cohen M, et al: Coupled scRNA-Seq and intracellular protein
activity reveal an immunosuppressive role of TREM2 in cancer. Cell.
182:872–885.e19. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Svensson V, Vento-Tormo R and Teichmann
SA: Exponential scaling of single-cell RNA-seq in the past decade.
Nat Protoc. 13:599–604. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Bai X, Li Y, Zeng X, Zhao Q and Zhang Z:
Single-cell sequencing technology in tumor research. Clin Chim
Acta. 518:101–109. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
McGinnis CS, Patterson DM, Winkler J,
Conrad DN, Hein MY, Srivastava V, Hu JL, Murrow LM, Weissman JS,
Werb Z, et al: MULTI-seq: Sample multiplexing for single-cell RNA
sequencing using lipid-tagged indices. Nat Methods. 16:619–626.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Kashima Y, Sakamoto Y, Kaneko K, Seki M,
Suzuki Y and Suzuki A: Single-cell sequencing techniques from
individual to multiomics analyses. Exp Mol Med. 52:1419–1427. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Ando Y, Kwon AT and Shin JW: An era of
single-cell genomics consortia. Exp Mol Med. 52:1409–1418. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Landeira-Viñuela A, Díez P, Juanes-Velasco
P, Lécrevisse Q, Orfao A, De Las Rivas J and Fuentes M: Deepening
into intracellular signaling landscape through integrative spatial
proteomics and transcriptomics in a lymphoma model. Biomolecules.
11:17762021. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Du J, Qiu C, Li WS, Wang B, Han XL, Lin
SW, Fu XH, Hou J and Huang ZF: Spatial transcriptomics analysis
reveals that CCL17 and CCL22 are robust indicators of a suppressive
immune environment in angioimmunoblastic T cell lymphoma (AITL).
Front Biosci (Landmark Ed). 27:2702022. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Tripodo C, Zanardi F, Iannelli F, Mazzara
S, Vegliante M, Morello G, Di Napoli A, Mangogna A, Facchetti F,
Sangaletti S, et al: A spatially resolved dark-versus light-zone
microenvironment signature subdivides germinal center-related
aggressive B cell lymphomas. iScience. 23:1015622020. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Colombo AR, Hav M, Singh M, Xu A, Gamboa
A, Lemos T, Gerdtsson E, Chen D, Houldsworth J, Shaknovich R, et
al: Single-cell spatial analysis of tumor immune architecture in
diffuse large B-cell lymphoma. Blood Adv. 6:4675–4690. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Efremova M, Vento-Tormo R, Park JE,
Teichmann SA and James KR: Immunology in the Era of single-cell
technologies. Annu Rev Immunol. 38:727–757. 2020. View Article : Google Scholar : PubMed/NCBI
|