Introduction
Prostate cancer (PCa) is the second most common
cancer among men (1), and remains a
major health challenge owing to it rising incidence and mortality
rates (2). According to the World
Health Organization, PCa is the fourth most common cancer globally,
with ~1.4 million new cases and ~375,000 deaths reported in 2020
alone (3), emphasizing the urgent
need for improved prevention and diagnostic strategies. PCa
progression is highly dependent on the androgen receptor (AR),
which fuels tumor growth and survival (4). The mainstream treatment for localized
and metastatic PCa is androgen deprivation therapy (ADT), which
reduces circulating androgens and abrogates AR signalling to
prevent disease progression (5,6).
Despite treatment with ADT, AR signalling is re-activated in most
patients and these patients evade therapy-induced castration
conditions, resulting in the recurrence of PCa as
castration-resistant PCa (CRPC) (7,8).
Re-activation of AR signalling occurs despite low levels of
androgens in CRPC. Thus, the AR plays a central role in mediating
tumour survival. Treatment with second-generation androgen receptor
pathway inhibitors (ARPIs) such as enzalutamide (ENZ; also known as
MDV3100), abiraterone and apalutamide has been successful in
managing CRPC tumors and increasing patient survival (9,10). The
competitive non-steroidal AR antagonist, ENZ, improves survival in
patients with non-metastatic CRPC (11,12).
Despite its potent AR pathway inhibition, the benefits of ENZ are
short-lived, and patients inevitably progress to metastatic CRPC
(mCRPC) (13,14). ARPI resistance represents a clinical
challenge due to the lack of third-line treatment options. Taken
together, these findings highlight the urgent need for new
therapeutic options for refractory patients with mCRPC, including
those resistant to second-generation ARPIs.
The progression of CRPC to ARPI resistance may be
mediated through adaptive responses that activate AR-signaling via
other pathways. A number of underlying mechanisms exist, including
the alteration of AR signalling via i) aberrant glucocorticoid
receptor upregulation (15), ii) AR
splice variants (such as AR-V7) (16,17),
iii) AR gene mutations (18), iv)
an increase in AR expression (19),
and v) enhancer amplification and duplication of the AR gene
(20–22). CRPC predominantly remains AR+
(23,24), and a subset of ENZ-resistant models
display AR reactivation (25,26),
which demonstrates the importance of AR signalling in mCRPC and
indicates that it remains a therapeutic vulnerability.
In the present study, the efficacy of ADA-308 was
explored, as a possible rigorous benchmark against established
anti-androgens, such as ENZ and darolutamide (ODM-201) (27). The mechanism of action of ADA-308
was investigated, particularly in terms of its AR inhibition
activity in both AR-sensitive adenocarcinoma (Adeno) and
ENZ-resistant cell models. Furthermore, the ability of ADA-308 to
inhibit AR nuclear translocation and its impact on proliferation
in vitro and on tumor growth in vivo were examined to
establish its potential as an anti-androgen therapeutic option.
Materials and methods
Compound
ADA-308 was synthesized by Aranda Pharma Ltd. and
manufactured by Jubilant Chemsys Ltd. ADA-308 boasts a notable
purity level of 99.8%, signifying its high quality and consistency.
The batch no. J763-Z01220-083 identifies the compound used in the
present study.
Cell lines and cell culture
treatments
Cell lines and cell culture treatments were
maintained under standard conditions of 37°C and 5% CO2. The LNCaP
cell line was obtained from ATCC. 49CENZR (49C
enzalutamide-resistant) and 49FENZR (49F enzalutamide-resistant)
were generated from LNCaP cells, as previously detailed by our
group (26,28). LNCaP and LNCaP-driven cell lines
were cultured in RPMI-1640 media (Gibco; Thermo Fisher Scientific,
Inc.; cat. no. 11875093) supplemented with 5% FBS (Gibco; Thermo
Fisher Scientific, Inc.; cat. no. A3160701). ENZ-resistant cell
lines were also cultured in 10 µmol/l ENZ. In addition, when
indicated, cell lines were treated with 10 µmol/l ENZ or 10 µmol/l
ADA-308 (Aranda Pharma Ltd.). For hormone stimulation with
synthetic androgen, cells were treated with 10 nM R1881
(MilliporeSigma; cat. no. 965-93-5).
In vivo study
The animal experiments adhered to protocols approved
by The Animal Care Committee at The University of British Columbia
(Vancouver, Canada; approval no. A16-0246; approval date,
12/15/2016). Mice were housed in ventilated cages (4 mice per cage)
under controlled conditions, including constant humidity (25–27%)
and temperature (21–22°C), with a 12-h light-dark cycle. The mice
were provided with unrestricted access to rodent chow diet and
water and experiments on the mice began between 6–8 weeks of age.
At the experimental or humane endpoint, mice were euthanized using
an inhalant anesthetic (3% isoflurane) followed by carbon dioxide
(50% of the cage volume per min). A secondary accepted physical
method of euthanasia (decapitation) was performed to prevent
revival. For castration, 2.5% isoflurane vaporizer and 2 l/min
oxygen were used for anesthesia, providing both the induction and
maintenance doses. A total of 12 mice were assigned per treatment
group. The mice weighed ~20 g at the start of the study and were
supplied by Envigo.
Male athymic mice were castrated and allowed to
recover from surgery for 3 days. Then, 2×106 49FENZR cells were
inoculated twice, once per site on the right and left flanks for
the first in vivo study using 25 or 50 mg/kg ADA-308 and
once on the right site for the second study using 12.5 or 25 mg/kg
ADA-308. The mice were recovered for 3 days then administered 10
mg/kg ENZ daily until the tumor volume reached 200 mm3. Next, ENZ
(10 mg/kg) was either continued (ENZ group) or switched to vehicle,
ADA-308 at 12.5, 25 or 50 mg/kg twice a day (BID), or ODM-201
(Orion Pharmaceuticals Corporation) at 50 mg/kg BID. All treatments
were administered orally (gavage) and all in vivo studies
utilized a common vehicle, a 2% Tween-0.5% carboxymethyl cellulose
sodium salt solution. The tumor volumes were measured three times
per week in a blinded fashion and calculated using the formula:
Volume=[π (length × width × height)]/6. Recruitment was conducted
in 10 cycles, the length of ENZ treatment ranging from 19 to 52
days. Mice were sacrificed at predetermined time points after
treatment, when the tumor volume reached 2,000 mm3, when tumors
reached >10% of the body weight or the body weight loss was
>15%, whichever came first. The maximum long diameter of a
single tumor was 21 mm and the maximum sum of the long diameter of
both the left and right tumors in a single mouse was 33.5 mm. The
maximum sum of the tumor volume of both the left and right tumors
in a single mouse was 2,425 mm3. While the humane endpoint was set
at a tumor volume of 2,000 mm3, the individual mouse in
question that exceeded the tumor size belonged to the ENZ treatment
group and had a tumor volume of 1,462 mm3 at the
2.5-week time point. Therefore, the mouse was not sacrificed at
that time. By the 3-week time point, when tumors were measured
again, the tumor volume had grown beyond the endpoint, resulting in
a measurement of 2,425 mm3, at which point the mouse was
then sacrificed.
Western blotting
Proteins were extracted from cells cultured in
vitro. The cells were washed once with 1X PBS and subsequently
lysed using RIPA buffer (Thermo Fisher Scientific, Inc.; cat. no.
PI89901) enriched with a 1X concentration of cOmplete EDTA-free
protease inhibitors cocktail (Roche Diagnostics; cat. no.
11836170001) and phosphatase inhibitors (PhosSTOP; Roche
Diagnostics; cat. no. 4906845001). Following protein quantification
using the BCA protein assay (Thermo Fisher Scientific, Inc.; cat.
no. 23225), the samples were subjected to a 5-min boiling step in
4X SDS sample buffer. The 4X SDS sample buffer contained 8–10% SDS,
200 mM Tris-HCl (pH 6.8), 40% glycerol, 0.02% Bromophenol Blue and
5% β-mercaptoethanol. Equal amounts of protein (40 µg per lane)
were resolved by SDS-PAGE using 10% polyacrylamide gels. The
proteins were transferred onto PVDF membranes, then the membranes
were blocked with Odyssey Blocking Buffer (LI-COR Biosciences; cat.
no. 15590545) at room temperature for 30 min and probed with
primary antibodies at the specified dilutions overnight at 4°C. The
membranes were washed three times with 1X TBST (2% Tween-20) for 10
min, then probed with the appropriate secondary antibody for 1 h at
room temperature. The membranes were washed three times with 1X
TBST for 10 min before visualization using a LI-COR Odyssey
Scanner. The immunoblotting utilized the following antibodies: AR
(clone D6F11; 1:1,000; Cell Signaling Technology, Inc.; cat. no.
5153) and prostate-specific antigen (PSA; clone D6B1; 1:5,000; Cell
Signaling Technology, Inc.; cat. no. 5365), with Vinculin (clone
hvin-1; 1:25,000; Cell Signaling Technology, Inc.; cat no. 4650)
serving as the loading control. The secondary antibodies included
IRDye 800CW donkey anti-rabbit (1:10,000; LI-COR Biosciences; cat.
no. 926-32213). Uncropped western blots are shown in Fig. S1.
Reverse transcription-quantitative PCR
(RT-qPCR)
Cells were plated in 100-mm plates at a density of
4.0×105 cells/plate in RPMI media supplemented with 10% FBS and 1%
penicillin/streptomycin (pen/strep). The following day, cells were
treated with either vehicle (DMSO, final concentration 0.1%), ENZ
(10 µM) or ADA-308 (concentrations of 1, 2, 5, 7.5 or 10 µM; final
concentration in 10 ml). After 72 h of treatment, the cells were
washed with 1X PBS and detached in PBS/5 mM EDTA/sodium vanadate,
pelleted (centrifuged at 1,200 × g for 5 min at 4°C) and
resuspended in TRIzol (Thermo Fisher Scientific, Inc.; cat. no.
15596026) for RNA extraction. For reverse transcription, cDNA
synthesis was performed using SuperScript™ IV Reverse Transcriptase
(SSIV RT; Thermo Fisher Scientific, Inc.; cat. no. 18090010),
according to the manufacturer's protocol. Briefly, 0.2 µg RNA was
mixed with oligo(dT)20 primers (Invitrogen; Thermo Fisher
Scientific, Inc.; cat. no. 18418020) and dNTPs (Invitrogen; Thermo
Fisher Scientific, Inc. cat. no. 10297018), and the mixture was
incubated at 65°C for 5 min. After chilling on ice, the buffer, DTT
and SuperScript IV enzyme were added. The reaction was carried out
at 23°C for 10 min, followed by 50°C for 30 min, and terminated at
80°C for 10 min. cDNA was then used for qPCR analysis using SYBR™
Green PCR Master Mix (Thermo Fisher Scientific, Inc.; cat. no.
4309155), according to the manufacturer's instructions. Primers and
cDNA templates were added to 386-well plates in triplicate. The
expression of each gene was normalized to the expression of GAPDH
and the 2-∆∆Cq method (29) was
used to quantify the change in expression from vehicle (DMSO)
treatment. Experiments were repeated twice and the mean ± SEM of
the independent experiments are shown. The primer sequences were as
follows: GAPDH forward, GGAGCGAGATCCCTCCAAAAT; GAPDH reverse,
GGCTGTTGTCATACTTCTCATGG; PSA/kallikrein-3 (KLK3) forward,
CACAGCCTGTTTCATCCTGA; KLK3 reverse, AGGTCCATGACCTTCACAG; homeobox
protein Nkx-3.1 (NKX3.1) forward, GGACTGAGTGAGCCTTTTGC; NKX3.1
reverse, CAGCCAGATTTCTCCTTTGC; FK506 binding protein 5 (FKBP5)
forward, TCCCTCGAATGCAACTCTCT; FKBP5 reverse, GCCACATCTCTGCAGTCAAA;
transmembrane protease, serine 2 (TMPRSS2) forward,
TGGTAGTGTCCCCAGCCTAC; TMPRSS2 reverse, AAAGCAGCTGAAATAGGCCA; AR
forward, TACCAGCTCACCAAGCTCCT; and AR reverse,
GCTTCACTGGGTGTGGAAAT. The temperature protocol used for the qPCR
reaction was as follows: Initial Denaturation at 95°C for 10 min;
amplification cycles (35 cycles): Denaturation at 95°C for 15 sec,
annealing at 60°C for 30 sec and extension at 72°C for 30 sec;
final extension at 72°C for 5 min.
Microscopy
Cells were plated in RPMI media supplemented with 5%
charcoal-stripped serum (Thermo Fisher Scientific, Inc.; cat. no.
A3382101) on poly-L-lysine-coated coverslips at a density of 1×105
cells/well. The following day, the cells were pretreated with ENZ
(10 µM), ADA-308 (10 µM) or DMSO (0.1%) for 24 h. Then, the cells
were treated with either DMSO or the AR agonist, R1881, at a
concentration of 10 nM for 20 min. The cells were fixed with 100%
ice-cold methanol for 10 min, followed by 1X PBS washes. The cells
were then incubated with anti-AR antibody (1:1,000; clone 441;
Santa Cruz Biotechnology, Inc.; cat. no. sc-7305) for 1.5 h at room
temperature, followed by washes with 1X PBS to remove unbound
antibodies. Next, the cells were incubated with a secondary
anti-mouse Alexa 488-conjugated antibody (1:1,000; Invitrogen;
Thermo Fisher Scientific, Inc.; cat. no. A21202) for 45 min at room
temperature. The cells were washed with 1X PBS to remove unbound
secondary antibodies, then DAPI (Thermo Fisher Scientific, Inc.;
cat. no. D1306) coverslips were mounted on slides to stain the
nucleus. Fluorescent images were collected using a ×60 oil
immersion objective, FV3000RS confocal microscope equipment and
Olympus FV31S-SW software (version 2.3.2.169; Olympus
Corporation).
Cell proliferation
Cells were seeded at a density of 2,000 cells per
well in 96-well plates in RPMI-1640 media supplemented with 5% FBS
and treated with either vehicle [DMSO (0.1%)], ENZ (10 µM), ADA-308
(10 µM) or ODM-201 (10 µM). Each treatment condition was set up in
8 wells. The plates were placed in the IncuCyte live-cell analysis
system (Essen Bioscience), and images were acquired every 12 h for
7 days. IncuCyte software (v2020C; Essen Bioscience) was used to
analyze the cell confluency automatically over time.
Cell cycle
Cells were plated in 100-mm plates at a density of
2×105 cells/plate in RPMI media supplemented with 10% FBS and 1%
pen/strep. The following day, the cells were treated with either
vehicle (DMSO at 0.1%), ENZ (10 µM) or ADA-380 (concentrations of
1, 2, 5, 7.5 or 10 µM; final concentration in 10 ml). After 72 h of
treatment, the cells were washed with 1X PBS and detached in PBS/5
mM EDTA/sodium vanadate, pelleted by centrifuging at 1,200 × g for
5 min at 4°C, fixed and permeabilized in 70% ice-cold ethanol for
30 min and then stored at −30°C for a minimum of 24 h. The cells
were pelleted by centrifuging at 1,200 × g for 10 min at 4°C and
washed in PBS, then stained in propidium iodide (PI;
MilliporeSigma; cat no. P4864) solution (50 µg/ml PI, 0.1 mg/ml
RNAse, 0.05% Triton X-100, 1X PBS) for 40 min at 37°C. Finally, the
cells were washed and strained before flow cytometry analysis. Data
were acquired by FACS on a Canti II (BD Biosciences). Data were
analyzed using FlowJo software (version 10.4.2; FlowJo LLC).
Representative histograms are shown in Fig. S2.
Luciferase assay
Cells were seeded in 12-well plates at a density of
1×105 cells/well in RPMI media supplemented with 10% FBS and 1%
pen/strep. The following day, the cells were transfected with 0.2
µg of the probasin RR3 luciferase reporter using TransIT-2020
(Mirus Bio, LLC) in Opti-MEM media (Gibco; Thermo Fisher
Scientific, Inc.) following the manufacturer's protocol. The
Probasin ARR3 tk-luc reporter was kindly provided by Dr Martin
Gleave's Lab at Vancouver Prostate Centre (Vancouver, Canada)
(30). After 24 h, the Opti-MEM
transfection mix was removed and replaced with 10 µM (final) of the
compound (ENZ, ODM-201 or ADA-308) in RPMI media supplemented with
10% FBS and 1% pen/strep in triplicate. The following day, the
wells were washed once with pre-warmed 1X PBS, then incubated with
200 µl of 1X Passive lysis buffer (Promega Corporation; supplier
no. E1941; cat. no. PAE1941) at room temperature with shaking for
30 min and frozen at −80°C for 45 min. The plates were thawed, the
lysate was collected in microcentrifuge tubes and the debris was
cleared via centrifugation at 1,200 × g for 10 min at 4°C. Next, 50
µl (per well) of the supernatant was added to 96-well white, flat
bottom plates, then 75 µl luciferase assay buffer (Promega
Corporation; cat. no. E1910) was automatically injected per well.
After 30 sec of incubation, signal was detected by a luminescent
plate reader (Tecan infinite M200Pro). Fluorescence units were
normalized to the protein concentration per sample (BCA assay) and
calculated relative to the control condition [DMSO for LNCaP; ENZ
(10 µM) for 49CENZR and 49FENZR]. Experiments were repeated three
times, and the mean ± SEM of the independent experiments was
calculated.
RNA-sequencing (RNA-seq)
Cells were grown in RPMI-1640 media supplemented
with 5% FBS and DMSO (0.1%), ENZ (10 µM) or ADA-308 (10 µM). Total
RNA was isolated from the cells after 72 h of treatment using the
PureLink RNA Mini Kit (Thermo Fisher Scientific, Inc.). The library
was generated using the NEBnext Ultra ii Stranded RNA Library Prep
Kit (New England BioLabs, Inc.; cat. E7770S), the quality of the
RNA samples was assessed by measuring the 230/260 and 260/280 ratio
using a NanoDrop spectrophotometer (Thermo Fisher Scientific,
Inc.), ensuring values were >1.8 and 2, respectively. Sequencing
was performed on an Illumina NextSeq 500 (42×42-bp paired-end
reads) by the University of British Columbia Sequencing +
Bioinformatics Consortium (Vancouver, Canada), targeting 20 million
reads per sample. Data was de-multiplexed using bcl2fastq2
Conversion Software (version 2.20; Illumina, Inc.), and read
sequences were aligned to the human reference genome, hg38, using
STAR aligner (version 2.7.8a) (31). Assembly and differential expression
were estimated using Cufflinks software (version 2.2.1) (32), available through the Illumina
BaseSpace Sequence Hub. Gene expression data (raw count data) were
normalized using ‘DESeq’ (33) in
Rstudio (version 4.1.2; http://cran.r-project.org/), and subsequently, log2
was transformed. Unsupervised clustering was generated using R, and
data were visualized using the R ‘ggplot’ program or GraphPad Prism
(version 8; Dotmatics). The significance of the expression level
differences between the treatment samples was determined using an
unpaired t-test in GraphPad Prism or R.
Chromatin
immunoprecipitation-sequencing (ChIP-seq) data analysis
ChIP-seq Fastq files were downloaded from the Gene
Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/). Publicly available
AR ChIP-seq datasets [GSM1069669 and GSM1236925, from Chen et
al (34)] used in the present
study from were downloaded from GSE43791. Publicly available
control LNCaP RNA-seq [GSM4777223 and GSM4777224, from Davies et
al (26)] data were downloaded
from GSE138460. Data was processed using FastQC (version 0.11.9)
(35) for quality control analysis.
Adapter sequences were removed using Cutadapt (version 1.18;
http://cutadapt.readthedocs.io/en/stable/) and reads
were aligned to the human genome reference, hg38, using BWA-MEM
software (version 0.7.17) (36).
SAM files were converted to BAM files using SAMtools software
(version 1.1.2) (37). MACS2
(version 2.2.7.1) (38) was used to
call peaks with a false discovery rate (FDR) of 0.05 using the
narrow peak caller for AR-bound genes. DeepTools (version 2.30.0)
(39) was used to visualize data,
and BEDtools (version 2.28.0) (40)
generated shared and unique peaks between dihydrotestosterone (DHT)
and ENZ AR ChIP-seq samples. The peaks were annotated using HOMER
(version 3.0; http://homer.ucsd.edu/homer/).
Gene ontology and pathway
analysis
Pathway analysis was conducted using gene set
enrichment analysis (GSEA) software, available from the Broad
Institute (Massachusetts Institute of Technology) and gProfiler
(41). This analysis aimed to
discern the functions associated with differentially expressed
genes within the Molecular Signatures Database (version 7.1)
(42). The GSEA tool was utilized
in classic mode to identify significantly enriched biological
pathways. Pathways exhibiting enrichment with a nominal P<0.05
and FDR <0.25 were considered statistically significant. For
single-sample GSEA, gProfiler, a web server designed for functional
enrichment analysis and the conversion of gene lists, was
utilized.
Statistical analysis
Statistical analysis was conducted using Microsoft
Excel and GraphPad Prism software (version 8; Dotmatics). All
presented in vitro experiments were independently replicated
a minimum of three times. To analyze data variance between multiple
groups, a one-way ANOVA followed by the Dunnet's test were used to
perform multiple comparisons within each group compared with the
control group, which is depicted in bar charts. For longitudinal
profiling experiments, a two-tailed unpaired Student's t-test was
performed to determine the statistical difference at the final time
point. Visualization was performed using GraphPad Prism 8 or
Rstudio (version 4.1.2; http://cran.r-project.org/). P<0.05 was considered
to indicate a statistically significant difference.
Results
Introduction to ADA-308
ADA-308, a novel arylamide compound, emerged
following an extensive and meticulous design process involving the
synthesis and evaluation of over 650 carefully crafted structures,
comprising >220 profiled small molecules and various chemical
scaffolds. In preliminary pharmacokinetic studies (unpublished
data), ADA-308 exhibited notable characteristics in mice. When
administered orally at 100 mg/kg in mice, ADA-308 displayed a
plasma half-life (T1/2) of 10.9 h. In comparison, oral
administration of ADA-308 at a 30 mg/kg dose in rats resulted in a
plasma T1/2 of 12.6 h. ADA-308 was designed to address
treatment resistance observed with other AR antagonists. The
compound was designed considering optimized binding affinity to the
AR, improved pharmacokinetic properties and its ability to retain
AR antagonism in CRPC cells (such as when AR is upregulated or
mutated). In addition to ENZ-resistant conditions (such as F876L AR
mutation), ADA-308 was screened in a panel of CRPC AR mutants
(T877A and W741L), where it retained its antagonistic activity,
highlighting its potential as a versatile and practical treatment
option.
ADA-308 suppresses AR transcriptional
activity in Adeno and ENZ-resistant cell lines
To assess the impact of ADA-308 treatment on AR
signalling, the prostate Adeno cell line, LNCaP, was treated with
ADA-308. A dose-dependent decrease in the expression of PSA, a
canonical AR target, was observed, which was similar to that
observed following ENZ treatment (Fig.
1A). The mRNA expression of other canonical AR targets also
decreased in a dose-dependent manner, comparable to ENZ (Fig. 1B and Table SI). Furthermore, using an
androgen-responsive luciferase reporter linked to the probasin
(43) promoter, known for its
robust AR-specific and tissue-specific regulation (43), it was observed that ADA-308
significantly and efficiently reduced AR activity. This was similar
to the results achieved with ENZ or ODM-201 (high-affinity AR
antagonists) (27) (Fig. 1C).
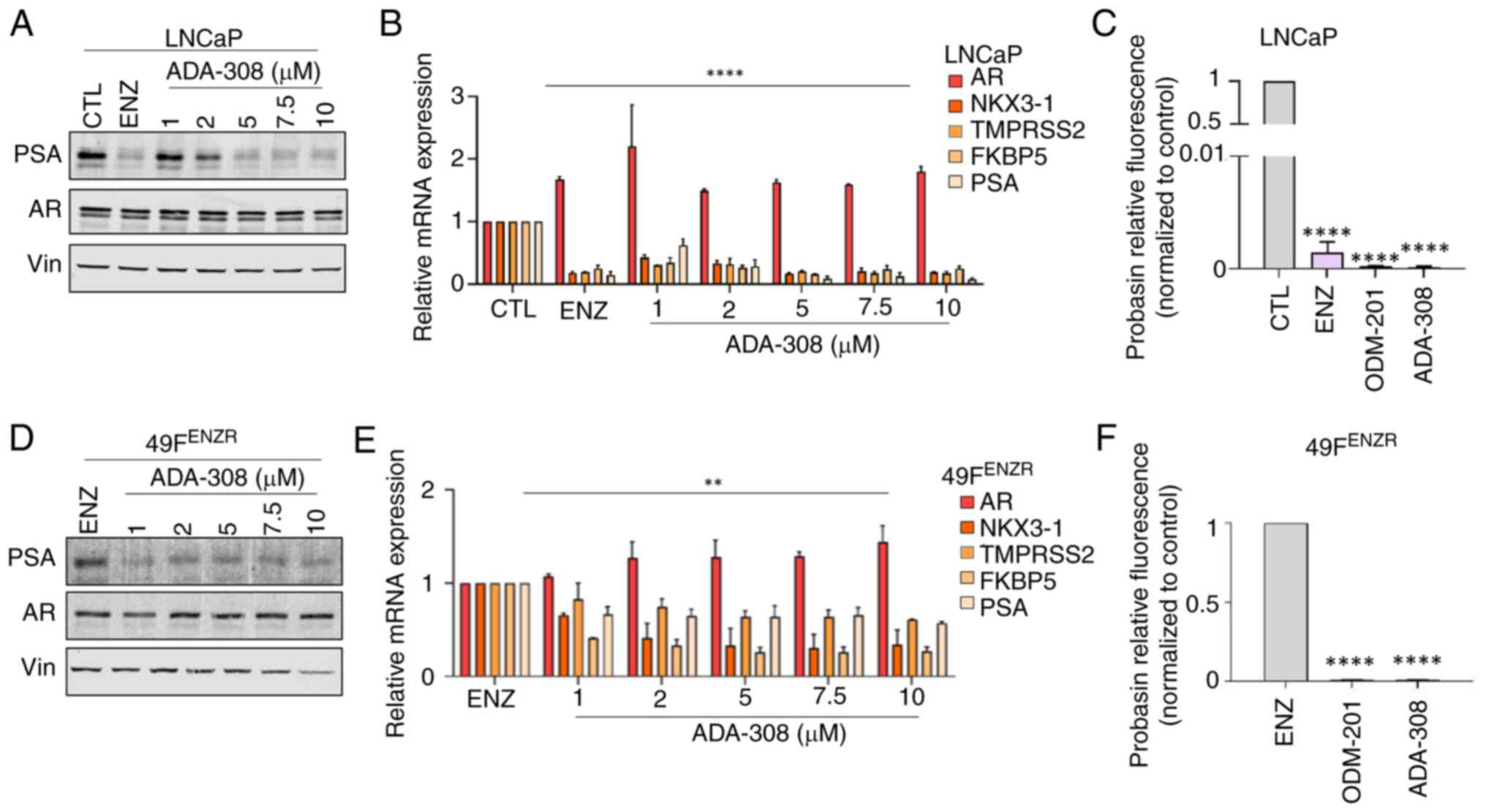 | Figure 1.ADA-308 decreases AR activity in
adenocarcinoma and ENZ-resistant prostate cancer cell lines. (A)
Western blot shows that treatment with ADA-308 inhibited PSA
(canonical AR target) expression in ENZ-sensitive LNCaP cells in a
dose-dependent manner (1–10 µM). ENZ (10 µM) was used as a positive
CTL. Cells were treated for 72 h before protein lysate was
harvested (n=3 independent biological replicates). Vin was used as
the loading CTL. (B) ADA-308 decreased the transcription of AR
target genes in the LNCaP cell line, as shown by RT-qPCR. Cells
were treated for 72 h with 10 µM ENZ or 1–10 µM ADA-308 before RNA
was extracted (n=3 independent biological replicates). (C) ADA-308
at 10 µM inhibited AR transactivation, as measured by luciferase
assay with R1881-induced activation of probasin AR reporter in
LNCaP cells (n=2 independent biological replicates). Data shows
relative fluorescence normalized to the CTL (DMSO). (D) Western
blot shows that treatment with ADA-308 inhibited PSA expression in
ENZ-resistant 49FENZR cells in a dose-dependent manner (1–10 µM).
ENZ (10 µM) was used as the CTL. Cells were treated for 72 h before
protein lysate was harvested (n=3 independent biological
replicates). Vin was used as the loading CTL. (E) ADA-308 decreased
the transcription of AR target genes in the 49FENZR cell line as
shown by RT-qPCR. Cells were treated for 72 h with 1–10 µM ADA-308
or 10 µM ENZ before RNA was extracted (n=2 independent biological
replicates). (F) ADA-308 at 10 µM inhibited AR transactivation, as
measured by luciferase assay with R1881-induced activation of
probasin AR reporter in 49FENZR cells. Data shows relative
fluorescence normalized to ENZ (n=2 independent biological
replicates). All data were analyzed using a one-way ANOVA to assess
the variance between dosages. Post hoc comparisons were performed
using Dunnett's test to compare each treatment group to the
control. Data are presented as the mean±standard deviation.
**P<0.01, ****P<0.0001. All exact P-values are listed in
Table SI. 49FENZR, 49F
ENZ-resistant; AR, androgen receptor; CTL, control; ENZ,
enzalutamide; PSA, prostate-specific antigen; RT-qPCR, reverse
transcription-quantitative PCR; Vin, vinculine. |
CRPC is often treated with the potent AR antagonist,
ENZ, which frequently leads to ENZ resistance through the
re-activation of the AR signalling axis. To model ENZ-resistant
disease, with re-activation of AR signalling, our lab previously
generated ENZ-resistant cell lines, 49CENZR and 49FENZR, by
serially passaging the PCa Adeno cell line, LNCaP, in castrated
mice treated with ENZ (28). These
cell lines are derived from PSA+ tumors and retain PSA expression
(44). These cell lines harbour the
AR F876L activating mutation (45),
a rare mutation in the early stages of the disease that is
frequently observed in CRPC (46)
and ENZ-resistant tumors (47,48).
By altering the ligand binding pocket of AR, F876L allows other
steroid hormones (such as corticosteroids and anti-androgens) to
activate AR (49), rendering ENZ an
agonist that drives phenotypic resistance (47,48).
To explore the potential of re-targeting AR signaling in these
models, the effect of ADA-308 on AR-dependent genes was
investigated. The findings revealed that treatment with ADA-308
exhibited a dose-dependent reduction in PSA expression in both
49CENZR and 49FENZR (Figs. 1D and
S3A). This reduction in AR
activity was reflected by decreased mRNA expression of canonical AR
target genes (Figs. 1E, S3B, and Table SI) and a significant decrease in
probasin luciferase activity (Figs.
1F and S3C). Notably,
treatment of LNCaP with either ENZ, ADA-308 or ODM-201 resulted in
a reduction in the PSA mRNA level (Fig. S3D), with no differences observed
between the different compounds. Taken together, these data
demonstrated that ADA-308 acts as an AR signaling inhibitor in
Adeno, particularly in ENZ-resistant cell line models.
ADA-308 inhibits AR nuclear
localization in Adeno and ENZ-resistant cell lines
AR, a nuclear transcription factor that belong to
the steroid hormone receptor superfamily (50), is activated upon binding of
androgens (51). In the absence of
a ligand, AR primarily resides in the cytoplasm and often form
complexes with heat shock protein chaperones (52). However, in the presence of ligands,
AR undergo homodimerization, translocates to the nucleus and
attaches to androgen response elements to initiate transcription
(53).
Notably, in the Adeno cell line, LNCaP, and the
ENZ-resistant cell lines, 49CENZR and 49FENZR, AR was predominantly
localized to the cytoplasm. However, AR translocates to the nucleus
upon stimulation with synthetic androgen (R1881).
Immunofluorescence microscopy showed that treatment with ADA-308
inhibited AR nuclear translocation in the LNCaP cell line, similar
to the effect observed for ENZ (Fig. 2A
and B), suggesting that both ADA-308 and ENZ effectively
hindered the androgen-induced nuclear translocation of AR. In
addition, AR was observed in the cytoplasm and nucleus of the
ENZ-resistant cell lines, 49CENZR and 49FENZR. However, treatment
with R1881 increased the ratio of nuclear AR to cytoplasmic AR
(Fig. 2C and D). Notably, ADA-308
treatment markedly prevented the androgen-induced nuclear
translocation of AR, whereas ENZ treatment failed to do so
(Figs. 2C, 2D and S4A). These results suggested that,
following the development of ENZ resistance, ADA-308 exerted its
inhibitory effect on AR signaling by preventing AR nuclear
translocation. These data highlight the potential of ADA-308 as an
antagonist of mutated AR and warrant further investigation into its
clinical response, particularly in ENZ-resistant tumors.
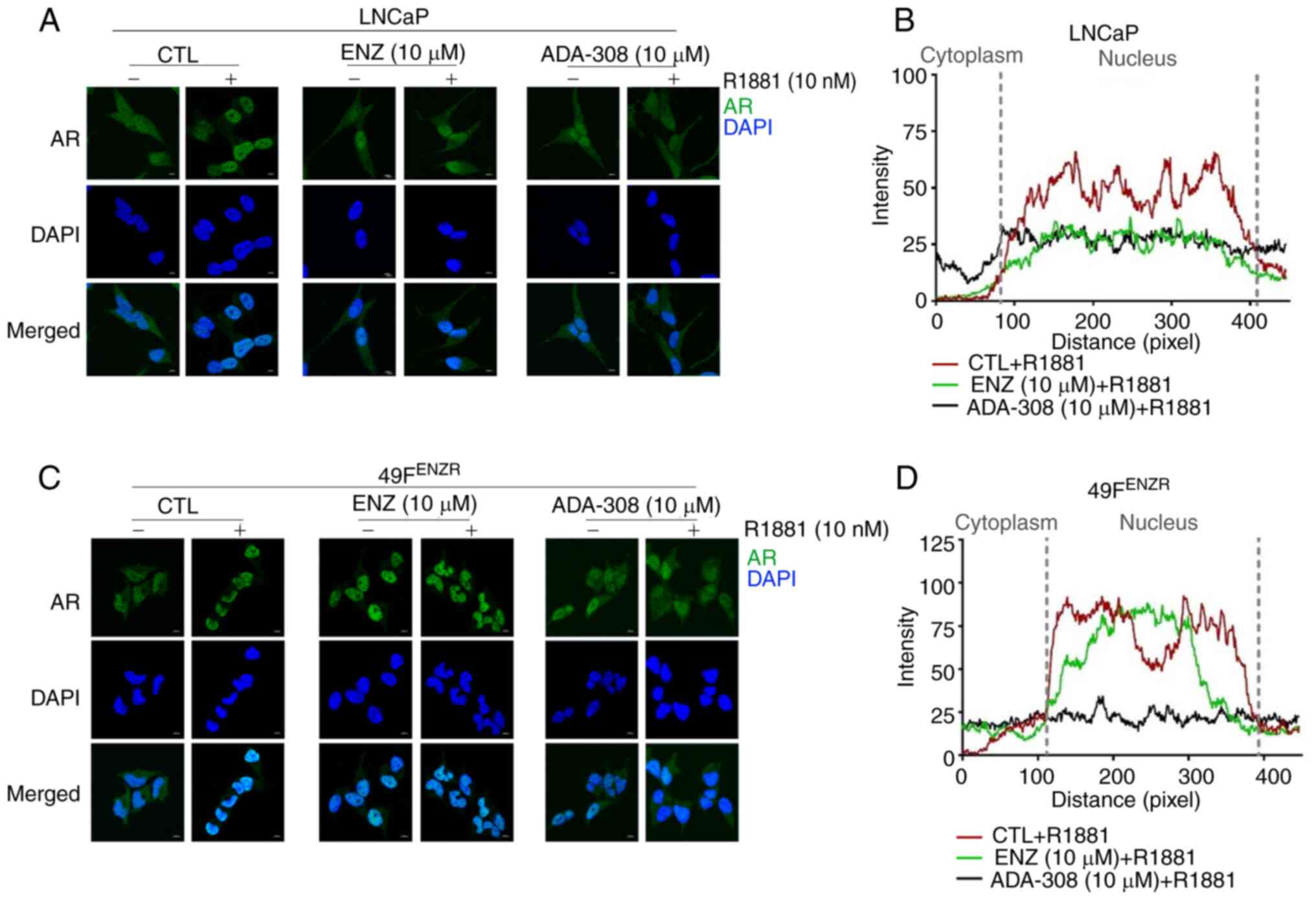 | Figure 2.ADA-308 inhibits AR nuclear
translocation in adenocarcinoma and ENZ-resistant prostate cancer
cell lines. (A) Immunofluorescence of LNCaP treated with DMSO
(CTL), AR antagonists ENZ (10 µM) or ADA-308 (10 µM) for 24 h with
or without a 20-min R1881 (10 nM) treatment; scale bar, 10 µm. AR
is shown in green and DAPI in blue. (B) Co-localization of AR and
the nuclei of LNCaP cells in the immunofluorescence data, as
measured by Zen software. Data are shown as DMSO (red), ENZ (green)
and ADA-308 (navy) plus R1881. (C) Immunofluorescence of 49FENZR
cells treated with DMSO (CTL), AR antagonists ENZ (10 µM) or
ADA-308 (10 µM) for 24 h with or without a 20-min R1881 (10 nM)
treatment; scale bar, 10 µm. AR is shown in green and DAPI in blue.
(D) Co-localization of AR and nuclei in 49FENZR cells in the
immunofluorescence data, as measured by Zen software. Data are
shown as DMSO (red), ENZ (green) and ADA-308 (navy) plus R1881.
49FENZR, 49F ENZ-resistant; AR, androgen receptor; CTL, control;
ENZ, enzalutamide. |
ADA-308 inhibits proliferation in
vitro
Next, the anti-proliferative properties of ADA-308
and its impact on the cell cycle were assessed. A reduction in the
proliferation rate of LNCaP cells was observed upon treatment with
either ADA-308 or ENZ, with ADA-308 exhibiting a more pronounced
suppression of cell proliferation than ENZ (Fig. 3A). Moreover, ADA-308 treatment
resulted in a modest increase in G0/G1 arrest; a ~9% increase in
the G0/G1 cell population was observed, which was comparable to the
~8% increase following ENZ treatment (Figs. 3C and S4). Next, the effect of ADA-308 on
ENZ-resistant cell lines was evaluated. Similar to the observations
in the Adeno cell line, ADA-308 notably suppressed ENZ-resistant
cell proliferation (Figs. 3B and
S4B). However, it was observed
that ODM-201 had a more profound effect on the cell proliferation
(Fig. S4B). In addition, a
significant increase in the G0/G1 cell population in the
ENZ-resistant cells after ADA-308 treatment was observed (Figs. 3D, S4C).
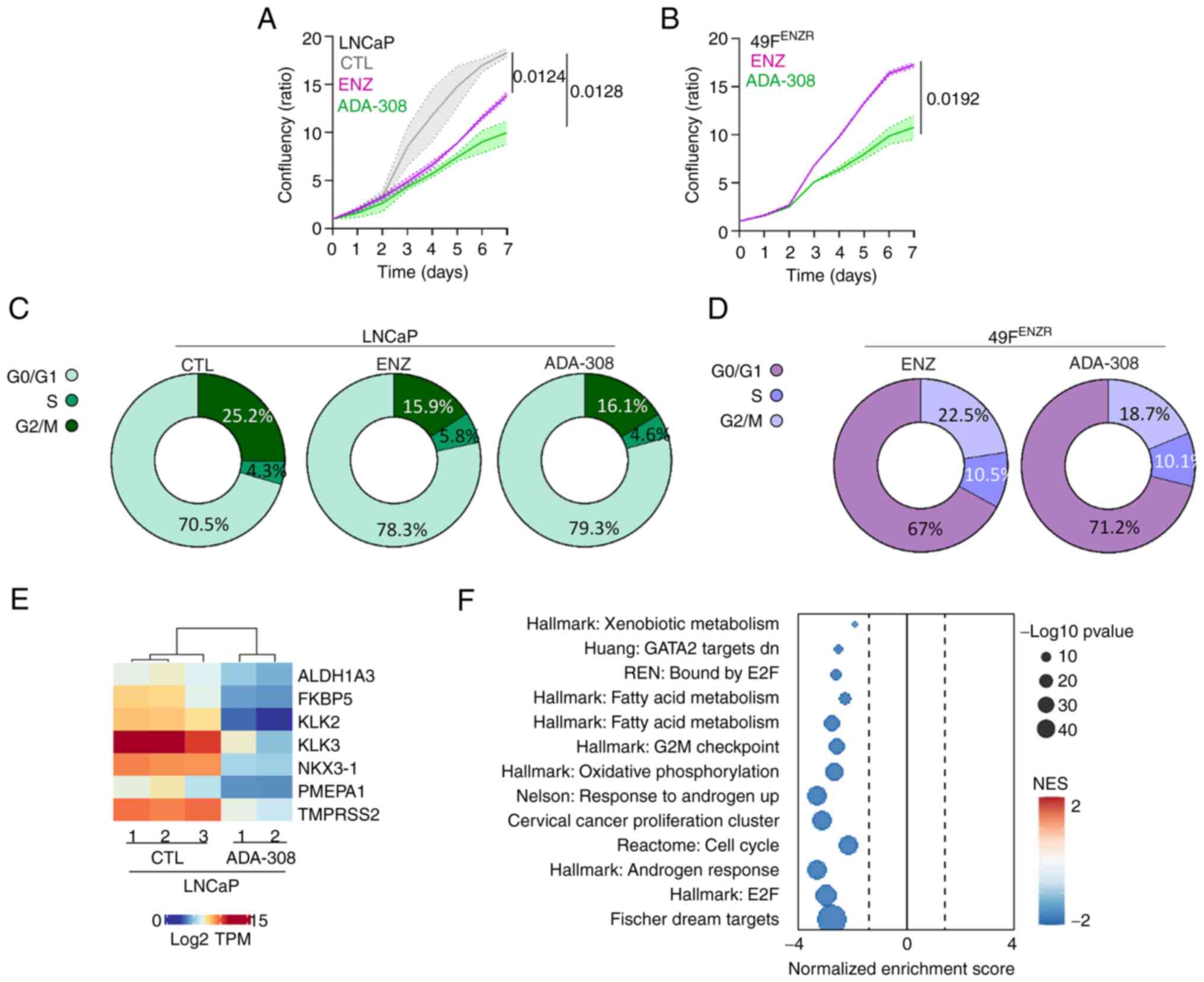 | Figure 3.ADA-308 induces G1 accumulation and
inhibits proliferation in vitro. (A) LNCaP and (B) 49FENZR
cell lines were treated with DMSO, ENZ (10 µM) or ADA-308 (10 µM)
for 7 days, and proliferation was measured using IncuCyte and
reported as confluency ratio over day 0. n=3 independently
biological replicates; data are presented as the mean ± standard
deviation. Significance was evaluated at the endpoint using
unpaired, two-tailed Student's t-test or one-way ANOVA. Post hoc
comparisons were performed using Dunnett's test to compare each
treatment group to the control. (LNCaP ENZ vs. CTL, P=0.0124; LNCaP
ADA-308 vs. CTL, P=0.0128; 49FENZR ADA-308 vs. ENZ, P=0.0192). (C)
LNCaP and (D) 49FENZR cell lines were treated with CTL, ENZ (10 µM)
or ADA-308 (10 µM) for 72 h, and the cell cycle was assessed using
flow cytometry. Two-tailed unpaired t-test or one-way ANOVA
followed by Dunnet's test was used to compare treatment to the CTL
group (LNCaP ENZ vs. CTL: G0/G1, P=0.003; S, P=0.00029; G2/M,
P=0.179; LNCaP ADA-308 vs. CTL: G0/G1, P=0.0002; S, P=0.0001; G2/M,
P= 0.319; 49FENZR ADA-308 vs. CTL: G0/G1, P=0.008; S, P=0.011;
G2/M, P=0.125); n=3 independently biological replicates. See
Figure S2 for the representative
histograms. (E) LNCaP was treated with ADA-308 (10 µM) for 72 h and
samples were collected for RNA-sequencing. The heatmap shows the
log2TPM values of canonical androgen receptor target genes. (F)
Gene Set Enrichment Analysis identified pathways differentially
regulated in LNCaP treated with ADA-308 (10 µM) for 72 h compared
with the CTL. 49FENZR, 49F ENZ-resistant; CTL, control; ENZ,
enzalutamide; NES, normalized enrichment score; TPM, transcripts
per million. |
To understand the biological impact of ADA-308,
LNCaP cells were treated with ADA-308 and RNA-seq was performed.
First, the changes in AR signalling induced by ADA-308 treatment
were assessed. A notable inhibition of AR activity was observed as
evidenced by a marked reduction in the expression of canonical AR
targets (Fig. 3E). GSEA was
performed to identify a range of pathways altered by ADA-308
treatment. As expected, the downregulation of AR-regulated pathways
following ADA-308 treatment was observed. Notably, ADA-308
significantly inhibited pathways crucial for cell proliferation and
cycle progression (Fig. 3F). These
observations highlighted the promising anti-proliferative
properties of ADA-308 in Adeno in vitro, particularly in the
context of ENZ-resistance models that are resistant to existing
treatments.
ADA-308 modulates AR-bound target
genes and associated pathways
To evaluate the efficacy of ADA-308 in comparison
with ENZ, RNA-seq of LNCaP cells treated with either ADA-308 or ENZ
was conducted. Unsupervised clustering revealed that the treated
samples clustered together and were distinct from those of the
control group (Fig. 4A). Although a
significant number of differentially expressed genes after
treatment with these AR inhibitors was observed (Fig. 4B), the difference between ADA-308
and ENZ was not statistically significant (Fig. 4C). Subsequent analysis identified
1,081 genes that were downregulated by both ADA-308 and ENZ
treatments (Fig. 4D). GSEA revealed
that these commonly downregulated genes were associated with
pathways regulating the cell cycle, proliferation and the androgen
response (Fig. 4E and Table SII). This was consistent with our
previous findings demonstrating that ENZ treatment led to reduced
proliferation and induced G0/G1 arrest in LNCaP cell lines
(54,55), but notably highlighting the
effectiveness of ADA-308 in the context of ENZ-resistant models
where ENZ was less responsive (Fig.
3A-D).
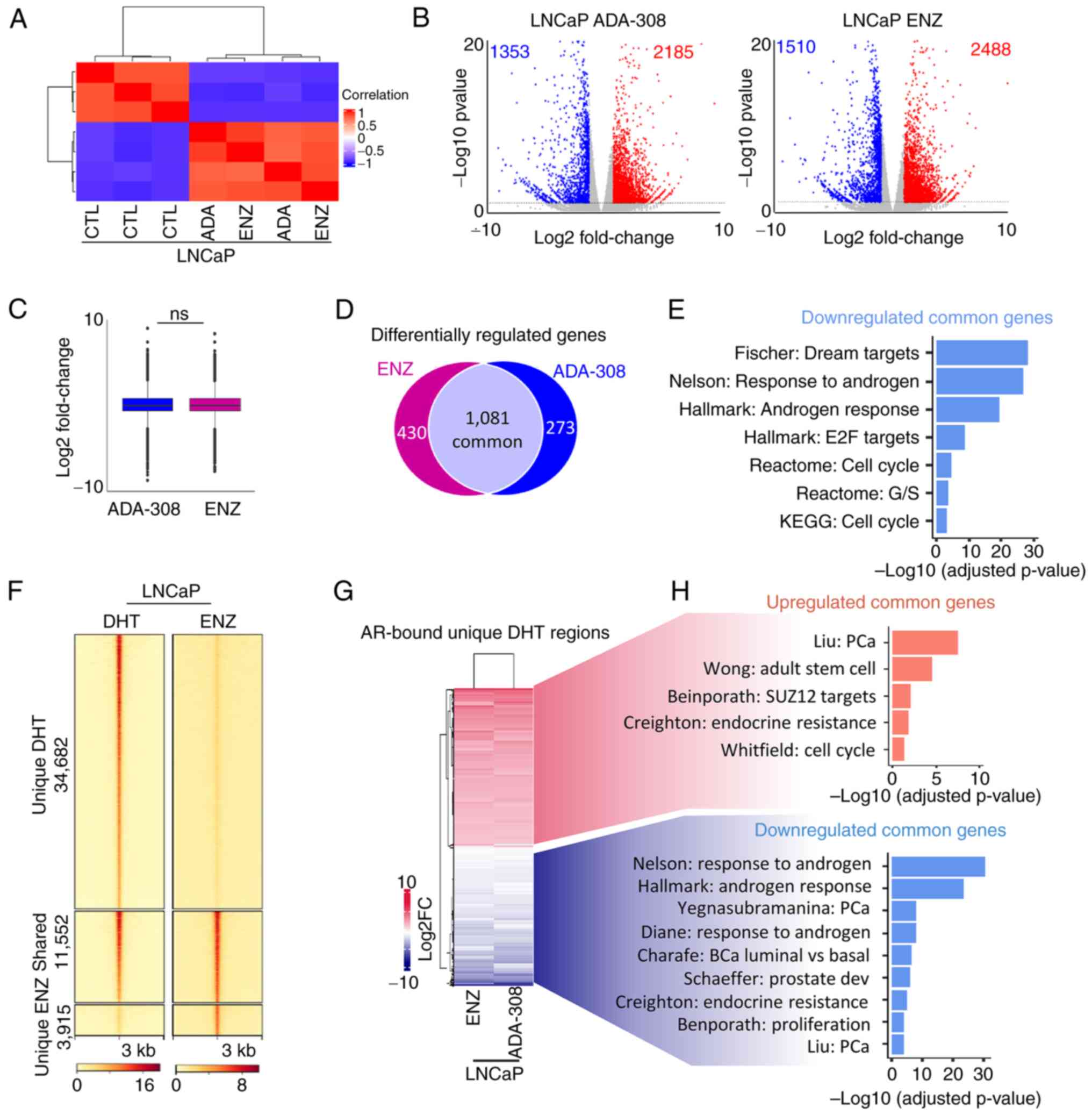 | Figure 4.Effectiveness of ADA-308 is
comparable to ENZ. (A) Unsupervised clustering using RNA-sequencing
data from LNCaP treated with ENZ (10 µM) or ADA-308 (10 µM) for 72
h shows a correlation between the CTL and treatment groups. (CTL
n=3 and treatment n=2 independent biological replicates). (B)
Volcano plot shows genes differentially regulated after treatment
with ADA-308 (10 µM) (left) or ENZ (10 µM) (right). Each dot
represents 1 gene. Blue dots represent genes downregulated >1
log2FC with P<0.05, and red dots represent genes upregulated
>1 log2FC with P<0.05. (C) The box plot shows an average
expression of all differentially regulated genes following ENZ (10
µM) or ADA-308 (10 µM) treatment compared with the CTL, presented
as log2FC. (D) Number of differentially regulated genes following
ENZ (10 µM) or ADA-308 (10 µM) treatment compared with the CTL.
Genes common between the two treatment groups are shown in the gray
section, P<0.05. (E) Gene Set Enrichment Analysis was performed
on commonly downregulated genes between ENZ (10 µM) or ADA-308 (10
µM) treatment compared with the CTL with P<0.05. Data are
presented as -log10 of the adjusted P-value. (F) A heatmap of AR
binding intensity in LNCaP cells treated with DHT or ENZ is
presented as a fold change over input, with each horizontal line
representing a 3 kb locus. Clusters are shown as regions unique to
DHT treatment (regions lost AR binding after ENZ treatment),
regions shared between DHT and ENZ treatment and regions unique to
ENZ (regions gained AR binding after ENZ treatment). (G) Heatmap
shows expression of all AR-bound annotated genes in unique DHT
regions in ENZ (10 µM) or ADA-308 (10 µM). Presented as log2FC. (H)
Pathways associated with commonly upregulated genes in ENZ (10 µM)
and ADA-308 (10 µM). Red, common upregulated genes in ENZ and
ADA-308; blue common downregulated genes in ENZ and ADA-308, using
gProfiler. Data are presented as -log10 of the adjusted P-value,
P<0.05. AR, androgen receptor; CTL, control; DHT,
dihydrotestosterone; ENZ, enzalutamide; FC, fold change; ns, not
significant. |
To delve deeper, publicly available AR ChIP-seq data
were leveraged (34). The regions
bound by the AR in the presence of DHT or ENZ were examined.
Notably, a substantial number of AR-bound regions (34,682 peaks)
were lost following ENZ treatment (Fig.
4F), consistent with previous reports indicating that ENZ
reduces AR chromatin binding and nuclear localization (56) and in alignment with aforementioned
observations (Fig. 2A and B).
However, despite ENZ treatment, ~15,000 regions remained bound by
AR. The AR-bound regions that were lost following ENZ treatment
were specifically focused on, and 12,382 genes within this region
were identified (Fig. 4F).
Following integration of the RNA-seq data, a significant
association between ADA-308 and ENZ-regulated AR target genes was
revealed (Fig. 4G). Moreover, the
GSEA results shed light on the functional consequences of these
alterations, with AR-bound genes upregulated following treatment
associated with stemness, whereas downregulated genes were linked
to androgen signaling, luminal phenotype and proliferation
(Fig. 4H). Collectively, these data
suggested that ADA-308 exerted effects comparable to those of ENZ
in modulating critical AR-bound target genes and their associated
pathways.
ADA-308 reduces tumor growth in
vivo
Investigation into the effects of ADA-308 revealed
its ability to inhibit cell proliferation in vitro. To
assess the in vivo pharmacodynamics activity of ADA-308,
castrated mice harboring 49FENZR ENZ-resistant xenograft tumors
were treated with ADA-308, ENZ or ODM-201 for comparison. For this,
male athymic mice were castrated and allowed to recover from
surgery. Then, ENZ-resistant 49FENZR cells were inoculated, and
mice were administered ENZ daily until the tumor volume reached 200
mm3. Thereafter, the treatment regimens were adjusted, with ENZ
either continued or replaced with vehicle, ADA-308 at 25 mg/kg BID,
ADA-308 at 50 mg/kg BID or ODM-201 at 50 mg/kg BID (Fig. 5A-C and Table SIII). The mice were treated for up
to 8 weeks or until the tumor volume reached 1,500 mm3.
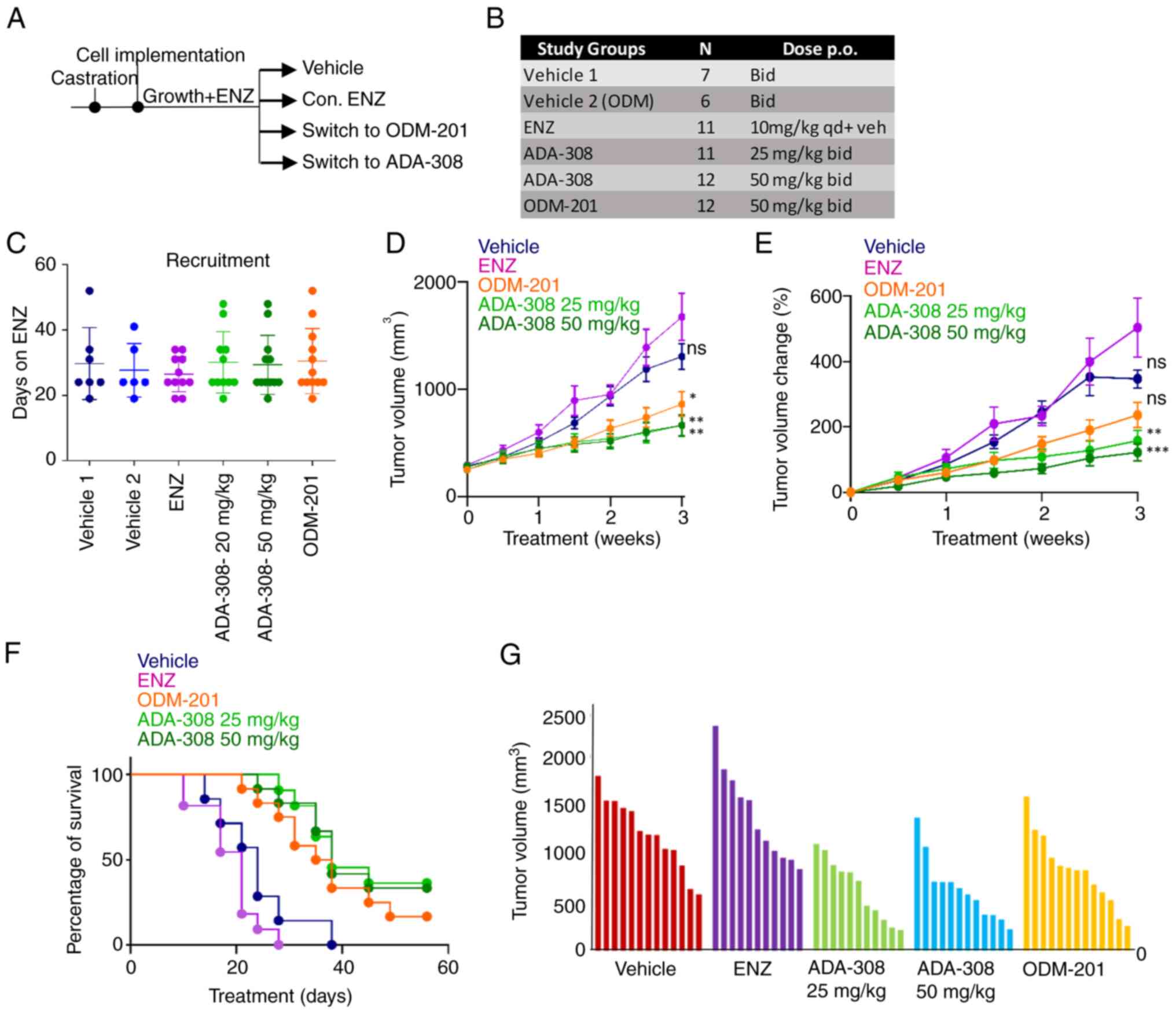 | Figure 5.ADA-308 reduces serum PSA and tumour
growth in ENZ-resistant cells in vivo. (A) Schematic of the
in vivo study. (B) In vivo study treatment groups,
number of mice recruited to each group and the respective doses.
(C) In vivo recruitment details are shown as days on ENZ
before recruitment to the respective treatment group. Castrated
Nu/Nu mice were inoculated with bilateral 49FENZR tumors, and tumor
dimensions were measured biweekly. Mice were assigned to vehicle
group 1, vehicle group 2, continuum on ENZ (10 mg/kg), two doses of
ADA-308 (25 mg/kg and 50 mg/kg) or ODM-201 (50 mg/kg). (D) Tumor
volume shown as mm3 (P-values were calculated at the endpoint,
comparing ENZ with the vehicle: 0.0307; ODM-201 with the vehicle:
0.0369; ADA-308 25 mg/kg with the vehicle: 0.0335; ADA-308 50 mg/kg
with the vehicle: 0.0302. P-values were calculated using one-way
ANOVA statistical test followed by Dunnett's test). (E) Tumor
volume change presented as a percentage over 3 weeks of treatment.
(P-value was calculated at the endpoint, comparing ENZ with the
vehicle: 0.2665; ODM-201 with the vehicle: 0.0261; ADA-308 25 mg/kg
with the vehicle: 0.0118; ADA-308 50 mg/kg with the vehicle:
0.0144. P-values were calculated using one-way ANOVA statistical
test followed by Dunnett's test). (F) Cancer-specific survival
indicates the percentage of mice with a tumour size <1,500 mm3
at a given treatment day. (G) Tumor volume (mm3) for the individual
mice in each treatment group after 3 weeks of treatment. Vehicle
groups 1 and 2 are combined. *P<0.05, **P<0.01,
***P<0.001. See Table SI for
the statistical test results. Bid, twice a day; ENZ, enzalutamide;
ns, not significant; PSA, prostate-specific antigen; qd, four times
a day. |
The reduction in proliferation rates observed in
vitro translated into a notable in vivo antitumor
response. It was observed that both doses of ADA-308 (25 or 50
mg/kg) exhibited improved antitumor responses in the ENZ-resistant
cell model compared with ENZ or ODM-201. Notably, in the
ENZ-treated group, most mice reached the study endpoint by 3 weeks
(Fig. 5D). The percentage change in
tumor volume after treatment with ADA-308 was significantly lower
(Fig. 5E and Table SIV), leading to higher survival
rates (Fig. 5F and Table SIII). Notably, prior ENZ
administration did not compromise the efficacy of ADA-308. In
addition, testing lower doses of ADA-308 (12.5 mg/kg BID) resulted
in a significantly reduced tumor volume in ENZ-resistant tumors
(Fig. S5A-C). Overall, these data
elucidated the in vivo efficacy of ADA-308 and its superior
capacity to inhibit tumor growth in ENZ-resistant 49FENZR xenograft
models.
Discussion
It is now understood that CRPC retains its androgen
sensitivity, both in the early stages of the disease as well as
following the successful treatment with next-generation ARPIs
(9,57,58).
This dependence on the AR for growth (59–61)
highlights the continued significance of the AR as a therapeutic
target in PCa (62). However, the
response to second-generation ARPIs is often only temporary, and
resistance poses an unavoidable challenge. As a result, several AR
antagonists including ARN-509 (47)
and ODM-201 (27), have been
developed and evaluated for inhibition of AR activity. Therefore,
development of alternative and novel AR-targeted therapies is of
paramount importance.
The ADA-308 compound was originally designed to
overcome the treatment resistance to other AR antagonists in
advanced PCa. The present study demonstrated that ADA-308 can
potentially reduce AR activity in ENZ-sensitive and ENZ-resistant
preclinical models. The investigation encompassed two distinct PCa
cell models: LNCaP (representing Adeno) and the ENZ-resistant
49FENZR and 49CENZR cell lines (which no longer responded to ENZ
treatment). Administration of ADA-308 in these models resulted in a
significant inhibition of AR signalling and the accumulation of
cells in the G0/G1 phase of the cell cycle, a response comparable
to that of ENZ in LNCaP cells. It was therefore demonstrated that
ADA-308 is a very potent AR inhibitor in PCa research models
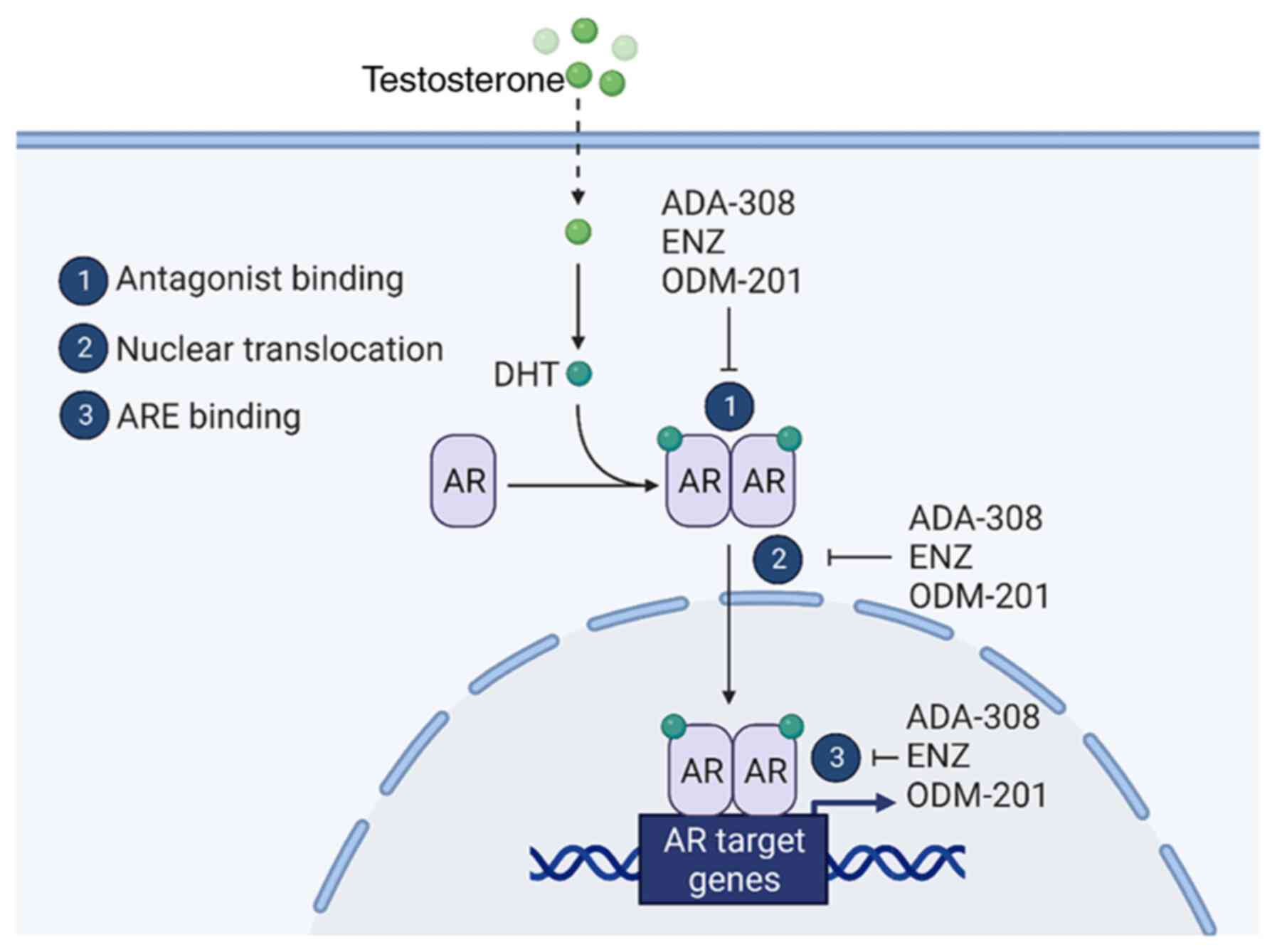
including those resistant to ENZ. Mechanistically, it was shown
that the mechanism of action of ADA-308 closely parallels that of
ENZ and ODM-201 (Fig. 6). Notably,
ADA-308 hindered androgen-induced AR nuclear localization in LNCaP
cells, which is a critical step in AR activation and targeted gene
transcription. In ENZ-resistant (49FENZR and 49CENZR) cell lines,
ENZ significantly failed to inhibit androgen-induced AR nuclear
localization, while ADA-308 prevented this effect. Moreover,
comparing the effect of ADA-308 to ENZ on the transcriptome of
LNCaP cells, the data revealed that ADA-308 was comparable to ENZ
in suppressing genes regulated by the AR or those associated with
proliferation. Notably, upon ADA-308 treatment of LNCaP cells, an
increased expression of AR-bound genes associated with the stemness
pathway was observed, similar to ENZ treatment. This raises a
noteworthy concern regarding whether treatment with ADA-308 can
induce lineage plasticity. Lineage plasticity has been postulated
to contribute to the failure of ARPIs in PCa, representing an
established mechanism of treatment resistance associated with the
loss of luminal lineage, and an induction of alternative programs
including stem cell-like phenotypes (26,63–65).
Therefore, it is important to evaluate whether ADA-308 induces
lineage plasticity.
In the present study, ADA-308 demonstrated a
superior in vitro anti-proliferative effect compared with
ENZ in ENZ-resistant cell line models. Moreover, the presented
in vivo study provided compelling evidence that ADA-308
reduced tumor growth in ENZ-resistant models. The antitumor effect
of ADA-308 was accompanied by an increase in overall survival.
Collectively, these findings suggested that ADA-308 may emerge as a
promising and viable candidate for future clinical development in
CRPC, particularly in an ENZ-resistant context where ENZ treatment
has failed, thereby offering a viable treatment strategy in the
evolving landscape of PCa therapy. Finaly, a more comprehensive
understanding of the safety profile, long-term effects and
potential resistance mechanisms of ADA-308 is essential as we
consider its transition into clinical development.
Although the present study provided valuable
insights into the therapeutic potential of ADA-308 in PCa,
particularly in overcoming resistance to other AR antagonists such
as ENZ, there are some limitations to the findings. One limitation
of the present study is the limited number of models, which may not
fully represent the genetic and phenotypic diversity of PCa
observed in a broader patient population. Additionally, the in
vivo studies were conducted exclusively in mouse models, which,
despite their utility, cannot perfectly mimic the complex human
tumor microenvironment and immune interactions. Notably, ARPIs can
lead to the development of lineage plasticity, a mechanism of
resistance in which cells alter their lineage to acquire an
alternative lineage that is often associated with stem-cell and
neuronal characteristics. Therefore, future studies are needed to
better characterize whether ADA-308 treatment leads to the
activation of resistance mechanisms, including lineage plasticity.
Additionally, longitudinal studies monitoring long-term outcomes
and potential side effects are essential to ensure that ADA-308 can
provide sustainable benefits for the treatment of PCa. In addition,
while it was shown that ADA-308 reduced PSA expression similar to
ODM-201 and that the in vivo effects of the compounds were
similar, without further investigation regarding the long-term
effect of ADA-308, we cannot comment on whether ADA-308 will be a
preferred option for treatment of PCa to ODM-201.
In the present study, the significant efficacy of
ADA-308 in suppressing AR signalling and reducing proliferation
in vitro and in vivo was highlighted. These findings
are particularly noteworthy and relevant given the growing
occurrence of resistance to potent ARPIs (66–68)
and the limited number of therapeutic options following the
development of resistance. The ability of ADA-308 to inhibit AR
activity in models that have developed resistance to ENZ suggests
its potential as an effective agent to follow ARPI resistance.
However, while there is potential for ADA-308 in PCa, the
commercialization of the program in PCa became challenging for
Aranda Pharma Ltd. due to changes in clinical practice (such as the
sequential use of second-generation AR inhibitors is not
recommended when one fails) and high competition in the market.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
We thank all the members of the Zoubeidi laboratory
(University of British Columbia, Vancouver, Canada) for their
valuable input in designing and progressing this research.
Specifically, Dr Dwaipayan Ganguli for performing peak calling,
quality control, annotation and visualization of the ChIP-seq data
and Dr Joshua Scurll for RNA-seq processing and quality control. We
also thank the Biomedical Research Centre Sequencing Core
(University of British Columbia, Vancouver, Canada) for the RNA-seq
processing and the Animal Core Facility (Vancouver Prostate Centre,
Vancouver, Canada) for the animal study.
Funding
This research was supported by funding from Aranda Pharma Ltd as
well as the Prostate Cancer Foundation Young Investigator Award (to
SN).
Availability of data and materials
The RNA-seq data generated in the present study may
be found in the GEO database under the accession no. GSE267309 or
at the following URL: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE267309.
All other data generated in the present study may be requested from
the corresponding author.
Authors' contributions
SN, FJ, SK, OS, NT, MK, AM and AZ confirm the
authenticity of all the raw data, conceived this study and took
responsibility for the quality of the data. AM and MK contributed
to the study design. AM, SN and FJ participated in the analysis and
interpretation of data and prepared all figures. FJ and SK
performed all the in vitro experiments and acquired data. NT
performed the proliferation assay and assisted in the revision of
this manuscript. OS performed the in vivo experiments. SN
wrote the manuscript. AZ, AM and FJ reviewed and edited the
manuscript. All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
All animal experiments were performed in accordance
with the procedures and protocols of the Laboratory Animal Center
of the University of British Columbia (Vancouver, Canada; approval
no. A16-0246; approval date, 12/15/2016).
Patient consent for publication
Not applicable.
Competing interests
Aranda Pharma Ltd. owns the IP of ADA-308. AM and MK
are shareholders of Aranda Pharma Ltd. All other authors declare
that they have no competing interests.
Glossary
Abbreviations
Abbreviations:
|
ENZ
|
enzalutamide
|
|
ARPIs
|
androgen receptor pathway
inhibitors
|
|
PCa
|
prostate cancer
|
|
CRPC
|
castration-resistant PCa
|
|
ENZR
|
enzalutamide-resistant
|
|
Adeno
|
adenocarcinoma
|
References
|
1
|
Bergengren O, Pekala KR, Matsoukas K,
Fainberg J, Mungovan SF, Bratt O, Bray F, Brawley O, Luckenbaugh
AN, Mucci L, et al: 2022 Update on prostate cancer epidemiology and
risk factors-a systematic review. Eur Urol. 84:191–206. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhang W, Cao G, Wu F, Wang Y, Liu Z, Hu H
and Xu K: Global burden of prostate cancer and association with
socioeconomic status, 1990-2019: A systematic analysis from the
global burden of disease study. J Epidemiol Glob Health.
13:407–421. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
World Health Organization (WHO), .
Prostate cancer statistics. WHO; Geneva: 2024
|
|
4
|
Heinlein CA and Chang C: Androgen receptor
in prostate cancer. Endocr Rev. 25:276–308. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Seikkula H, Boström PJ, Seppä K,
Pitkäniemi J, Malila N and Kaipia A: Survival and mortality of
elderly men with localized prostate cancer managed with primary
androgen deprivation therapy or by primary observation. BMC Urol.
20:252020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kokorovic A, So AI, Serag H, French C,
Hamilton RJ, Izard JP, Nayak JG, Pouliot F, Saad F, Shayegan B, et
al: Canadian urological association guideline on androgen
deprivation therapy: Adverse events and management strategies. Can
Urol Assoc J. 15:E307–E322. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vellky JE and Ricke WA: Development and
prevalence of castration-resistant prostate cancer subtypes.
Neoplasia. 22:566–575. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kirby M, Hirst C and Crawford ED:
Characterising the castration-resistant prostate cancer population:
A systematic review. Int J Clin Pract. 65:1180–1192. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Scher HI, Fizazi K, Saad F, Taplin ME,
Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, et
al: Increased survival with enzalutamide in prostate cancer after
chemotherapy. N Engl J Med. 367:1187–1197. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
de Bono JS, Logothetis CJ, Molina A,
Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB Jr, Saad F,
et al: Abiraterone and increased survival in metastatic prostate
cancer. N Engl J Med. 364:1995–2005. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nevedomskaya E, Baumgart SJ and Haendler
B: Recent advances in prostate cancer treatment and drug discovery.
Int J Mol Sci. 19:13592018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mateo J, Smith A, Ong M and de Bono JS:
Novel drugs targeting the androgen receptor pathway in prostate
cancer. Cancer Metastasis Rev. 33:567–579. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Crona DJ, Milowsky MI and Whang YE:
Androgen receptor targeting drugs in castration-resistant prostate
cancer and mechanisms of resistance. Clin Pharmacol Ther.
98:582–589. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Abida W, Cyrta J, Heller G, Prandi D,
Armenia J, Coleman I, Cieslik M, Benelli M, Robinson D, Van Allen
EM, et al: Genomic correlates of clinical outcome in advanced
prostate cancer. Proc Natl Acad Sci USA. 116:11428–11436. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Arora VK, Schenkein E, Murali R, Subudhi
SK, Wongvipat J, Balbas MD, Shah N, Cai L, Efstathiou E, Logothetis
C, et al: Glucocorticoid receptor confers resistance to
antiandrogens by bypassing androgen receptor blockade. Cell.
155:1309–1322. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Antonarakis ES, Lu C, Wang H, Luber B,
Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, et
al: AR-V7 and resistance to enzalutamide and abiraterone in
prostate cancer. N Engl J Med. 371:1028–1038. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cato L, de Tribolet-Hardy J, Lee I,
Rottenberg JT, Coleman I, Melchers D, Houtman R, Xiao T, Li W, Uo
T, et al: ARv7 represses tumor-suppressor genes in
castration-resistant prostate cancer. Cancer Cell. 35:401–413.e6.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Joseph JD, Lu N, Qian J, Sensintaffar J,
Shao G, Brigham D, Moon M, Maneval EC, Chen I, Darimont B and Hager
JH: A clinically relevant androgen receptor mutation confers
resistance to second-generation antiandrogens enzalutamide and
ARN-509. Cancer Discov. 3:1020–1029. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Einstein DJ, Arai S and Balk SP: Targeting
the androgen receptor and overcoming resistance in prostate cancer.
Curr Opin Oncol. 31:175–182. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Takeda DY, Spisák S, Seo JH, Bell C,
O'Connor E, Korthauer K, Ribli D, Csabai I, Solymosi N, Szállási Z,
et al: A somatically acquired enhancer of the androgen receptor is
a noncoding driver in advanced prostate cancer. Cell.
174:422–432.e13. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Quigley DA, Dang HX, Zhao SG, Lloyd P,
Aggarwal R, Alumkal JJ, Foye A, Kothari V, Perry MD, Bailey AM, et
al: Genomic hallmarks and structural variation in metastatic
prostate cancer. Cell. 175:8892018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Viswanathan SR, Ha G, Hoff AM, Wala JA,
Carrot-Zhang J, Whelan CW, Haradhvala NJ, Freeman SS, Reed SC,
Rhoades J, et al: Structural alterations driving castration-
resistant prostate cancer revealed by linked-read genome
sequencing. Cell. 174:433–447.e19. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bluemn EG, Coleman IM, Lucas JM, Coleman
RT, Hernandez-Lopez S, Tharakan R, Bianchi-Frias D, Dumpit RF,
Kaipainen A, Corella AN, et al: Androgen receptor
pathway-independent prostate cancer is sustained through FGF
signaling. Cancer Cell. 32:474–489.e6. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li Q, Deng Q, Chao HP, Liu X, Lu Y, Lin K,
Liu B, Tang GW, Zhang D, Tracz A, et al: Linking prostate cancer
cell AR heterogeneity to distinct castration and enzalutamide
responses. Nat Commun. 9:36002018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
He Y, Wei T, Ye Z, Orme JJ, Lin D, Sheng
H, Fazli L, Jeffrey Karnes R, Jimenez R, Wang L, et al: A
noncanonical AR addiction drives enzalutamide resistance in
prostate cancer. Nat Commun. 12:15212021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Davies A, Nouruzi S, Ganguli D, Namekawa
T, Thaper D, Linder S, Karaoğlanoğlu F, Omur ME, Kim S, Kobelev M,
et al: An androgen receptor switch underlies lineage infidelity in
treatment-resistant prostate cancer. Nat Cell Biol. 23:1023–1034.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Moilanen AM, Riikonen R, Oksala R, Ravanti
L, Aho E, Wohlfahrt G, Nykänen PS, Törmäkangas OP, Palvimo JJ and
Kallio PJ: Discovery of ODM-201, a new-generation androgen receptor
inhibitor targeting resistance mechanisms to androgen
signaling-directed prostate cancer therapies. Sci Rep. 5:120072015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bishop JL, Thaper D, Vahid S, Davies A,
Ketola K, Kuruma H, Jama R, Nip KM, Angeles A, Johnson F, et al:
The master neural transcription factor BRN2 Is an androgen
receptor-suppressed driver of neuroendocrine differentiation in
prostate cancer. Cancer Discov. 7:54–71. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yeh S, Kang HY, Miyamoto H, Nishimura K,
Chang HC, Ting HJ, Rahman M, Lin HK, Fujimoto N, Hu YC, et al:
Differential induction of androgen receptor transactivation by
different androgen receptor coactivators in human prostate cancer
DU145 cells. Endocrine. 11:195–202. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dobin A, Davis CA, Schlesinger F, Drenkow
J, Zaleski C, Jha S, Batut P, Chaisson M and Gingeras TR: STAR:
Ultrafast universal RNA-seq aligner. Bioinformatics. 29:15–21.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Trapnell C, Roberts A, Goff L, Pertea G,
Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL and Pachter L:
Differential gene and transcript expression analysis of RNA-seq
experiments with TopHat and cufflinks. Nat Protoc. 7:562–578. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Love MI, Huber W and Anders S: Moderated
estimation of fold change and dispersion for RNA-seq data with
DESeq2. Genome Biol. 15:5502014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen Z, Lan X, Thomas-Ahner JM, Wu D, Liu
X, Ye Z, Wang L, Sunkel B, Grenade C, Chen J, et al: Agonist and
antagonist switch DNA motifs recognized by human androgen receptor
in prostate cancer. EMBO J. 34:502–516. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bittencourt SA: FastQC: A quality control
tool for high throughput sequence data. Babraham Bioinformatics.
2010.
|
|
36
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li H, Handsaker B, Wysoker A, Fennell T,
Ruan J, Homer N, Marth G, Abecasis G and Durbin R; 1000 Genome
Project Data Processing Subgroup, : The sequence alignment/map
format and SAMtools. Bioinformatics. 25:2078–2079. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang Y, Liu T, Meyer CA, Eeckhoute J,
Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W and
Liu XS: Model-based analysis of ChIP-Seq (MACS). Genome Biol.
9:R1372008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ramirez F, Ryan DP, Grüning B, Bhardwaj V,
Kilpert F, Richter AS, Heyne S, Dündar F and Manke T: deepTools2: A
next generation web server for deep-sequencing data analysis.
Nucleic Acids Res. 44:W160–W165. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Quinlan AR and Hall IM: BEDTools: A
flexible suite of utilities for comparing genomic features.
Bioinformatics. 26:841–842. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Reimand J, Arak T, Adler P, Kolberg L,
Reisberg S, Peterson H and Vilo J: g:Profiler-a web server for
functional interpretation of gene lists (2016 update). Nucleic
Acids Res. 44:W83–W89. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Maffey AH, Ishibashi T, He C, Wang X,
White AR, Hendy SC, Nelson CC, Rennie PS and Ausió J: Probasin
promoter assembles into a strongly positioned nucleosome that
permits androgen receptor binding. Mol Cell Endocrinol. 268:10–19.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Namekawa T, Ikeda K, Horie-Inoue K and
Inoue S: Application of prostate cancer models for preclinical
study: Advantages and limitations of cell lines, patient-derived
xenografts, and three-dimensional culture of patient-derived cells.
Cells. 8:742019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Borgmann H, Lallous N, Ozistanbullu D,
Beraldi E, Paul N, Dalal K, Fazli L, Haferkamp A, Lejeune P,
Cherkasov A and Gleave ME: Moving towards precision urologic
oncology: Targeting enzalutamide-resistant prostate cancer and
mutated forms of the androgen receptor using the novel inhibitor
darolutamide (ODM-201). Eur Urol. 73:4–8. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Waltering KK, Urbanucci A and Visakorpi T:
Androgen receptor (AR) aberrations in castration-resistant prostate
cancer. Mol Cell Endocrinol. 360:38–43. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy
DA, Doshi S, Yuan J, Kovats SG, Kim S, Cooke VG, et al: An F876L
mutation in androgen receptor confers genetic and phenotypic
resistance to MDV3100 (enzalutamide). Cancer Discov. 3:1030–1043.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liu H, Wang L, Tian J, Li J and Liu H:
Molecular dynamics studies on the enzalutamide resistance
mechanisms induced by androgen receptor mutations. J Cell Biochem.
118:2792–2801. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Veldscholte J, Ris-Stalpers C, Kuiper GG,
Jenster G, Berrevoets C, Claassen E, van Rooij HC, Trapman J,
Brinkmann AO and Mulder E: A mutation in the ligand binding domain
of the androgen receptor of human LNCaP cells affects steroid
binding characteristics and response to anti-androgens. Biochem
Biophys Res Commun. 173:534–540. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Dai C, Heemers H and Sharifi N: Androgen
signaling in prostate cancer. Cold Spring Harb Perspect Med.
7:a0304522017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Eder IE, Culig Z, Putz T, Nessler-Menardi
C, Bartsch G and Klocker H: Molecular biology of the androgen
receptor: From molecular understanding to the clinic. Eur Urol.
40:241–251. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Smith DF and Toft DO: Minireview: The
intersection of steroid receptors with molecular chaperones:
Observations and questions. Mol Endocrinol. 22:2229–2240. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Verrijdt G, Haelens A and Claessens F:
Selective DNA recognition by the androgen receptor as a mechanism
for hormone-specific regulation of gene expression. Mol Genet
Metab. 78:175–185. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhao J, Zhao Y, Wang L, Zhang J, Karnes
RJ, Kohli M, Wang G and Huang H: Alterations of androgen
receptor-regulated enhancer RNAs (eRNAs) contribute to enzalutamide
resistance in castration-resistant prostate cancer. Oncotarget.
7:38551–38565. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Li K, Guo Y, Yang X, Zhang Z, Zhang C and
Xu Y: ELF5-mediated AR activation regulates prostate cancer
progression. Sci Rep. 7:427592017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Rodriguez-Vida A, Galazi M, Rudman S,
Chowdhury S and Sternberg CN: Enzalutamide for the treatment of
metastatic castration-resistant prostate cancer. Drug Des Devel
Ther. 9:3325–3339. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Ryan CJ, Smith MR, de Bono JS, Molina A,
Logothetis CJ, de Souza P, Fizazi K, Mainwaring P, Piulats JM, Ng
S, et al: Abiraterone in metastatic prostate cancer without
previous chemotherapy. N Engl J Med. 368:138–148. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Fizazi K, Scher HI, Molina A, Logothetis
CJ, Chi KN, Jones RJ, Staffurth JN, North S, Vogelzang NJ, Saad F,
et al: Abiraterone acetate for treatment of metastatic
castration-resistant prostate cancer: Final overall survival
analysis of the COU-AA-301 randomised, double-blind,
placebo-controlled phase 3 study. Lancet Oncol. 13:983–992. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Mostaghel EA, Marck BT, Plymate SR,
Vessella RL, Balk S, Matsumoto AM, Nelson PS and Montgomery RB:
Resistance to CYP17A1 inhibition with abiraterone in
castration-resistant prostate cancer: Induction of steroidogenesis
and androgen receptor splice variants. Clin Cancer Res.
17:5913–5925. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Locke JA, Guns ES, Lubik AA, Adomat HH,
Hendy SC, Wood CA, Ettinger SL, Gleave ME and Nelson CC: Androgen
levels increase by intratumoral de novo steroidogenesis during
progression of castration-resistant prostate cancer. Cancer Res.
68:6407–6415. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Montgomery RB, Mostaghel EA, Vessella R,
Hess DL, Kalhorn TF, Higano CS, True LD and Nelson PS: Maintenance
of intratumoral androgens in metastatic prostate cancer: A
mechanism for castration-resistant tumor growth. Cancer Res.
68:4447–4454. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Lallous N, Dalal K, Cherkasov A and Rennie
PS: Targeting alternative sites on the androgen receptor to treat
castration-resistant prostate cancer. Int J Mol Sci.
14:12496–12519. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Nouruzi S, Ganguli D, Tabrizian N, Kobelev
M, Sivak O, Namekawa T, Thaper D, Baca SC, Freedman ML, Aguda A, et
al: ASCL1 activates neuronal stem cell-like lineage programming
through remodeling of the chromatin landscape in prostate cancer.
Nat Commun. 13:22822022. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Tabrizian N, Nouruzi S, Cui CJ, Kobelev M,
Namekawa T, Lodhia I, Talal A, Sivak O, Ganguli D and Zoubeidi A:
ASCL1 is activated downstream of the ROR2/CREB signaling pathway to
support lineage plasticity in prostate cancer. Cell Rep.
42:1129372023. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Beltran H, Hruszkewycz A, Scher HI,
Hildesheim J, Isaacs J, Yu EY, Kelly K, Lin D, Dicker A, Arnold J,
et al: The role of lineage plasticity in prostate cancer therapy
resistance. Clin Cancer Res. 25:6916–6924. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Beltran H, Prandi D, Mosquera JM, Benelli
M, Puca L, Cyrta J, Marotz C, Giannopoulou E, Chakravarthi BV,
Varambally S, et al: Divergent clonal evolution of
castration-resistant neuroendocrine prostate cancer. Nat Med.
22:298–305. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Aggarwal R, Huang J, Alumkal JJ, Zhang L,
Feng FY, Thomas GV, Weinstein AS, Friedl V, Zhang C, Witte ON, et
al: Clinical and genomic characterization of treatment-emergent
small-cell neuroendocrine prostate cancer: A multi-institutional
prospective study. J Clin Oncol. 36:2492–2503. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Linder S, Hoogstraat M, Stelloo S,
Eickhoff N, Schuurman K, de Barros H, Alkemade M, Bekers EM,
Severson TM, Sanders J, et al: Drug-induced epigenomic plasticity
reprograms circadian rhythm regulation to drive prostate cancer
toward androgen independence. Cancer Discov. 12:2074–2097. 2022.
View Article : Google Scholar : PubMed/NCBI
|