Introduction
Cribriform morular thyroid carcinoma (CMTC) is a
type of TC associated with familial adenomatous polyposis (FAP),
which can also at times occur sporadically (1). This rare tumor was first identified in
patients with FAP as a distinctive follicular neoplasm different
from conventional follicular TC (FTC) and papillary TC (PTC),
usually presenting with multiple and bilateral tumor foci (2). Later, the sporadic counterpart of this
neoplasm was reported as a subtype of PTC (cribriform morular
variant) (3). In fact, both the
familial and sporadic forms of this tumor were designated as the
cribriform-morular variant of PTC in the 4th edition of the
classification of thyroid tumors of the World Health Organization
(WHO) (4). More recently, the
peculiar combination of growth patterns of CMTC has been associated
with the permanent activation of the Wnt/β-catenin signaling
pathway (5). Consequently, this
tumor has been included as a distinct TC in the most recent WHO
classification of endocrine and neuroendocrine tumors, within the
group of thyroid tumors of uncertain histogenesis (1).
As this new tumor entity remains poorly understood
(1,6–9), for
the present review, the MEDLINE-PubMed database (https://pubmed.ncbi.nlm.nih.gov) was searched for
entries added up to January 2024 with the search terms
‘cribriform’, ‘morular’ and ‘thyroid’ to evaluate the
epidemiological, clinicopathological, immunohistochemical and
molecular features of CMTC as well as its possible histogenesis.
Only papers written in English were reviewed. At the same time, the
study aimed to underscore the molecular characteristics of this
peculiar neoplasm as a basis for proposing an approach that is both
preventive and therapeutic.
Epidemiological characteristics
CMTC accounts for 0.16–0.5% of all cases of TC
(10,11) and 0.16–0.66% of the cases of PTC
(3,12,13).
Although a case of CMTC has been reported in a 15-year-old female
with hypothyroidism secondary to neck radiotherapy due to Hodgkin's
disease (14), no increased
prevalence of CMTC was detected in a population exposed to ionizing
radiation post-Chernobyl (11).
SARS-CoV-2 infection has been reported in one sporadic case of CMTC
and lymphocytic thyroiditis (15).
CMTC has been reported in 0.4–2.6% of patients with
FAP (16,17), but these percentages increased to
3–12% when ultrasonography (US) was used (17,18),
and it was detected in up to 16% of patients with FAP when US in
combination with fine needle aspiration cytology (FNAC) was carried
out (19). A higher risk and
incidence of CMTC in Hispanic patients with FAP have also been
reported (20). About 60% of cases
of CMTC occur in the setting of FAP and in 40% of these cases, it
is the first clinical manifestation of FAP (13,16,18,21–23).
Cases of CMTC and a conventional subtype of PTC in different
members of the same family sharing the same adenomatous polyposis
coli (APC) germline mutation have been described (18,24,25).
It is possible that cases of conventional PTC in these families are
actually cases of incidental PTC not related to the APC
germline mutation.
Clinical findings
Most patients with CMTC are euthyroid young women
(female-to-male ratio, ~61:1), and the mean age at diagnosis was
determined to be 25 years (range, 8–69 years), both for sporadic
and for FAP-associated CMTC (5,12,26).
CMTC may be incidentally detected during physical and/or US
examination with or without FNAC (19) or present as a painless mass,
dysphagia and/or hoarseness (14,27,28).
During US screening, most nodules are observed to be well-defined,
oval to round, hypoechoic and solid without calcifications (with
benign-looking features) (17,18,29,30).
While most cases of familial CMTC are multifocal (and bilateral) at
diagnosis, most sporadic cases present as single nodules (5,26,31).
Because congenital hypertrophy of the retinal pigment epithelium
(CHRPE) is reported to occur in up to 80% of individuals with FAP
(32,33), and ophthalmoscopic examination can
help to clinically confirm hereditary cases of CMTC (34).
In addition to colorectal adenomatous polyps and
cancer (33–35), the coexistence of TC with
medulloblastoma (36), stomach
polyps (37), duodenal polyps
(36,38), ampullary neoplasms (25,37),
hepatoblastoma (39), adrenal
adenoma (25), endometrial cancer
(40), osteomas (35), lipomas (35), sebaceous and epidermoid cysts
(35), desmoid tumors (25,35,38) as
well as bilateral breast fibromatosis in the context of silicone
prosthetics (41) have been
described in patients with FAP.
Pathological features
CMTC is encapsulated or well demarcated and often
partially lobulated by sclerotic septa (2,3).
Histologically, the tumor exhibits ‘an intricate blending of
cribriform, follicular, papillary, trabecular and solid patterns of
growth, with morular (squamoid) areas’ (3) (Fig.
1). In different cases of CMTC and even within the same tumor,
the percentage of these different growth patterns is variable. The
cribriform pattern is composed of tumor cells without interposed
stroma that merge with tubulo-glandular follicles (devoid of
colloid), as well as with papillae. The papillae are lined by
cuboidal or tall cells, often mimicking the tall-cell or
columnar-cell subtypes of PTC (3,8). There
is also continuity with areas of the trabecular pattern,
reminiscent of the hyalinizing trabecular tumor and areas of solid
patterns with spindle-like cells (1,3,5).
Psammoma bodies are rare. Nuclei are round to oval, clear and can
show irregular contours, grooves and pseudoinclusions. Morular
structures are nests of nonkeratinized cells having biotin-rich
nuclei with characteristic chromatin clearance (1,3,5).
Ultrastructurally, these clear morular nuclei have numerous aligned
quasiparallel filaments (10,42,43).
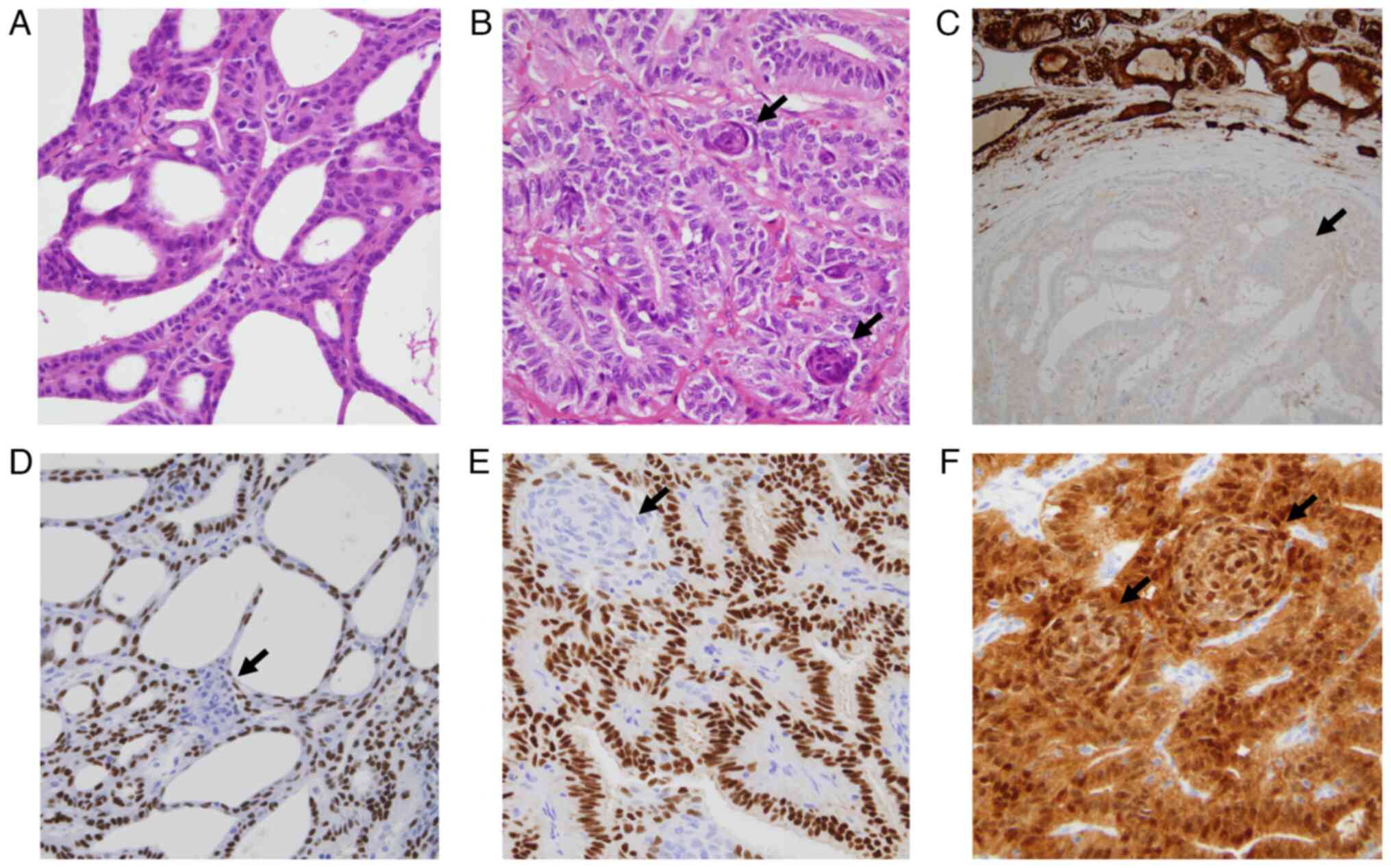 | Figure 1.Microscopic features of morular
cribriform thyroid carcinoma. (A) Characteristic cribriform and
follicular tumor growth pattern without colloid. (B) Tumor area
with predominance of trabecular and follicular patterns (without
colloid) and presence of morular structures (arrows). (C)
Well-defined tumor area showing predominance of the cribriform
pattern, a morular (squamoid) structure (arrow), papillae, lack of
colloid and thyroglobulin negativity. (D) Cribriform and papillary
tumor area with nuclear positivity for TTF1/NKX2-1 except in the
morular structure (arrow). (E) Diffuse nuclear positivity for
estrogen receptors in the areas of the tumor with a trabecular
growth pattern and negativity in the morular component (arrow). (F)
Tumor area with predominance of the trabecular, follicular growth
pattern and morular structures (arrows) showing diffuse nuclear and
cytoplasmic positivity for β-catenin (original magnification: A, C
and D, ×200×; B, E and F, ×400. Stains: A and B, hematoxylin-eosin;
C, thyroglobulin; D, TTF1/NKX2-1; E, estrogen receptors; and F,
β-catenin). TTF1/NKX2-1, NK2 homeobox 1. |
There are usually <5 mitotic figures per 2
mm2. Vascular and capsular invasion have been reported
in 30 and 40% of tumors, respectively (2,3,5,44–47).
Isolated cases with marked stromal hyalinization and calcification
(48), adamantinous-like pattern
(44), adenoid cystic
carcinoma-like areas (49) or focal
squamous differentiation (9) have
been reported.
According to the criteria of the new edition of the
WHO classification of thyroid tumors (1,6), rare
cases of ‘high-grade CMTC’ with necrosis and/or high proliferative
activity have also been reported (7,9,46,50–52).
Immunohistochemical profile
Strong diffuse nuclear and cytoplasmic
immunostaining for β-catenin is the hallmark of CMTC (1,5,7)
(Fig. 1). The immunohistochemical
profile of CMTC is summarized in Table
I. Tumor cells are always immunoreactive for thyroid
transcription factor-1 (TTF1)/NK2 homeobox 1 (NKX2-1) (clones
8G3G7/1 and SPT24) and keratin (KRT) (7), as well as negative for thyroglobulin
(Tg), calcitonin and KRT20 (1,5,53).
Variable staining for paired box-8 (PAX8) (clones MRQ-50 and SP348)
has been detected (5,7,8).
Strong positivity for estrogen receptor (ER) (Fig. 1) and progesterone receptor (PR) is
highly characteristic, but focal reactivity for androgen receptors
has also been detected (1,5,7,11,53).
The Ki-67 proliferative index is usually <5% (1,5).
 | Table I.Immunohistochemical profile of
morular cribriform thyroid carcinoma. |
Table I.
Immunohistochemical profile of
morular cribriform thyroid carcinoma.
| Marker | Main tumor
cells | Morular
structures | (Refs.) |
|---|
| β-catenin | + | + | (1,5,7,8,56) |
| TTF1/NKX2-1 | + | - | (1,5,7–9,56) |
| PAX8 | -/+a | - | (1,5,7,8) |
| Thyroglobulin | - | - | (1,5,8,56) |
|
Thyroperoxidase | - | - | (8) |
| Calcitonin | - | - | (1,5) |
| CEA | - | - | (5,8) |
| Chromogranin | -b | - | (5,44) |
| Synaptophysin | -b | - | (5,44) |
| β-estrogen
receptors | + | - | (1,5,8,11) |
| Progesterone
receptors | + | - | (1,5,8,11) |
| Androgen
receptors | +/- | - | (5) |
| KRT5 | - | + | (1,7) |
| KRT7 | + | +/- | (5,8) |
| KRT19 | + | - | (5) |
| KRT20 | - | - | (1,5,8) |
| KRT5/6 | + | + | (1,8) |
| KRT (clone
AE1/AE3) | + | + | (5) |
| KRT (clone
34βE12) | + | + | (1,5) |
| KRT (clone
CAM5.2) | + | + | (5) |
| EMA | + | NS | (1,3,5) |
| Vimentin | + | - | (5) |
| CA 19.9 | - | + | (5) |
| CA 125 | - | - | (5) |
| CDX2 | - | + | (1,5,8,56,57) |
| CD5 | - | + | (1,7–9) |
| CD10 | -/+ | + | (1,5,7–9) |
| CD117 (c-KIT) | -/+ | - | (5,8) |
| E-cadherin | + | -/+ | (1,5) |
| HBME1 | +/- | - | (5,7) |
| Galectin-3 | + | -/+ | (5,8) |
| APC | + | NS | (51) |
| LEF-1 | + | NS | (54) |
| BCL-2 | + | + | (5,44) |
| p27 | + | NS | (5,44) |
| Rb | + | NS | (3,5) |
| β-hCG | + | NS | (5,55) |
| NIS | - | - | (8) |
| EGFR | - | - | (5,7,8,44) |
| p53 | +/-c | NS | (7,8) |
| p63 | - | - | (5,7,8) |
| p40 | -d | - | (7–9) |
| Calretinin | - | - | (5) |
| WT1 | - | - | (1,5,44) |
| SATB2 | - | - | (8) |
Morular structures also show aberrant nuclear and
cytoplasmic positivity for β-catenin (Fig. 1) and can be easily identified in the
tumor by their characteristic positivity for caudal type homeobox 2
(CDX2), CA19.9, CD10, CD5 and KRT5 (3,5,7,56,57).
Follicular lineage
CMTC has been initially considered a distinct type
of follicular cell neoplasm associated with FAP (2). According to its nuclear
characteristics and the immunohistochemical data (particularly the
KRT profile), this tumor was later considered a variant of PTC
(3,4). Of note, RET rearrangements, a
molecular alteration typical of PTC, have also been described in
certain cases of CMTC (44,40,58).
Despite the features shared by PTC and CMTC, our group postulated
that the latter deserves to be considered its own tumor category,
CMTC, based on its ‘peculiar primitive endodermal (intestinal-like)
type phenotype and permanent activation of the wingless
(Wnt/β-catenin) signaling pathway’ (5). Different researchers have proposed
that CMTC arises due to germline or somatic APC mutations or
because of somatic mutations in functionally equivalent genes
related to the Wnt/β-catenin signaling pathway (5,58–60).
The immunohistochemical profile (KRT pattern,
positivity for TTF1, variable staining for PAX8 and negativity for
calcitonin and CEA), fits with a primary non-neuroendocrine
epithelial neoplasm of the thyroid gland. A conclusive follicular
cell derivation has not been demonstrated, however, due to
negativity for Tg (both at the protein and mRNA levels),
thyroperoxidase and solute carrier family 5 member 5 (NIS)
(8).
A putative thymic/ultimobranchial pouch-related
differentiation has also been proposed due to the lack of Tg in the
tumor cells, the occasional negativity for PAX8 and the phenotype
of the morular component, positivity for KRT5 and CD5 and
negativity for p63, p40, TTF1 and PAX8 (7). Others (8), however, considered that the negativity
for p40 and p63 described by Boyraz et al (7) in the same series contradicts their own
hypothesis due to the recognized positivity for p40 and p63 in both
thymic tissue and intrathyroidal remains of the ultimobranchial
body (solid cell nests) (61). A
relation to the ultimobranchial body has also been proposed in a
unique case of CMTC based on positivity for p40 in the poorly
differentiated component (9); in
this case, however, the negativity for p40 in the differentiated
component of the tumor (including the morulae) as well as the total
negativity for CD5 argue against both the ultimobranchial and the
thymic derivations, respectively. Another case, however, supports
an origin from follicular cells (or their endodermal precursors)
based on the cytopathological similarities of this tumor with
classic PTC (8).
Morular structures (positive for CD10 and CDX2) have
been the subject of different interpretations (62–64).
Of note, they are characteristically present in a series of tumors
such as fetal adenocarcinoma of the lung (65), pancreatoblastoma (66), mesonephric-like adenocarcinoma of
the ovary (67), colorectal polyps
(68) and others (69), all of which share the permanent
activation of the Wnt/β-catenin pathway. It is recognized that the
activation of Wnt/β-catenin signaling, through transcription factor
CDX2, activates small intestine gene expression at low levels and
colonic gene expression at higher levels (70). These mechanisms of embryonic
intestinal induction would explain both the blockage in the
terminal/follicular differentiation of follicular cells (or their
precursor cells) in CMTC, as well as its phenotype. CMTC would be
another example of TC with endodermal/intestinal-like
(non-committed) differentiation (71). In fact, the immunophenotype of CMTC
cells fits into the progenitor stem cell phase in its continuum
toward thyroid follicular cells, according to a recent study
(72).
In summary, the molecular alterations linked to the
constitutive activation of the Wnt/β-catenin pathway are consistent
with the development and phenotype of the CMTC from follicular
cells (5). Additional points
supporting an origin of CMTC from follicular cells (or their
precursor cells) are as follows (8): a) The cytoarchitectural and
immunohistochemical similarities with other neoplasms of follicular
lineage, particularly with PTC; and b) the multicentricity of CMTC
associated with patients with FAP (usually with molecularly
different tumors), which is easier to explain when they are derived
from different follicular cells (or precursor thyroid cells) than
from multiple intrathyroid thymic or branchial remnants. Of note, a
light increase in Tg levels along with a slight iodine uptake in
pulmonary metastases of CMTC after treatment with 131I
has been described (73).
Furthermore, a rapid increase in serum Tg levels parallel to
histologically confirmed lung metastases has also been reported in
another case of CMTC after treatment with 131I and a
selective inhibitor of receptor tyrosine kinases (lenvatinib)
(74). It is still necessary to
carry out functional studies on Wnt/β-catenin pathway inactivation
in primary CMTC cell cultures to confirm (or discard) this
hypothesis (8).
Pathogenesis
FAP is an autosomal dominant genetic disorder caused
by germline mutations in the APC gene (34). The constitutive activation of the
Wnt/β-catenin signaling pathway has a central role in the
pathogenesis of CMTC, mainly through inactivating mutations in the
APC, CTNNB1 and AXIN1 genes (5) (Fig.
2). Additional molecular alterations may have a synergistic
effect on this pathway, while sex hormones appear to play a
promoting role in CMTC development, which is elaborated on further
below.
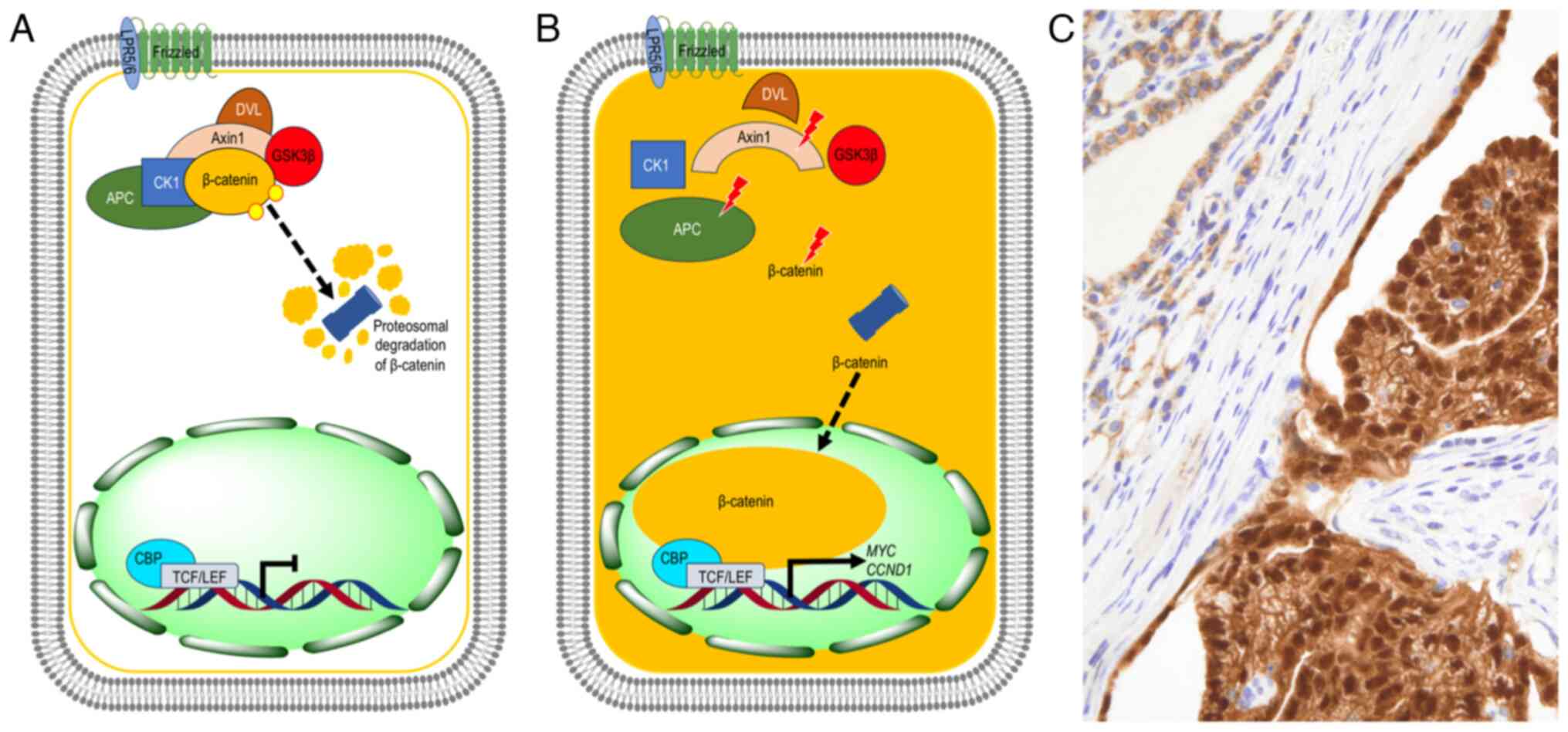 | Figure 2.Schematic representation of the WNT
signaling pathway in CMTC. (A) In normal thyroid cells, the
canonical (β-catenin-dependent) WNT signaling is in the ‘off’ state
because the APC protein, together with the scaffolding
protein Axin, serine/threonine kinases CK1 and GSK3β, and
β-catenin, form a ‘destruction complex’ that phosphorylates
β-catenin, thus promoting its ubiquitination and proteasomal
degradation. (B) In CMTC cells, different somatic mutations in
APC, CTNNB1 and/or AXIN1 genes alter the destruction
complex, impairing β-catenin phosphorylation and degradation in the
proteasome. β-catenin accumulates in the cytoplasm and subsequently
translocates to the nucleus, where it binds to the TCF/LEF family
to activate the transcription of target genes involved in
proliferation and loss of differentiation, such as MYC and
CCND1. (C) Localization of β-catenin in CMTC. Tumor cells
(right) show characteristic nuclear and cytoplasmic positivity for
β-catenin, while non-tumor cells (left) show membrane positivity
with discrete cytoplasmic immunostaining (original magnification,
×400). CMTC, cribriform morular thyroid carcinoma; TCF/LEF, T-cell
factor/lymphoid enhancer factor; CCND1, cyclin D1; APC, APC
regulator of WNT signaling pathway; CK1, casein kinase 1; GSK3β,
glycogen synthase kinase 3β; DVL, disheveled; CBP, cyclic adenosine
monophosphate responsive element binding protein 1 binding protein;
LRP5/6, low-density lipoprotein receptor-related protein 5/6. |
CMTC and FAP
More than 85% of germline APC mutations in
patients with CMTC have been detected in exon 15 (codons 463 to
1,387), in the same genomic location usually associated with CHRPE
(39). This area also includes a
hotspot (codon 1,061) for CMTC and hepatoblastoma (25,35,37,39),
but mutations in codons 140, 159, 161, 213, 278, 302, 313, 325,
332, 418, 471, 499, 554, 578, 582, 593, 625, 654, 698, 704, 737,
769, 778, 804, 834, 848, 935, 937, 938, 964, 976, 977, 979, 993,
1,062, 1,068, 1,073, 1,105, 1,110, 1,157, 1,275, 1,307, 1,309,
1,394, 1,465 and 2,092 have also been described (33,56,75,76).
When comparing the prevalence of APC mutations in patients
with FAP and TC in relation to the prevalence of such mutations in
unselected individuals with FAP, a higher risk of CMTC exists in
the population harboring APC mutations proximal to the 5′
end (proximal to codon 528) as well as in the established high-risk
group with mutation at codon 1,061 (75). It is noteworthy that part of the
β-catenin binding sites and the axin binding sites are outside the
mutation cluster region (codons 1,286 to 1,513) of the APC
gene (7).
When Wnt/β-catenin signaling is not activated, the
APC protein, together with the scaffolding protein Axin,
serine/threonine kinases CK1 and glycogen synthase kinase (GSK)3β
and β-catenin, form a ‘destruction complex’ that phosphorylates
β-catenin, thus promoting its ubiquitination and proteosomal
degradation (Fig. 2). APC
mutations lead to a truncated APC protein unable to bind to the
destruction complex, which prevents β-catenin phosphorylation and
leads to its accumulation in the cytoplasm (77) (Fig.
2). Therefore, the accumulated β-catenin translocates to the
nucleus where it binds to T-cell factor (TCF)/lymphoid enhancer
factor family DNA-binding proteins triggering the constitutive
expression of Wnt target genes (MYC, cyclin D1, AXIN2 and
Dickkopf 1) involved in proliferation, invasion and loss of
differentiation, as well as in oncogenesis (77). The Wnt/β-catenin pathway plays a key
role in the maintenance of the intestinal stem cell niche (78) and it is well known that activation
of this signaling pathway by APC mutation is sufficient to
induce intestinal epithelial hyperproliferation and polyposis
(78), matching both the phenotype
and the risk of CMTC in patients with FAP.
As the function-inactivating germline mutation of
the APC gene can be partially compensated for by the other
allele, an additional somatic mutation of the APC gene or
another phenotypically equivalent gene (second hit), is required
for tumor development (25,52). In fact, in patients with FAP, CMTC
is frequently multifocal and shows different somatic APC
mutations in each tumor (79). A
missense AXIN1 somatic mutation in exon 7 was detected by
our group in a case of CMTC associated with FAP (80). In other cases of familial CMTCs,
second-hit somatic mutations in the lysine methyltransferase 2D
(KMT2D) and KMT2C genes have been reported (25). Of note, both KMT2D and
KMT2C have been shown to be transcriptional regulators of
the ER gene (81).
Sporadic CMTC
In cases of CMTC without APC germline
mutation, the coexistence of two different oncogenic somatic
variants of the APC gene has been detected (8,82). In
a sporadic case of CTCM with a single somatic APC mutation
at codon 1,309, the dominant negative effect of this mutation
agrees with the two-hit Knudson hypothesis (59).
The existence of sporadic cases of CMTC with
missense somatic mutations of exon 3 of the β-catenin gene
(CTNNB1) lacking mutations in APC or loss of
heterozygosity near the APC gene (83), have confirmed the key role of the
constitutive activation of the Wnt/β-catenin pathway in the
development of CMTC. In these cases, mutations in exon 3 of
CTNNB1 were located at codons 29, 22, 39, 44, 49, 54 and 56.
In a way similar to that described for the APC gene, the
multicentric CMTCs showed different somatic mutations of
CTNNB1 (83), additionally
supporting an independent origin of each of the foci of these
multifocal tumors.
In another sporadic case of CMTC, a missense
AXIN1 somatic mutation, has been identified in exon 1
(80).
Additional molecular alterations in
FAP-associated CMTC
In addition to germline APC mutations,
certain cases of familial CMTC showed molecular alterations
typically described in follicular-derived TC. In fact,
coiled-coil domain containing 6::RET and nuclear receptor
coactivator 4::RET rearrangements have been reported in two
CMTC cases (III-1 and III-11) from a family (kindred #1) with
FAP-associated CMTC (40), and
RET translocations have also been described in other series
(44,58). A somatic K-RAS mutation
(p.Gln61Lys) was detected in FAP-associated CMTC (84). An activating
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit
α (PIK3CA) mutation and copy number gains of BRCA2,
fibroblast growth factor (FGF23), FGFR1 and
PIK3CB have been detected exclusively in the high-grade
component of a FAP-associated CMTC, with tumor protein
(TP)53 mutation in the well-differentiated component
and concurrent APC and telomerase reverse transcriptase
(TERT) promoter mutations in both tumor components (9).
Additional molecular alterations in
sporadic CMTC
Somatic KRAS mutation was reported in one
case of sporadic CMTC (80). The
same mutation in exon 9, codon 545 of the PIK3CA gene
(p.Glu545Ala) was detected in 3 cases of sporadic CMTC (85). TERT promoter mutation was
described in sporadic CMTC (47).
As has been confirmed in conventional follicular cell
differentiated TC (86), as well as
in cases with FAP, sporadic CMTC cases with TERT promoter
mutations have been associated with aggressive behavior (47). Alterations of TP53 do not
appear to be an early event in CMTC tumorigenesis (8,40).
Sex hormones as a growth promoter of
CMTC
ERs may have a role in follicular-lineage TC
through genomic (ER-α and ER-β isoforms) and non-genomic pathways,
stimulating both the PI3K/AKT/mTOR and the RAS/RAF/MAPK signaling
pathways, resulting in increased reactive oxygen species production
and cell cycle progression/proliferation through the modulation of
cyclin D1, as well as angiogenesis and migration (87). Since genetic alterations in the
PI3K/AKT/mTOR pathway mainly promote the transformation of TC with
follicular growth pattern (FTC and follicular subtype of PTC) and
alterations in the MAPK pathway are associated with PTC with a
papillary growth pattern (88), the
activation of both pathways by estrogens is consistent with the
mixed (papillary and follicular) growth pattern of CMTC.
Circulating estrogen levels would produce activation of
PI3K/AKT/mTOR and the RAS/RAF/MAPK pathways, which, acting
synergistically with the Wnt/β-catenin pathway, would explain the
notable predominance of CMTC in women, even among patients with FAP
(5).
Recent studies using human colonic epithelial cells
demonstrated that activation of ERα but not ERβ increased the
protein levels of cyclin D1, proliferating cell nuclear antigen and
β-catenin, indicating increased proliferation and migration
(89). Consequently, an analogous
activation mechanism may be hypothesized to be present in CMTC
cells, given their ‘intestinal/non-committed’ phenotype and their
richness in ERα, producing and thus further increasing the levels
of β-catenin. Since ERβ is overexpressed in PTC stem cells
(90), estrogens could also
participate in the maintenance of the non-committed status of CMTC
cells through the effect on ERβ.
Estrogen-related receptor γ (ERRγ) acts as a tumor
suppressor in gastrointestinal cancers through inhibition of the
Wnt/β-catenin pathway (91). In
preclinical models, it was recently shown that an inverse agonist
of ERRγ, GSK5182, enhances the function of NIS protein via the
modulation of ERRγ and MAPK signaling, thereby leading to increased
responsiveness to radioiodine (RAI) in previously RAI-refractory
PTC cells (92). This suggests that
estrogens could also participate in the lack of expression of
follicular differentiation markers in CMTC cells.
Prognostic markers
CMTC generally behaves more indolently than PTC
(26,93), with ~12% of cases having lymph node
metastases at diagnosis (3,16,18,27,36,45–47,55,58,93,94)
and 5% have distant metastases (mainly to the lung, bone and brain)
(10,36,44,46,47,51,55,73,74,93)
(Table II). To our knowledge,
mortality due to CMTC has been reported in only 4 patients
(10,16,36,44).
| Table II.Main clinicopathological features of
patients with cribriform morular thyroid carcinoma and distant
metastasis. |
Table II.
Main clinicopathological features of
patients with cribriform morular thyroid carcinoma and distant
metastasis.
| Author(s),
year | Sex/age, years | FAP | Relevant tumor
findings | Distant
metastasis | Follow-up,
months | Outcome | (Refs.) |
|---|
| Okamoto et
al, 1995 | F/29 | NS | TS: 50 mm | Bone and lungs | 45.6 | Died with
widespread metastases | (10) |
| Okamoto et
al, 1995 | F/36 | NS | TS: 17 mm | Lungs | 92.4 | Alive with
metastases | (10) |
| Perrier et
al, 1998 | F/36 | Yes | Two local
recurrences at 6 and 84 months after the initial surgery | Extensive
mediastinal adenopathy | 85 | Treatment after TT
with 131I and T4 suppression. Died of cardiac
tamponade due to tumor infiltration. | (16) |
| Fenton et
al, 2001 | F/20 | Yes | Recurrence in the
right supraclavicular fossaa 29 years after TT | Extensive
metastasis in the cervical spine (C2-C4, T1 and T2), 360 months
after TT | 360 | Initial TT followed
by 131I. After radiotherapy sessions on the metastases,
the patient developed basal pneumonia and died. | (36) |
| Cameselle-Teijeiro
et al, 2009 | M/42 | Yes | Angioinvasive
neoplasm with positivity for chromogranin and synaptophysin in 40%
of tumor cells. 7 MF/10 HPF. Ki-67 index: 60%. APC
p.Thr1493Thr RET rearrangements | Bilateral lung
metastases at diagnosis | 23 | After diagnosis,
radiolabeled somatostatin analogue therapy
(dotatoc-90Yttrium, 5 GBq) was attempted with partial
response to treatment. Developed multiple brain metastases treated
with palliative whole-brain radiotherapy. The patient continued to
deteriorate and died. | (44) |
| Nakazawa et
al, 2013 | F/35 | No | TS: 90 mm. Poorly
differentiated features (50%). Necrosis and prominent venous
invasion. 4 MF/10 HPF. Ki-67 index: 15–20%. APC
p.Cys520Tyr_fsX534 | Multiple osteolytic
metastases (pelvis and left femur), bilateral lung
metastasesa and hilar
lympha-denopathya at
diagnosis | 9 | TT followed by
external radiotherapy (23 Gy) and therapy with 131I.
Alive with multiple bone and pulmonary metastases. | (46) |
| Alikhan et
al, 2015 | F/25 | Yes | Tumor infiltration
of surgical margins. Positivity for β-hCG in tumor cells. Tumor
recurrence and cervical lymph node metastasis 12 months after
TT. | Left pleural
effusion and pleural metastases 36 months after TT | 49 | TT followed by 30
mCi 131I. An additional dose of 100 mCi 131I
12 months later. Palliative surgical debulking of the tumor in the
neck was performed 48 months after TT. | (55) |
| Oh et al,
2017 | F/45 | No | TS: 27 mm.
Lymphatic and venous tumor invasion with perithyroid soft tissue
infiltration. Metastasis in 5 central compartment lymph nodes.
TERTp (C228T) | Bone metastasis
(right humerus, left seventh rib, C7, L2 and L5 vertebrae, and left
iliac bone 16 months after diagnosis | 48 | TT with central
lymphadenectomy followed by 150 mCi 131I. Partial
resection of left 7th rib and curettage of the right distal humerus
and palliative radiotherapy (3,000 cGy) for bone metastases. Second
ablation with 200 mCi 131I. Patient is alive multiple
metastases and continues to be with followed up. | (47) |
| Tsuji et al,
2018 | F/28 | No | TS: 91 mm. 6 MF/10
HPF. Ki-67 index: 40·2% (primary tumor) and 22.1% (lung
metastases) | Multiple bilateral
metastases 36 months after TT | 36 | TT with cervical
lymph node dissection followed by thyrotropin suppression,
radioactive iodine ablation and sorafenib therapies. | (51) |
| Akaishi et
al, 2018 | F/37 | NS | TS: 95 mm. Presence
of lymphatic and venous tumor invasion | Lung
metastases | 84 | Treated with 3
sessions of high doses of 131I. Alive with lung
metastases. | (93) |
| Ito et al,
2019 | F/24 | Yes | TS: 87 mm. Ki-67
index: 50% (primary tumor) and 70% (lung metastases). No lymph node
metastases. | Multiple bilateral
metastases 42 months after TT | 70 | TT with
prophylactic central node dissection and right modified radical
neck dissection followed by 100 mCi 131I 45 months after
surgery. Subsequently, treatment with lenvatinib for 24 months with
partial and sustained response of lung metastases during
follow-upb. | (74) |
| Laforga et
al, 2020 | F/45 | NS | TS: 55 mm. | Multiple bilateral
metastases 84 months after TT | 84 | TT followed by 99.5
mCi 131I. Sixty months later, ultrasound findings and
slightly increased Tg levels led to a right neck dissection that
identified metastasis in 2 lymph nodes. Then, repeated treatment
with 131I (147.5 mC1) resulted in slight iodine uptake
in pulmonary metastases (histologically confirmed). | (73) |
Although cases of encapsulated CMTC with both a
high Ki-67 percentage and apoptotic cell count have a favorable
prognosis (95), tumors with
necrosis and/or a high proliferative index (high-grade CMTC)
usually behave aggressively (1,44,46,51).
In agreement with the immunohistochemical negativity of CMTC cells
for Tg, radioactive iodine whole-body scans show no iodine-avid
lesions (47,74), and the serum Tg levels without
anti-Tg antibodies are not reliable for following up CMTC patients
(47,55,73,96).
Curiously, in a case with lung metastases, Tg levels were low for
tumor volume, but the associated Tg doubling time was short
(74). Uptake of
octreotide-111Indium by the CMTC cells has been
described in two cases (36,44).
Therapeutic and preventive approaches
This review confirms that the permanent activation
of the Wnt/β-catenin signaling pathway constitutes the key
pathogenetic event for the development of CMTC. High expression of
sex hormone receptors (mainly ERα) in tumor cells would act
synergistically in young women promoting tumor development through
the PI3K/AKT/mTOR and the RAS/RAF/MAPK signaling pathways.
Different somatic mutational events (e.g., RET
rearrangements, or KRAS, PIK3CA, TERT or TP53
mutations) may further potentiate the development and/or
progression of CMTC. These molecular events constitute the basis
for the establishment of targeted therapies but given their current
state of development, it is reasonable to propose surgical
treatment as an initial approach. This treatment should follow the
same surgical approach as that currently recommended for PTC
(97), with the specifications
described below. The role of radioactive isotopes, hormone therapy
and the so-called precision medicine treatments are also
discussed.
At the same time, all patients with CMTC should be
informed and investigated in relation to the FAP syndrome (97), including genetic counseling with
screening for colonic tumors and other extracolonic manifestations
of the disease in FAP-positive cases.
Surgical treatment
Hemithyroidectomy would be sufficient in cases of
sporadic CMTC without high-risk data (Fig. 3). Total thyroidectomy would be
indicated in high-risk cases (high-grade tumors, widely invasive
tumors, those with extensive lymphatic or venous vascular invasion
and/or with extrathyroidal extension), as well as in all cases of
CMTC associated with FAP due to their association with
multicentricity and bilaterality. Prophylactic and therapeutic
central neck dissection should be performed with the same criteria
as those currently used for the treatment of differentiated TC
(DTC) of a follicular lineage (97).
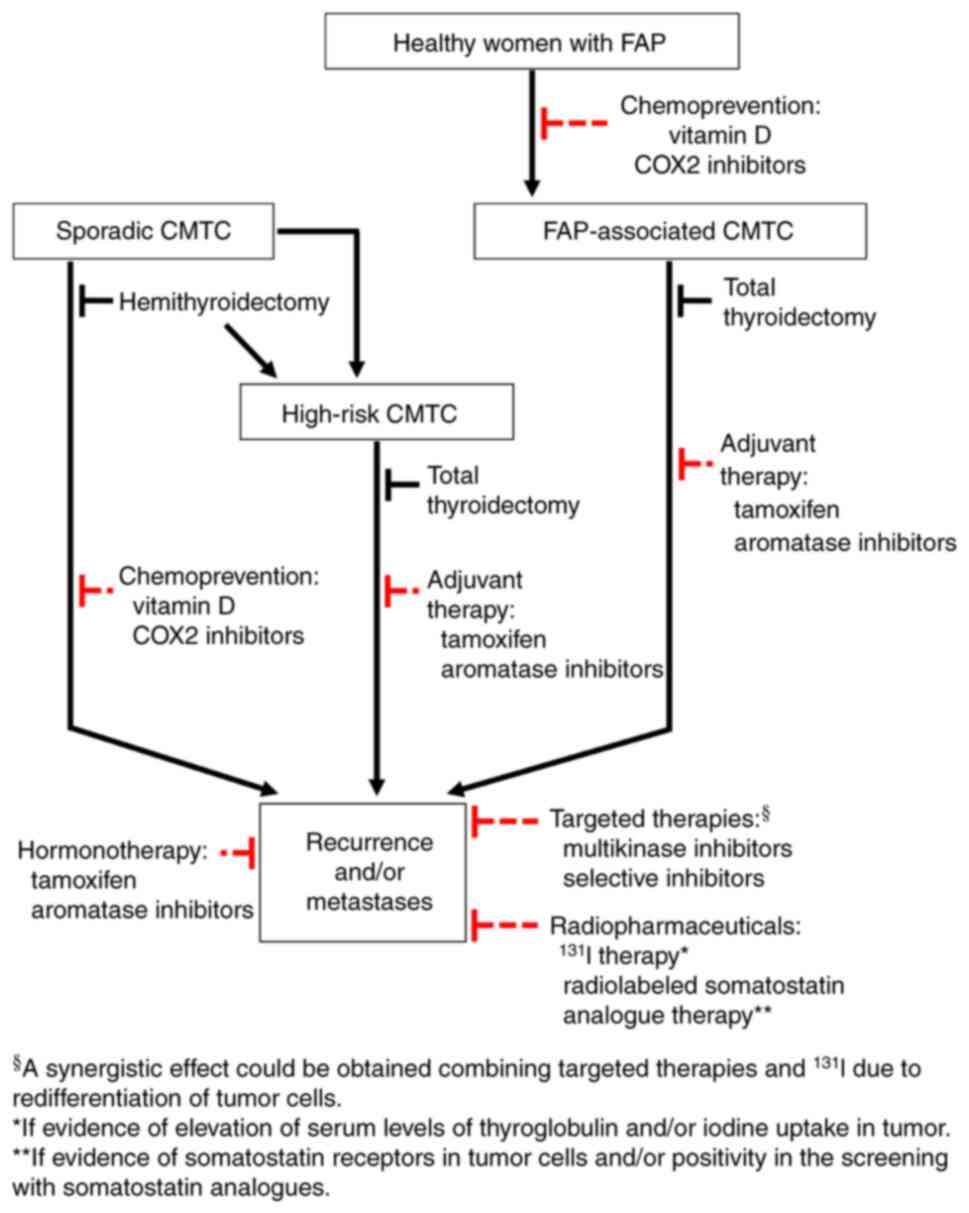 | Figure 3.Proposal of algorithm for possible
treatment and prevention of CMTC. Hemithyroidectomy should be the
treatment of choice in cases of sporadic CMTC without high-risk
data; however, due to multicentricity, total thyroidectomy should
be indicated in FAP-associated cases. Although clinical evidence is
still scarce, it may be hypothesized that for FAP-associated CMTC
and high-risk CMTC, adjuvant therapy with tamoxifen or aromatase
inhibitors could be beneficial. In all cases with recurrence and/or
metastases, it may be suggested that hormonotherapy (tamoxifen or
aromatase inhibitors), targeted therapies (multikinase or selective
inhibitors) and/or radiotherapy (131I treatment or
radiolabeled somatostatin analogues) are used. Vitamin D or COX2
inhibitors could hypothetically play a chemopreventive role in
healthy women with FAP, as well as in sporadic cases after
hemithyroidectomy. A dotted red line was used to indicate the
therapeutic proposals where clinical evidence is limited. CMTC,
cribriform morular thyroid carcinoma; FAP, familial adenomatous
polyposis; COX, cyclooxygenase. |
Radiopharmaceutical therapy
RAI with 131I has occasionally been used
in CMTC (see Table II). However,
the use of this isotope in CMTC is controversial, given its
uncertain histogenesis and the consistent immunohistochemical
negativity of tumor cells for Tg. A few isolated cases with
elevation of serum Tg levels (73,74),
as well as with slight iodine uptake in pulmonary metastases, have
been described (73). Recent data
suggest that multikinase inhibitors (MKIs) have the potential for
re-establishing 131I uptake in selected patients with
RAI refractory metastatic DTC (mDTC) of follicular lineage
(98,99). These drugs may also enhance
131I uptake in these mDTCs on a diagnostic and/or
post-therapy radioiodine scan, as well as increase the likelihood
of a better therapeutic effect for the administered 131I
activity (99). Therefore, a
divergence between increasing Tg and structural response may be a
reliable biomarker of redifferentiation in certain TCs of
follicular lineage refractory to radioactive iodine (100). Whether the increase in Tg
associated with lenvatinib treatment in a patient with metastatic
CMTC (74) represents an example of
redifferentiation deserves further investigation. If these findings
could be demonstrated in more patients with CMTC, both the
follicular lineage of the CMTC and an opportunity for additional
131I therapy would be confirmed.
A partial response to radiolabeled somatostatin
analogue therapy (90Yttrium-dotatoc) was reported in a
patient with an aggressive CMTC who showed neuroendocrine
differentiation in 40% of the tumor cells (44) (Table
II). Intense uptake of 111Indium-labelled octreotide
was detected in lung and lymph node metastases from another patient
with chromogranin- and synaptophysin-negative CMTC (46). The expression of somatostatin
receptors in cell cultures and tumor tissue of non-medullary
thyroid cancers has been described (101), but limited information is
available about these receptors and their possible clinical utility
in CMTC (44).
Hormone therapy
Levothyroxine and thyroid stimulating hormone
(TSH)
There is no rational basis for TSH suppression in
patients with CMTC until their follicular lineage has been
demonstrated and/or the existence of TSH receptors in their tumor
cells has been confirmed. Replacement treatment with levothyroxine,
however, should be indicated in patients with CMTC after total
thyroidectomy to prevent hypothyroidism; in addition, TSH
suppression could be considered given the existence of isolated
cases with elevated Tg and/or uptake of 131I in
metastases (73,74,93)
(Table II).
Antiestrogens
The use of drugs with antiestrogenic activity
widely used in hormone-dependent breast cancer, such as tamoxifen
and aromatase inhibitors (102),
could imply therapeutic repositioning in patients undergoing
surgery with high-risk CMTC, as well as when there is tumor
recurrence and/or metastasis (Fig.
4); however, there are currently insufficient clinical data in
this regard.
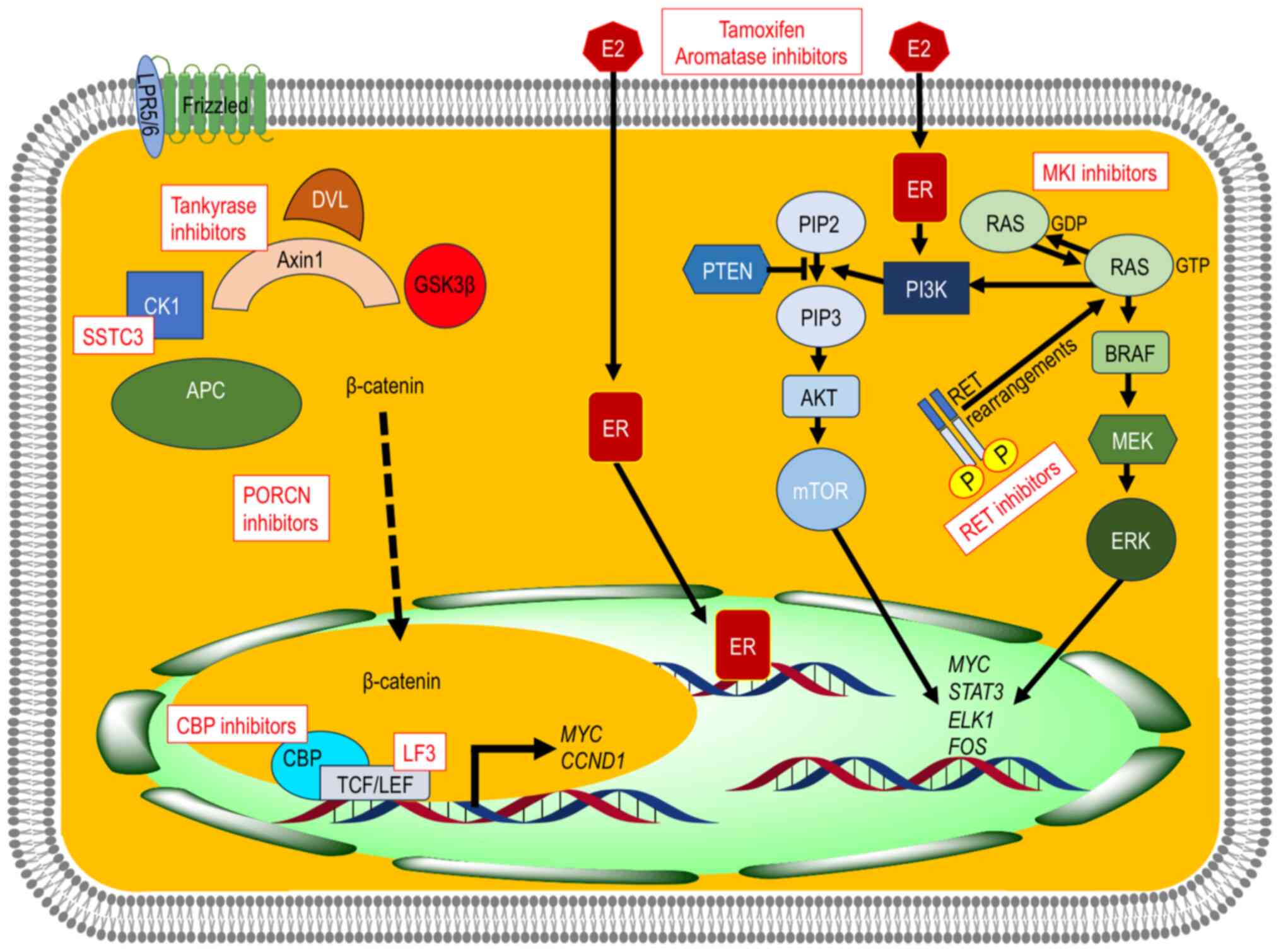 | Figure 4.Schematic illustration of the
rationale for possible targeted therapies in CMTC. Blocking the
constitutive activation of the Wnt/β-catenin signaling pathway
could play an essential role in the treatment of CMTC through the
degradation of β-catenin by stabilizing Axin2 (tankyrase
inhibitors), CK1 agonists (SSTC3), inhibitors targeting
β-catenin-TFC transcription complex (LF3), CBP inhibitors, as well
as porcupine inhibitors, but data on clinical efficacy and safety
are still insufficient. E2 via ER would have a tumor-promoting role
through the PI3K/AKT/mTOR and the RAS/RAF/MAPK signaling pathways,
which would justify treatment with both antiestrogens (tamoxifen
and aromatase inhibitors) and MKI inhibitors. The detection of
additional somatic events (e.g. RET rearrangements) could be
subsidiary to treatment with specific inhibitors. CMTC, morular
cribriform thyroid carcinoma; E2, estrogens; ER, estrogen
receptors; MKI, multikinase; TCF/LEF, T-cell factor/lymphoid
enhancer factor; CTNNB1, β-catenin 1; CCND1, cyclin D1; APC, APC
regulator of WNT signaling pathway; CK1, casein kinase 1; GSK3β,
glycogen synthase kinase 3β; DVL, disheveled; CBP, cyclic adenosine
monophosphate responsive element binding protein 1 binding protein;
LRP5/6, low-density lipoprotein receptor-related protein 5/6; PI3K,
phosphatidylinositol 3-kinase, AKT, serine/threonine kinase; mTOR,
mechanistic target of rapamycin kinase; RAS, small GTPase; RAF,
Raf-1 proto-oncogene, serine/threonine kinase; MAPK,
mitogen-activated protein kinase. |
The efficacy of preventive therapy with selective
ER modulators or aromatase inhibitors in women with germline
APC gene mutation (FAP) may be questionable, however. The
preventive benefits in these patients do not seem clear, since they
would mainly be young women who need long periods of treatment with
drugs that have serious adverse effects (102–104).
Targeted therapy
Targeting the Wnt/β-catenin pathway
Blocking the constitutive activation of the
Wnt/β-catenin pathway constitutes the theoretical cornerstone of
targeted treatment of CMTC (Fig.
4). These therapies should be aimed at restoring the β-catenin
destruction complex and/or neutralizing the TCF/β-catenin
transcription complex, since the initial component of this
signaling cascade, i.e., the Wnt ligand/receptor interface, usually
does not participate in the development of CMTC (see above). An
antineoplastic effect of tankyrase inhibitors has been demonstrated
through the degradation of β-catenin by stabilizing axin 2
(105), and CK1 agonists such as
SSTC3 have been shown to inhibit the growth of colorectal cancer in
mice (106). Inhibitors targeting
β-catenin-TFC transcription complex such as LF3 and ICG-001
(107) as well as CREB binding
protein inhibitors have shown antineoplastic efficacy with minimal
off-target effects (108), but
most of these data still come from preclinical models.
Small-molecule compounds targeting the Wnt/β-catenin cascade have
been used to inhibit cancer stem cells (109). Unfortunately, phase I/II studies
using some of these molecules, such as WNT9748 (a first-in-class
Porcupine inhibitor) in colorectal cancer have not shown improved
antitumor efficacy (110), and
data from another study with niclosamide, an anti-helminthic agent
inhibitor of the Wnt/β-catenin pathway in colorectal cancer
(111) were not published.
Therefore, insufficient data exist to support the therapeutic
efficacy and safety of therapies targeting the Wnt/β-catenin
pathway in CMTC.
Cyclooxygenase (COX)1 and COX2 inhibitors may
inhibit the Wnt/β-catenin pathway in cancer cells; they could
therefore be used for adjuvant therapy or chemoprevention of CMTC
in a manner analogous to that used in Lynch syndrome and FAP
(112), but clinical data are
still lacking.
Unfortunately, despite these preclinical data,
randomized clinical trials have not demonstrated a clear impact of
vitamin D supplementation on colorectal cancer incidence,
progression or mortality (113).
Vitamin D receptor increases the levels of E-cadherin in the plasma
membrane and decreases the levels of β-catenin in the nucleus in
differentiated TC (114).
Therefore, it is possible that vitamin D could play a
chemopreventive role in women with FAP.
Other precision therapy
Although the evidence is scarce, a sustained
response to treatment with lenvatinib was observed in a patient
with lung metastases of CMTC during follow-up of the patient
(74) (Table II). The clinical response to this
MKI is consistent with the signaling of the PI3K/AKT/mTOR and the
RAS/RAF/MAPK pathways induced by estrogens in CMTC. Lenvatinib,
sorafenib, cabozantinib and other MKIs have shown antiangiogenic
activity and could be used as treatments for CMTC with progression
after surgery in a manner analogous to that of differentiated TC
(114) (Fig. 4). Furthermore, selective RET
inhibitors (i.e. selpercatinib and pralsetinib) may be used in
cases of CMTC with RET rearrangements (115). Thus, although the clinical
evidence is still insufficient, the detection of somatic mutations
through next-generation sequencing could represent a precision
therapeutic alternative in CMTC.
Limitations
In this review, hypotheses are proposed in an
attempt to understand the nature of CMTC and establish a
therapeutic approach. As most of the information comes from
isolated cases or small series, the level of evidence is limited.
Therefore, additional validations and appropriate clinical trials
are still necessary.
Conclusions
CMTC is the TC associated with FAP. This rare tumor
usually occurs in young women with multiple lesions or as a single
nodule in sporadic cases. Constitutive activation of the
WNT/β-catenin signaling pathway plays a key role in the distinctive
growth pattern of CMTC and its immunohistochemical profile. Strong
nuclear and cytoplasmic positivity for β-catenin is the hallmark of
CMTC. Tumor cells are also positive for ER and PR and for
TTF1/NKX2-1, but negative for Tg and calcitonin. Although the
histogenesis of CMTC is controversial, the presence of a follicular
or follicular precursor cell lineage may be postulated, whose block
in differentiation would be secondary to WNT/β-catenin signaling.
In FAP cases, total thyroidectomy is the treatment of choice due to
the frequent multifocality and bilaterality of CMTC. Despite the
central role of the Wnt/β-catenin signaling pathway in the
pathogenesis of this tumor, more studies seem necessary to be able
to translate the targeting of this signaling pathway into clinical
practice. Although there is still little clinical evidence, in
cases of CMTC recurrence and metastasis, targeted treatments with
MKIs or more selective inhibitors could be useful. And although
sufficient clinical data are also lacking, adjuvant antiestrogenic
therapy could also be useful in women undergoing surgery with
high-risk CMTC as well as when there is tumor recurrence and/or
metastasis. The chemopreventive role of vitamin D and COX
inhibitors in women with FAP requires additional investigation.
Acknowledgements
Not applicable.
Funding
This work was supported by the Instituto de Salud Carlos III
(ISCIII), Spain (grant nos. PI19/01316 and PI23/00722), co-funded
by the European Union (EU).
Availability of data and materials
Not applicable.
Authors' contributions
All authors contributed to the conception of the
review and interpretation of the data. SCG: Review of the
literature, writing-original draft and editing. IAN and MSA:
critical review and editing. JMCT: Review of the literature,
writing-original draft and editing, and funding acquisition. All
authors have read and approved the final manuscript. Data
authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
The images in Figs.
1 and 2C were obtained from
cases in the TIROCHUS collection. TIROCHUS is a registered
collection of human tissues at the ISCIII national biobank (no.
c.0003960 to JMCT) of the Department of Pathology of the Hospital
Clínico Universitario (Santiago de Compostela, Spain). Informed
consent was obtained from each patient with a specific signed
document updated to the current legislation.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Erickson LA, Mete O, Cameselle-Teijeiro
JM, LiVolsi V, Sobrinho-Simoes M and Jung CK: Cribriform morular
thyroid carcinoma. WHO Classification of Tumours Editorial Board.
Endocrine and neuroendocrine tumours [Internet] Lyon (France):
International Agency for Research on Cancer; 2022, [2023 03 07].
(WHO classification of tumours series, 5th edition. Vol. 10).
Available from:. https://tumourclassification.iarc.who.int/chapters/53
|
|
2
|
Harach HR, Williams GT and Williams ED:
Familial adenomatous polyposis associated thyroid carcinoma: A
distinct type of follicular cell neoplasm. Histopathology.
25:549–561. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cameselle-Teijeiro J and Chan JK:
Cribriform-morular variant of papillary carcinoma: A distinctive
variant representing the sporadic counterpart of familial
adenomatous polyposis-associated thyroid carcinoma? Mod Pathol.
12:400–411. 1999.PubMed/NCBI
|
|
4
|
Lloyd RV, Osamura RY, Klöppel G and Rosai
J: WHO classification of tumours of endocrine organs. 4th edition.
IARC; Lyon: 2017
|
|
5
|
Cameselle-Teijeiro JM, Peteiro-González D,
Caneiro-Gómez J, Sánchez-Ares M, Abdulkader I, Eloy C, Melo M,
Amendoeira I, Soares P and Sobrinho-Simões M: Cribriform-morular
variant of thyroid carcinoma: A neoplasm with distinctive phenotype
associated with the activation of the WNT/β-catenin pathway. Mod
Pathol. 31:1168–1179. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Baloch ZW, Asa SL, Barletta JA, Ghossein
RA, Juhlin CC, Jung CK, LiVolsi VA, Papotti MG, Sobrinho-Simões M,
Tallini G and Mete O: Overview of the 2022 WHO classification of
thyroid neoplasms. Endocr Pathol. 33:27–63. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Boyraz B, Sadow PM, Asa SL, Dias-Santagata
D, Nosé V and Mete O: Cribriform-morular thyroid carcinoma is a
distinct thyroid malignancy of uncertain cytogenesis. Endocr
Pathol. 32:327–335. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Echegoyen-Silanes A, Pineda-Arribas JJ,
Sánchez-Ares M, Cameselle-García S, Sobrino B, Ruíz-Ponte C,
Piso-Neira M, Anda E and Cameselle-Teijeiro JM: Cribriform morular
thyroid carcinoma: A case report with pathological,
immunohistochemical, and molecular findings suggesting an origin
from follicular cells (or their endodermal precursors). Virchows
Arch. 482:615–623. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dettmer MS, Hürlimann S, Scheuble L,
Vassella E, Perren A and Wicke C: Cribriform morular thyroid
carcinoma-ultimobranchial pouch-related? Deep molecular insights of
a unique case. Endocr Pathol. 34:342–348. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Okamoto Y, Kashima K, Daa T, Yokoyama S,
Nakayama I and Noguchi S: Morule with biotin-containing
intranuclear inclusions in thyroid carcinoma. Pathol Int.
45:573–579. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lam AK and Fridman M: Characteristics of
cribriform morular variant of papillary thyroid carcinoma in
post-Chernobyl affected region. Hum Pathol. 74:170–177. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hirokawa M, Maekawa M, Kuma S and Miyauchi
A: Cribriform-morular variant of papillary thyroid
carcinoma-cytological and immunocytochemical findings of 18 cases.
Diagn Cytopathol. 38:890–896. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Levy RA, Hui VW, Sood R, Fish S, Markowitz
AJ, Wong RJ and Guillem JG: Cribriform-morular variant of papillary
thyroid carcinoma: An indication to screen for occult FAP. Fam
Cancer. 13:547–551. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Or Koca A and Güler Şimşek G:
Post-radiotherapy cribriform-morular thyroid carcinoma. J Clin Lab
Anal. 37:e248192023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lahbacha B, Chaabane A, Nechi S, Mfarrej
MK, Douggaz A, Kharrat G and Chelbi E: Cribriform-morular thyroid
carcinoma: A case report with review of the literature. Ear Nose
Throat J. 14556132311523322023.(Epub ahead of print). PubMed/NCBI
|
|
16
|
Perrier ND, van Heerden JA, Goellner JR,
Williams ED, Gharib H, Marchesa P, Church JM, Fazio VW and Larson
DR: Thyroid cancer in patients with familial adenomatous polyposis.
World J Surg. 22:738–743. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jarrar AM, Milas M, Mitchell J, Laguardia
L, O'Malley M, Berber E, Siperstein A, Burke C and Church JM:
Screening for thyroid cancer in patients with familial adenomatous
polyposis. Ann Surg. 253:515–521. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Herraiz M, Barbesino G, Faquin W,
Chan-Smutko G, Patel D, Shannon KM, Daniels GH and Chung DC:
Prevalence of thyroid cancer in familial adenomatous polyposis
syndrome and the role of screening ultrasound examinations. Clin
Gastroenterol Hepatol. 5:367–373. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Uchino S, Ishikawa H, Miyauchi A, Hirokawa
M, Noguchi S, Ushiama M, Yoshida T, Michikura M, Sugano K and Sakai
T: Age- and gender-specific risk of thyroid cancer in patients with
familial adenomatous polyposis. J Clin Endocrinol Metab.
101:4611–4617. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Casellas-Cabrera N, Díaz-Algorri Y,
Carlo-Chévere VJ, González-Pons M, Rodríguez-Mañón N, Pérez-Mayoral
J, Bertrán-Rodríguez C, Soto-Salgado M, Giardiello FM,
Rodríguez-Quilichini S and Cruz-Correa M: Risk of thyroid cancer
among Caribbean Hispanic patients with familial adenomatous
polyposis. Fam Cancer. 15:267–274. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kurihara K, Shimizu S, Chong J, Hishima T,
Funata N, Kashiwagi H, Nagai H, Miyaki M and Fukayama M: Nuclear
localization of immunoreactive beta-catenin is specific to familial
adenomatous polyposis in papillary thyroid carcinoma. Jpn J Cancer
Res. 91:1100–1102. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tomoda C, Miyauchi A, Uruno T, Takamura Y,
Ito Y, Miya A, Kobayashi K, Matsuzuka F, Kuma S, Kuma K and Kakudo
K: Cribriform-morular variant of papillary thyroid carcinoma: clue
to early detection of familial adenomatous polyposis-associated
colon cancer. World J Surg. 28:886–889. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ito Y, Miyauchi A, Ishikawa H, Hirokawa M,
Kudo T, Tomoda C and Miya A: Our experience of treatment of
cribriform morular variant of papillary thyroid carcinoma;
difference in clinicopathological features of FAP-associated and
sporadic patients. Endocr J. 58:685–689. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cetta F, Moltoni L, Barellini L, Monti M
and Gotti G: Familial adenomatous polyposis-associated papillary
thyroid carcinoma shows an indolent course and usually, but not
always, belongs to the cribriform-morular variant of papillary
thyroid carcinoma. Acta Cytol. 56:107–108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nieminen TT, Walker CJ, Olkinuora A,
Genutis LK, O'Malley M, Wakely PE, LaGuardia L, Koskenvuo L, Arola
J, Lepistö AH, et al: Thyroid carcinomas that occur in familial
adenomatous polyposis patients recurrently harbor somatic variants
in APC, BRAF, and KTM2D. Thyroid. 30:380–388. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Park J, Kim JW, Park H, Park SY, Kim TH,
Kim SW, Oh YL and Chung JH: Multifocality in a patient with
cribriform-morular variant of papillary thyroid carcinoma is an
important clue for the diagnosis of familial adenomatous polyposis.
Thyroid. 29:1606–1614. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yamashita T, Hosoda Y, Kameyama K, Aiba M,
Ito K and Fujimoto Y: Peculiar nuclear clearing composed of
microfilaments in papillary carcinoma of the thyroid. Cancer.
70:2923–2928. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dalal KM, Moraitis D, Iwamoto C, Shaha AR,
Patel SG and Ghossein RA: Clinical curiosity: Cribriform-morular
variant of papillary thyroid carcinoma. Head Neck. 28:471–476.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hizawa K, Iida M, Yao T, Aoyagi K, Oohata
Y, Mibu R, Yamasaki K, Hirata T and Fujishima M: Association
between thyroid cancer of cribriform variant and familial
adenomatous polyposis. J Clin Pathol. 49:611–613. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chong Y, Shin JH, Oh YL, Han BK and Ko EY:
Cribriform-morular variant of papillary thyroid carcinoma:
Ultrasonographic and clinical characteristics. Thyroid. 23:45–49.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guilmette J and Nosé V: Hereditary and
familial thyroid tumours. Histopathology. 72:70–81. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rehan S and Aye K: In patients with a
positive family history of familial adenomatous polyposis can the
condition be diagnosed from the presence of congenital hypertrophy
of the retinal pigment epithelium detected via an eye examination:
A systematic review. Clin Exp Ophthalmol. 48:98–116. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xu M, Zheng Y, Zuo Z, Zhou Q, Deng Q, Wang
J and Wang D: De novo familial adenomatous polyposis associated
thyroid cancer with a c.2929delG frameshift deletion mutation in
APC: A case report and literature review. World J Surg Oncol.
21:732023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dinarvand P, Davaro EP, Doan JV, Ising ME,
Evans NR, Phillips NJ, Lai J and Guzman MA: Familial adenomatous
polyposis syndrome: An update and review of extraintestinal
manifestations. Arch Pathol Lab Med. 143:1382–1398. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Truta B, Allen BA, Conrad PG, Kim YS, Berk
T, Gallinger S, Bapat B, Terdiman JP and Sleisenger MH: Genotype
and phenotype of patients with both familial adenomatous polyposis
and thyroid carcinoma. Fam Cancer. 2:95–99. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fenton PA, Clarke SE, Owen W, Hibbert J
and Hodgson SV: Cribriform variant papillary thyroid cancer: A
characteristic of familial adenomatous polyposis. Thyroid.
11:193–197. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee S, Hong SW, Shin SJ, Kim YM, Rhee Y,
Jeon BI, Moon WC, Oh MR and Lim SK: Papillary thyroid carcinoma
associated with familial adenomatous polyposis: Molecular analysis
of pathogenesis in a family and review of the literature. Endocr J.
51:317–323. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kashiwagi H, Konishi F, Kanazawa K and
Miyaki M: Sisters with familial adenomatous polyposis affected with
thyroid carcinoma, desmoid tumour and duodenal polyposis. Br J
Surg. 83:2281996. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cetta F, Montalto G, Gori M, Curia MC,
Cama A and Olschwang S: Germline mutations of the APC gene in
patients with familial adenomatous polyposis-associated thyroid
carcinoma: Results from a European cooperative study. J Clin
Endocrinol Metab. 85:286–292. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Soravia C, Sugg SL, Berk T, Mitri A, Cheng
H, Gallinger S, Cohen Z, Asa SL and Bapat BV: Familial adenomatous
polyposis-associated thyroid cancer: A clinical, pathological, and
molecular genetics study. Am J Pathol. 154:127–135. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Silva S, Lage P, Cabral F, Alves R,
Catarino A, Félix A and André S: Bilateral breast fibromatosis
after silicone prosthetics in a patient with classic familial
adenomatous polyposis: A case report. Oncol Lett. 16:1449–1454.
2018.PubMed/NCBI
|
|
42
|
Kameyama K, Mukai M, Takami H and Ito K:
Cribriform-morular variant of papillary thyroid carcinoma:
Ultrastructural study and somatic/germline mutation analysis of the
APC gene. Ultrastruct Pathol. 28:97–102. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nesland JM: Ultrastructural and molecular
analysis of cribriform-morular variant of papillary thyroid
carcinoma. Ultrastruct Pathol. 28:532004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cameselle-Teijeiro J, Menasce LP, Yap BK,
Colaco RJ, Castro P, Celestino R, Ruíz-Ponte C, Soares P and
Sobrinho-Simões M: Cribriform-morular variant of papillary thyroid
carcinoma: Molecular characterization of a case with neuroendocrine
differentiation and aggressive behavior. Am J Clin Pathol.
131:134–142. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jung CK, Choi YJ, Lee KY, Bae JS, Kim HJ,
Yoon SK, Son YI, Chung JH and Oh YL: The cytological, clinical, and
pathological features of the cribriform-morular variant of
papillary thyroid carcinoma and mutation analysis of CTNNB1 and
BRAF genes. Thyroid. 19:905–913. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Nakazawa T, Celestino R, Machado JC,
Cameselle-Teijeiro JM, Vinagre J, Eloy C, Benserai F, Lameche S,
Soares P and Sobrinho-Simões M: Cribriform-morular variant of
papillary thyroid carcinoma displaying poorly differentiated
features. Int J Surg Pathol. 21:379–389. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Oh EJ, Lee S, Bae JS, Kim Y, Jeon S and
Jung CK: TERT Promoter mutation in an aggressive cribriform morular
variant of papillary thyroid carcinoma. Endocr Pathol. 28:49–53.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sahu A, Shahin M, Jain P, Sultania M and
Ayyanar P: Cribriform morular thyroid carcinoma: A rare case and
associated uncommon features. Int J Surg Pathol.
106689692312065722023.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Baloch ZW, Segal JP and Livolsi VA: Unique
growth pattern in papillary carcinoma of the thyroid gland
mimicking adenoid cystic carcinoma. Endocr Pathol. 22:200–205.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Mogoş V, Mogoş S, Sfarti C, Băcăuanu R,
Huţanu O, Cotea E, Ciobanu D, Tudorache C and Târcoveanu E:
Familial syndromic papillary thyroid carcinoma report of two cases.
Rev Med Chir Soc Med Nat Iasi. 116:1048–1054. 2012.PubMed/NCBI
|
|
51
|
Tsuji H, Yasuoka H, Nakamura Y, Hirokawa
M, Hiroshima T, Sakamaki Y, Miyauchi A and Tsujimoto M: Aggressive
cribriform-morular variant of papillary thyroid carcinoma: Report
of an unusual case with pulmonary metastasis displaying poorly
differentiated features. Pathol Int. 68:700–705. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Corean J, Furtado LV, Kadri S, Segal JP
and Emerson LL: Cribriform-morular variant of papillary thyroid
carcinoma with poorly differentiated features: A case report with
immunohistochemical and molecular genetic analysis. Int J Surg
Pathol. 27:294–304. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Cameselle-Teijeiro JM, Mete O, Asa SL and
LiVolsi V: Inherited follicular epithelial-derived thyroid
carcinomas: From molecular biology to histological correlates.
Endocr Pathol. 32:77–101. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Mohindra S, Sakr H, Sturgis C and Chute
DJ: LEF-1 is a sensitive marker of cribriform morular variant of
papillary thyroid carcinoma. Head Neck Pathol. 12:455–462. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Alikhan M, Koshy A, Hyjek E, Stenson K,
Cohen RN and Yeo KT: Discrepant serum and urine β-hCG results due
to production of β-hCG by a cribriform-morular variant of thyroid
papillary carcinoma. Clin Chim Acta. 438:181–185. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Lyu S, Zhong G, Chen H, Li J and Li M: The
first case of cribriform-morular thyroid carcinoma and FAP with APC
gene mutation in China: A case report and brief review. Case Rep
Gastrointest Med. 2023:62224322023.PubMed/NCBI
|
|
57
|
Chang HY, Lim MY and Bundele MM:
Cribriform morular thyroid carcinoma: A recently reclassified
entity. ANZ J Surg. 93:2257–2259. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Cetta F, Curia MC, Montalto G, Gori M,
Cama A, Battista P and Barbarisi A: Thyroid carcinoma usually
occurs in patients with familial adenomatous polyposis in the
absence of biallelic inactivation of the adenomatous polyposis coli
gene. J Clin Endocrinol Metab. 86:427–432. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Cameselle-Teijeiro J, Ruiz-Ponte C, Loidi
L, Suarez-Peñaranda J, Baltar J and Sobrinho-Simoes M: Somatic but
not germline mutation of the APC gene in a case of
cribriform-morular variant of papillary thyroid carcinoma. Am J
Clin Pathol. 115:486–493. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kumamoto K, Ishida H, Ohsawa T, Ishibashi
K, Ushiama M, Yoshida T and Iwama T: Germline and somatic mutations
of the APC gene in papillary thyroid carcinoma associated with
familial adenomatous polyposis: Analysis of three cases and a
review of the literature. Oncol Lett. 10:2239–2243. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Manzoni M, Roversi G, Di Bella C, Pincelli
AI, Cimino V, Perotti M, Garancini M and Pagni F: Solid cell nests
of the thyroid gland: Morphological, immunohistochemical and
genetic features. Histopathology. 68:866–874. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Houghton O, Connolly LE and McCluggage WG:
Morules in endometrioid proliferations of the uterus and ovary
consistently express the intestinal transcription factor CDX2.
Histopathology. 53:156–165. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
McCluggage WG and Van de Vijver K: SATB2
is consistently expressed in squamous morules associated with
endometrioid proliferative lesions and in the stroma of atypical
polypoid adenomyoma. Int J Gynecol Pathol. 38:397–403. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Travaglino A, Raffone A, Russo D, Guadagno
E, Pignatiello S, Moretta P, Zullo F, Del Basso De Caro M, Insabato
L and Mascolo M: Does endometrial morular metaplasia represent
odontogenic differentiation? Virchows Arch. 479:607–616. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Gu Y, Zhang S, Liang X, Zhao H, Li X and
Lu J: Clinical and pathological characteristics and prognosis of
lung adenocarcinoma with high-grade fetal features: A retrospective
analysis. Int J Surg Pathol. 32:667–678. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Isobe T, Seki M, Yoshida K, Sekiguchi M,
Shiozawa Y, Shiraishi Y, Kimura S, Yoshida M, Inoue Y, Yokoyama A,
et al: Integrated molecular characterization of the lethal
pediatric cancer pancreatoblastoma. Cancer Res. 78:865–876. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Xu J, Park KJ, Rehrauer WM and Weisman PS:
Mesonephric-like adenocarcinoma of the ovary with squamoid morular
metaplasia, aberrant β-catenin expression, and concurrent FGFR2 and
CTNNB1 mutations: A case report. Virchows Arch. 484:147–150. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Lee HE, Chandan VS, Lee CT and Wu TT:
Squamoid morules in the pseudoinvasive foci of colonic polyp
morphologically mimic invasive carcinoma. Hum Pathol. 68:54–60.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Cameselle-Teijeiro J, Alberte-Lista L,
Chiarelli S, Buriticá C, Gonçalves L, González-Cámpora R and
Nogales FF: CD10 is a characteristic marker of tumours forming
morules with biotin-rich, optically clear nuclei that occur in
different organs. Histopathology. 52:389–392. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Sherwood RI, Maehr R, Mazzoni EO and
Melton DA: Wnt signaling specifies and patterns intestinal
endoderm. Mech Dev. 128:387–400. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Cameselle-Teijeiro J, Febles-Pérez C and
Sobrinho-Simões M: Papillary and mucoepidermoid carcinoma of the
thyroid with anaplastic transformation: A case report with
histologic and immunohistochemical findings that support a
provocative histogenetic hypothesis. Pathol Res Pract.
191:1214–1221. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Davies TF, Latif R, Sachidanandam R and Ma
R: The transient human thyroid progenitor cell: Examining the
thyroid continuum from stem cell to follicular cell. Thyroid.
31:1151–1159. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Laforga JB, Pedro T and Gasent JM:
Pulmonary metastasis of cribriform-morular variant of thyroid
carcinoma mimicking primary adenocarcinoma of the lung: A potential
pitfall. Diagn Cytopathol. 48:78–81. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Ito Y, Ishikawa H, Kihara M, Hirokawa M,
Kiyota N, Kasahara T and Miyauchi A: Control of lung metastases and
colon polyposis with lenvatinib therapy in a patient with
cribriform-morular variant of papillary thyroid carcinoma and an
APC gene mutation: A case study. Thyroid. 29:1511–1517. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Septer S, Slowik V, Morgan R, Dai H and
Attard T: Thyroid cancer complicating familial adenomatous
polyposis: Mutation spectrum of at-risk individuals. Hered Cancer
Clin Pract. 11:132013. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Domingues GAB, Kizys MML, Janovsky CCPS,
de Barros Maciel RM, Dias-da-Silva MR, Martins JRM, Camacho CP and
Cunha LL: The impact of the genetic background in a patient with
papillary thyroid cancer and familial adenomatous polyposis. Arch
Endocrinol Metab. 66:112–117. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Krausova M and Korinek V: Wnt signaling in
adult intestinal stem cells and cancer. Cell Signal. 26:570–579.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Mah AT, Yan KS and Kuo CJ: Wnt pathway
regulation of intestinal stem cells. J Physiol. 594:4837–4847.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Miyaki M, Iijima T, Ishii R, Hishima T,
Mori T, Yoshinaga K, Takami H, Kuroki T and Iwama T: Molecular
evidence for multicentric development of thyroid carcinomas in
patients with familial adenomatous polyposis. Am J Pathol.
157:1825–1827. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Cameselle-Teijeiro JM, Peteiro-González D,
Carreira M, Abdulkader I, Reyes-Santías R, Celestino R, Romero
Rojas A, Ruíz-Ponte C, Soares P, Casanueva F and Sobrinho-Simões M:
Molecular alterations in the cribriform-morular variant of
papillary thyroid carcinoma. Virchows Arch. 469 (1
Supp):S722016.
|
|
81
|
Hussain I, Deb P, Chini A, Obaid M, Bhan
A, Ansari KI, Mishra BP, Bobzean SA, Udden SMN, Alluri PG, et al:
HOXA5 expression is elevated in breast cancer and is
transcriptionally regulated by estradiol. Front Genet.
11:5924362020. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Aydemirli MD, van der Tuin K, Hes FJ, van
den Ouweland AMW, van Wezel T, Kapiteijn E and Morreau H: A unique
case of two somatic APC mutations in an early onset
cribriform-morular variant of papillary thyroid carcinoma and
overview of the literature. Fam Cancer. 19:15–21. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Xu B, Yoshimoto K, Miyauchi A, Kuma S,
Mizusawa N, Hirokawa M and Sano T: Cribriform-morular variant of
papillary thyroid carcinoma: A pathological and molecular genetic
study with evidence of frequent somatic mutations in exon 3 of the
beta-catenin gene. J Pathol. 199:58–67. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Giannelli SM, McPhaul L, Nakamoto J and
Gianoukakis AG: Familial adenomatous polyposis-associated,
cribriform morular variant of papillary thyroid carcinoma harboring
a K-RAS mutation: Case presentation and review of molecular
mechanisms. Thyroid. 24:1184–1189. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Kwon MJ, Rho YS, Jeong JC, Shin HS, Lee
JS, Cho SJ and Nam ES: Cribriform-morular variant of papillary
thyroid carcinoma: A study of 3 cases featuring the PIK3CA
mutation. Hum Pathol. 46:1180–1188. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Melo M, da Rocha AG, Vinagre J, Batista R,
Peixoto J, Tavares C, Celestino R, Almeida A, Salgado C, Eloy C, et
al: TERT promoter mutations are a major indicator of poor outcome
in differentiated thyroid carcinomas. J Clin Endocrinol Metab.
99:E754–E765. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Denaro N, Romanò R, Alfieri S, Dolci A,
Licitra L, Nuzzolese I, Ghidini M, Bareggi C, Bertaglia V, Solinas
C and Garrone O: The tumor microenvironment and the estrogen loop
in thyroid cancer. Cancers (Basel). 15:24582023. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Cancer Genome Atlas Research Network, .
Integrated genomic characterization of papillary thyroid carcinoma.
Cell. 159:676–690. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Wan H, Li J, Chen X, Sellers ZM and Dong
H: Divergent roles of estrogen receptor subtypes in regulating
estrogen-modulated colonic ion transports and epithelial repair. J
Biol Chem. 299:1050682023. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Li M, Chai HF, Peng F, Meng YT, Zhang LZ,
Zhang L, Zou H, Liang QL, Li MM, Mao KG, et al: Estrogen receptor β
upregulated by lncRNA-H19 to promote cancer stem-like properties in
papillary thyroid carcinoma. Cell Death Dis. 9:11202018. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Guo X, Yue L, Li M, Dai A, Sun J, Fang L,
Zhao H and Sun Q: Nuclear receptor estrogen-related receptor gamma
suppresses colorectal cancer aggressiveness by regulating
Wnt/β-catenin signaling. Carcinogenesis. 43:865–873. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Singh TD, Lee JE, Son KH, Lee BR, Kim SK,
Gulwani D, Sarangthem V and Jeon YH: An inverse agonist of
estrogen-related receptor gamma, GSK5182, enhances
Na+/I− symporter function in
radioiodine-refractory papillary thyroid cancer cells. Cells.
12:4702023. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Akaishi J, Kondo T, Sugino K, Ogimi Y,
Masaki C, Hames KY, Yabuta T, Tomoda C, Suzuki A, Matsuzu K, et al:
Cribriform-morular variant of papillary thyroid carcinoma: Clinical
and pathological features of 30 cases. World J Surg. 42:3616–3623.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Rossi ED, Revelli L, Martini M, Taddei A,
Pintus C, Panunzi C and Fadda G: Cribriform-morular variant of
papillary thyroid carcinoma in an 8-year-old girl: A case report
with immunohistochemical and molecular testing. Int J Surg Pathol.
20:629–632. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Hirokawa M, Matsuda K, Kudo T, Higuchi M,
Suzuki A, Takada N, Nakashima M and Miyauchi A: Cribriform-morular
variant of papillary thyroid carcinoma shows high Ki-67 labeling
indices, despite its excellent prognosis. Pathobiology. 86:248–253.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Fujimoto T, Hirokawa M, Ota H, Yabuta T,
Fukushima M, Kobayashi K, Amino N and Miyauchi A: Characteristic
sonographic features of cribriform papillary thyroid carcinoma for
differentiation from other thyroid nodules. J Med Ultrason (2001).
42:83–87. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Haugen BR, Alexander EK, Bible KC, Doherty
GM, Mandel SJ, Nikiforov YE, Pacini F, Randolph GW, Sawka AM,
Schlumberger M, et al: 2015 American thyroid association management
guidelines for adult patients with thyroid nodules and
differentiated thyroid cancer: The American thyroid association
guidelines task force on thyroid nodules and differentiated thyroid
cancer. Thyroid. 26:1–133. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Weber M, Kersting D, Riemann B,
Brandenburg T, Führer-Sakel D, Grünwald F, Kreissl MC, Dralle H,
Weber F, Schmid KW, et al: Enhancing radioiodine incorporation into
radioiodine-refractory thyroid cancer with MAPK inhibition
(ERRITI): A single-center prospective two-arm study. Clin Cancer
Res. 28:4194–4202. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Van Nostrand D, Veytsman I, Kulkarni K,
Heimlich L and Burman KD: Redifferentiation of differentiated
thyroid cancer: Clinical insights from a narrative review of
literature. Thyroid. 33:674–681. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Montes de Jesus FM, Espeli V, Paone G and
Giovanella L: Add-on radioiodine during long-term BRAF/MEK
inhibition in patients with RAI-refractory thyroid cancers: a
reasonable option? Endocrine. 81:450–454. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Carmona Matos DM, Jang S, Hijaz B, Chang
AW, Lloyd RV, Chen H and Jaskula-Sztul R: Characterization of
somatostatin receptors (SSTRs) expression and antiproliferative
effect of somatostatin analogues in aggressive thyroid cancers.
Surgery. 165:64–68. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Loibl S, André F, Bachelot T, Barrios CH,
Bergh J, Burstein HJ, Cardoso MJ, Carey LA, Dawood S, Del Mastro L,
et al: Early breast cancer: ESMO clinical practice guideline for
diagnosis, treatment and follow-up. Ann Oncol. 35:159–182. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Shete N, Calabrese J and Tonetti DA:
Revisiting estrogen for the treatment of endocrine-resistant breast
cancer: Novel therapeutic approaches. Cancers (Basel). 15:36472023.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Generali D, Berardi R, Caruso M, Cazzaniga
M, Garrone O, Minchella I, Paris I, Pinto C and De Placido S:
Aromatase inhibitors: The journey from the state of the art to
clinical open questions. Front Oncol. 13:12491602023. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Li B, Liang J, Lu F, Zeng G, Zhang J, Ma
Y, Liu P, Wang Q, Zhou Q and Chen L: Discovery of novel inhibitor
for WNT/β-catenin pathway by tankyrase 1/2 structure-based virtual
screening. Molecules. 25:16802020. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Li B, Orton D, Neitzel LR, Astudillo L,
Shen C, Long J, Chen X, Kirkbride KC, Doundoulakis T, Guerra ML, et
al: Differential abundance of CK1α provides selectivity for
pharmacological CK1α activators to target WNT-dependent tumors. Sci
Signal. 10:eaak99162017. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Fang L, Zhu Q, Neuenschwander M, Specker
E, Wulf-Goldenberg A, Weis WI, von Kries JP and Birchmeier W: A
small-molecule antagonist of the β-catenin/TCF4 interaction blocks
the self-renewal of cancer stem cells and suppresses tumorigenesis.
Cancer Res. 76:891–901. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Wiese M, Walther N, Diederichs C, Schill
F, Monecke S, Salinas G, Sturm D, Pfister SM, Dressel R, Johnsen SA
and Kramm CM: The β-catenin/CBP-antagonist ICG-001 inhibits
pediatric glioma tumorigenicity in a Wnt-independent manner.
Oncotarget. 8:27300–27313. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Chatterjee A, Paul S, Bisht B,
Bhattacharya S, Sivasubramaniam S and Paul MK: Advances in
targeting the WNT/β-catenin signaling pathway in cancer. Drug
Discov Today. 27:82–101. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Tabernero J, Van Cutsem E, Garralda E, Tai
D, De Braud F, Geva R, van Bussel MTJ, Fiorella Dotti K, Elez E, de
Miguel MJ, et al: A phase Ib/II study of WNT974 + encorafenib +
cetuximab in patients with BRAF V600E-mutant KRAS wild-type
metastatic colorectal cancer. Oncologist. 28:230–238. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Burock S, Daum S, Keilholz U, Neumann K,
Walther W and Stein U: Phase II trial to investigate the safety and
efficacy of orally applied niclosamide in patients with
metachronous or sychronous metastases of a colorectal cancer
progressing after therapy: The NIKOLO trial. BMC Cancer.
18:2972018. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Mraz KA, Hodan R, Rodgers-Fouche L, Arora
S, Balaguer F, Guillem JG, Jeter JM, Kanth P, Li D, Liska D, et al:
Current chemoprevention approaches in lynch syndrome and familial
adenomatous polyposis: A global clinical practice survey. Front
Oncol. 13:11418102023. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Peixoto RD, Oliveira LJC, Passarini TM,
Andrade AC, Diniz PH, Prolla G, Amorim LC, Gil M, Lino F,
Garicochea B, et al: Vitamin D and colorectal cancer-A practical
review of the literature. Cancer Treat Res Commun. 32:1006162022.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Filetti S, Durante C, Hartl DM, Leboulleux
S, Locati LD, Newbold K, Papotti MG and Berruti A; ESMO Guidelines
Committee. Electronic address, : simpleclinicalguidelines@esmo.org:
ESMO clinical practice guideline update on the use of systemic
therapy in advanced thyroid cancer. Ann Oncol. 33:674–684. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Gouda MA and Subbiah V: Precision oncology
with selective RET inhibitor selpercatinib in RET-rearranged
cancers. Ther Adv Med Oncol. 15:175883592311770152023. View Article : Google Scholar : PubMed/NCBI
|














