Introduction
Oral squamous cell carcinoma (OSCC), the most common
type of head and neck cancer, primarily occurs in the oral cavity
and lips (1). Data from the Global
Cancer Observatory revealed that the worldwide incidence and
mortality of OSCC in 2020 were ~380,000 (2.0%) and 180,000 (1.8%),
respectively, and are predicted to increase by 47% by 2040
(2). The standard treatment
approach for OSCC involves surgical resection followed by adjuvant
radiotherapy or chemoradiotherapy depending on the disease stage
(3). However, despite advancements
in treatment modalities, the 5-year survival rate of OSCC remains
stagnant, and the adverse effects of these therapies cannot be
ignored (4). For example, because
chemotherapy is primarily administered intravenously to
non-specific tissues in the body, it may cause collateral damage to
healthy tissues as it causes substantial toxicity to normal cells,
leading to severe side effects (5).
Moreover, even though irradiation is effective in treating tumor
cells by inducing senescence, it poses a potential risk of
carcinogenesis by inducing senescence in the surrounding normal
cells (6). Therefore, it is
necessary to develop alternative OSCC-specific therapies and
identify novel biomarkers for the treatment of OSCC. To accomplish
this, it is necessary to investigate the underlying mechanisms of
the disease.
Pituitary tumor-transforming gene 1 (PTTG1) is a
transcription factor ubiquitously expressed, excluding the testes,
and mainly participates in regulating sister chromatid separation
during cell division and aneuploidy (7,8). PTTG1
overexpression has been reported in various tumor types and is
involved in genetic instability, playing an oncogenic role in
tumorigenesis (9–11). PTTG1 induces DNA damage in cervical
cancer cells through its involvement in genome instability,
ultimately promoting cell apoptosis (12). Furthermore, PTTG1 overexpression
stimulates c-Myc expression by interacting with p53 and is
involved in sister chromatid separation or the inhibition of DNA
damage repair by inducing chromosomal instability (13–16).
Regarding its role in OSCC, Liao et al (17) reported that PTTG1 was markedly
overexpressed in the tissues of patients with OSCC. Moreover, Zhang
et al (18) demonstrated
that PTTG1 overexpression promotes epithelial-mesenchymal
transition (EMT), which results in OSCC cell migration and invasion
regulation. However, the precise role of PTTG1 in OSCC remains
unclear and requires further investigation.
Cellular senescence plays a vital role as a
tumor-suppressive mechanism during the initial stages of tumor
development (19). Currently, this
process is referred to as replicative senescence because of the
diverse intrinsic and extrinsic stresses that lead to permanent
cell cycle arrest (20,21). Senescence prevents the proliferation
of damaged cells and differs from apoptosis, which eliminates
irreparably damaged cells (22).
Senescent cells are commonly characterized by the accumulation of
intracellular senescence-associated beta-galactosidase (SA-β-gal)
along with morphological alterations, such as enlarged cell size
and a flattened shape (23,24). Senescent cells are also
characterized by activation of the p21/WAF1 and p16/INK4a signaling
pathways, the senescence-associated secretory phenotype (SASP), and
DNA damage, which are collectively referred to as senescent
phenotypes (25). Senescence was
initially considered to be associated with limited cell
proliferation in normal cell culture (26). A recent study revealed that cell
division cycle 25B (Cdc25B), a cell cycle regulator, triggers cell
senescence in a p53-dependent manner, leading to the inhibition of
DNA synthesis without affecting apoptosis in normal fibroblast
cells (27). Jung et al
(28) demonstrated that the loss of
PTEN by mTOR kinase leads to cell cycle inhibition and cellular
senescence induction in breast cancer cells through p53
phosphorylation. However, the potential association among
senescence, cell cycle arrest and tumor suppression in OSCC remains
poorly understood.
Therefore, the present study aimed to elucidate the
role of PTTG1 as an oncogene in OSCC progression and to elucidate
the underlying mechanism and impact of PTTG1 expression on cell
cycle, cell death and cellular senescence. The present study
highlights the significance of PTTG1 as a potential diagnostic and
post-treatment biomarker for patients with OSCC.
Materials and methods
Human tissues samples
Tumor and adjacent healthy tissue specimens from 32
patients (24 males and 8 females) with OSCC were obtained from
Ganenung-Wonju National University Dental Hospital (Gangneung),
Keimyung University Dongsan Hospital (Daegu), Pusan National
University (Pusan), Inje Paik University (Pusan), and Korea Biobank
Network members (Yongin). All tissue samples were obtained with
written informed consent of patients aged 50-75 years (mean age:
62) before surgery, and OSCC was histologically diagnosed by
pathologists using tissue fragments from the oral region. The
tissues were divided into two sections. One section was fixed in
10% formalin overnight at 25°C for immunohistochemical (IHC)
staining. The other section was frozen immediately in liquid
nitrogen, stored at −80°C and was used for real time-PCR and
Western blotting. In total, 32 paired individual samples were
randomly pooled into four samples, each containing an equal mass of
tissues, for analysis. The present study was approved (approval no.
GWNUIRB-2020-26-1) by the Committee for Ethical Review of Research
of Gangneung-Wonju National University (Gangneung-si, Korea) and
conducted from December 2020 to February 2023.
Cell culture and transfection
The p53 mutant human OSCC cell lines HSC-2, SCC-9
and YD-10B were obtained from the Japanese Collection of Research
Bioresources Cell Bank, the American Type Culture Collection and
the Korean Cell Line Bank, respectively. All OSCC cell lines were
cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen;
Thermo Fisher Scientific, Inc.) supplemented with 10% (w/v) fetal
bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin/streptomycin (P/S; Gibco; Thermo Fisher Scientific,
Inc.). The cell lines were incubated at 37°C with 5%
CO2.
For the transient knockdown of PTTG1, small
interfering (si)RNA (Invitrogen; Thermo Fisher Scientific, Inc) was
used to silence PTTG1 expression. A negative vehicle siRNA plasmid
was used as a control (Table SI).
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) was
used for transfection following the manufacturer's protocol.
Briefly, OSCC cell lines were transfected with 25 nM PTTG1 siRNA in
serum-free media until the cells reached 30–40% confluence. After 6
h of incubation at 37°C in a 5% CO2 atmosphere, the
medium was replaced with a culture medium, and the cells were
incubated overnight before harvesting.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from OSCC cells transfected
with the negative control vehicle or with the PTTG1 siRNA using the
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). The isolated RNA was used for cDNA synthesis using the
Accupower® RocketScript™️ Cycle RT PreMix kit (Bioneer,
Inc.) according to the manufacturer's protocol. The thermocycling
conditions for cDNA amplification were as follows: 50°C for 60 min,
95°C for 5 min, and hold at 4°C. The RT-qPCR was conducted using
the CFX96 Touch Real-Time PCR Detection System (Bio-Rad
Laboratories, Inc.) and the following thermocycling conditions:
95°C for 3 min, followed by 40 cycles of 95°C for 15 sec, 60°C for
30 sec, 95°C for 15 sec, 65°C for 5 sec, and 95°C for 30 sec. The
RT-qPCR was performed using primers specific for PTTG1
(forward, 5′-TGACTCAGGCTGGAAGATTTG-3′ and reverse,
5′-GGTGGGAGAAGCAAAGGTATAG-3′), p21 (forward,
5′-AGGTGGACCTGGAGACTCTCAG-3′ and reverse,
5′-TCCTCTTGGAGAAGATCAGCCG-3′) and GAPDH (forward,
5′-CAAAGTTGTCATGGATGACC-3′ and reverse, 5′-CCATGGAGAAGGCTGGGG-3′).
The 2−ΔΔCq method was used for the relative
quantification of gene expression (29). The mRNA levels of the target genes
were normalized to that of GAPDH. Independent experiments
were conducted in triplicates.
Western blotting
For protein isolation, OSCC samples (vehicle control
and siRNA) were lysed in Laemmli buffer supplemented with a
protease inhibitor cocktail (Roche Diagnostics). The protein
concentration was measured using a BCA kit (Thermo Fisher
Scientific, Inc.). An equal amount of proteins (50 µg) were
separated by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) using an 8–15% acrylamide gel at 100 V
for ~2 h and 30 min. The separated proteins were transferred to
0.45-µm nitrocellulose membranes (Bio-Rad Laboratories, Inc.) at 20
V overnight. The membranes were blocked in 5% (w/v) bovine serum
albumin (BSA; Sigma-Aldrich; Merck KGaA) dissolved in
phosphate-buffered saline (PBS) containing 0.01% Tween-20 (PBS-T)
for 1 h at room temperature (RT, 15–25°C) and then washed with
0.05% PBST. Next, the membranes were incubated with the following
primary antibodies overnight at 4°C: Rabbit anti-PTTG1 (cat. no.
GTX111938; GeneTex, Inc.), anti-p21 (cat. no. 2947; Cell Signaling
Technology, Inc.), anti-p53 (cat. no. 2527; Cell Signaling
Technology, Inc.), anti-retinoblastoma (Rb; cat. no. 9390; Cell
Signaling Technology, Inc.), anti-phosphorylated Rb (cat. no. 8180;
Cell Signaling Technology, Inc.), anti-proliferating cell nuclear
antigen (PCNA; cat. no. 2586; Cell Signaling Technology, Inc.),
mouse anti-Caspase-7 (Cas-7; cat no. sc-56063; Santa Cruz
Biotechnology, Inc.), anti-cleaved (c-) Cas-7 (cat. no. 8438; Cell
Signaling Technology, Inc.), anti-c-poly (ADP-ribose) polymerase
(PARP; cat. no. 5625; Cell Signaling Technology, Inc.), anti-PARP
(cat. no. 9542; Cell Signaling Technology, Inc.), mouse anti-cyclin
D1 (cat. no. sc-450; Santa Cruz Biotechnology, Inc.), mouse
anti-cyclin E (cat. no. sc-247; Santa Cruz Biotechnology, Inc.),
mouse anti-cyclin B1 (cat. no. sc-245; Santa Cruz Biotechnology,
Inc.), anti-phosphorylated histone H2AX (γH2AX; cat. no. 80312;
Cell Signaling Technology, Inc.), anti-phosphorylated ATR (cat. no.
2853; Cell Signaling Technology, Inc.) anti-ATR (cat. no. 2790;
Cell Signaling Technology, Inc.), anti-phosphorylated ataxia
telangiectasia mutant (p-ATM; cat. no. 13050; Cell Signaling
Technology, Inc.) and anti-ATM (cat. no. 2873; Cell Signaling
Technology, Inc.); all primary antibodies were diluted at 1:1,000
with BSA. GAPDH (cat. no. LF-PA0018; AbFrontier Co., Ltd.; 1:3,000
dilutions in BSA) was used as a loading control. After washing
three times with PBST, the membranes were incubated with
horseradish peroxidase-conjugated secondary antibodies against
rabbit (cat. no. 7074P2; Cell Signaling Technology, Inc.) or mouse
IgG (cat. no. 7076P2; Cell Signaling Technology, Inc.) at a
dilution of 1:5,000 in BSA for 1 h at RT. Protein bands were
visualized using a FUSION Solo S imaging system (Vilber China) with
an enhanced chemiluminescence reagent (cat. no. WBLUF0100; Merck
KGaA). Protein expression was quantified in triplicate using ImageJ
software (version 1.53a; National Institutes of Health), and the
intensity of the bands was calculated as fold change. The
expression levels of phosphorylated proteins were normalized to the
total form of each protein. This experiment was repeated
thrice.
IHC
Paraffin-embedded OSCC tissues were sectioned into
4-µm sections and placed onto coated slides and fixed in 4%
paraformaldehyde overnight at RT. Next, the sections were dewaxed
in xylene for 20 min and dehydrated in a graded ethanol series for
1 min each. After incubating in 3% H2O2 for
30 min, the samples were washed with PBS and incubated in sodium
sulfate buffer (pH 6.0) at RT for antigen retrieval. The samples
were blocked with 5% (w/v) BSA for 30 min at RT and washed three
times with PBS. Subsequently, the samples were incubated with
rabbit anti-PTTG1 antibody (cat. no. GTX111938; GeneTex, Inc.;
1:500 dilutions in BSA) at 4°C overnight. Tissues were washed three
times with PBS and incubated with biotinylated goat anti-rabbit
(1:1,00; cat. no. 7074P2; Cell Signaling Technology, Inc.) or
anti-mouse IgG (1:1,000; cat. no. 7076P2; Cell Signaling
Technology, Inc.) for 1 h at RT. The samples were washed thrice
with PBS and incubated with diaminobenzidine (DAB; Abcam).
Subsequently, the samples were counterstained with hematoxylin
(Dako; Agilent Technologies, Inc.) and dehydrated using a graded
ethanol and xylene series before mounting in Canada balsam mixture
(cat. no. 2525-4405; Sigma-Aldrich; Merck KGaA). All observations
were performed using an upright microscope (BX53; Olympus
Corporation) equipped with an objective lens (×10).
Immunofluorescence
To explore chromosomal damage in OSCC cells,
3×104 cells were cultured in Opti-MEM media (Gibco;
Thermo Fisher Scientific, Inc.) on coverslips and transfected with
25 nM vehicle control or siRNA-PTTG1 at a density of 30–40%. The
following day, the cells were transferred to culture media,
cultured overnight, fixed with 4% paraformaldehyde, permeabilized
with 0.5% Triton X-100 (Sigma-Aldrich; Merck KGaA) for 15 min at
RT, and then washed twice with PBS for 3 min. After incubation with
the primary antibody against γH2AX (1:500; cat. no. 80312; Cell
Signaling Technology, Inc.) at 4°C overnight, the coverslips were
washed with PBS and incubated with a secondary goat anti-mouse
antibody conjugated with Alexa Fluorescence 488 (1:1,000; cat. no.
A32723; Invitrogen; Thermo Fisher Scientific, Inc.). Images were
acquired using a confocal microscope (STELLARIS 5; Leica
Microsystems, Inc.) equipped with an oil objective lens (×63).
Z-stacks were used in three confocal images and merged using the
ImageJ software. The experiment was performed in triplicate.
5-Ethynyl-2′-deoxyuridine (EdU)
assay
To detect cell proliferation in OSCC, an EdU
staining Proliferation kit (cat. no. C10337; Fluor 488, Invitrogen;
Thermo Fisher Scientific, Inc.) was used following the
manufacturer's protocol. A total of 5×104 cells were
plated on 12-mm coverslips in serum-free media and incubated
overnight. The cells were transfected with vehicle control or
siRNA-PTTG1 at a density of 30–40% in Opti-MEM media (Gibco; Thermo
Fisher Scientific, Inc.) for 6 h and the culture medium was
changed. After 24 h, 2 µg/ml EdU was added, and the cells were
cultured in 5% CO2 at 37°C for 2 h. The cells were
fixed, permeabilized with 4% paraformaldehyde and 0.5% Triton X-100
(Sigma-Aldrich; Merck KGaA) for 15 min at RT, stained with 500 µl
Click-iT reaction buffer for 1 h at RT, and mounted with a
Fluorshield Mounting Medium containing DAPI (cat. no. ab104139;
Abcam) for 15 min at RT. For quantification, at least seven images
were randomly obtained using a fluorescence microscope (BX53;
Olympus Corporation) with a ×40 objective lens. The experiments
were performed in triplicate.
SA-β-gal staining
To investigate the cellular senescence of the OSCC
cells, a SA-β-gal staining kit (cat. no. 9860; Cell Signaling
Technology, Inc.) was used following the manufacturer's protocol.
The cells were seeded in 6-well plates at 5×104 cells
per well, allowed to attach overnight, treated with either vehicle
control or siRNA-PTTG1 at 25 µM in Opti-MEM media (Gibco; Thermo
Fisher Scientific, Inc.) for 6 h, and cultured overnight. The cells
were then fixed in the fixation solution for 15 min, washed twice
with PBS, and incubated with SA-β-gal staining solution for 12 h at
37°C. For quantification, at least seven images were randomly
acquired using an inverted light microscope (BX53; Olympus
Corporation) at ×40 magnification. Independent experiments were
conducted in triplicate.
Human OSCC xenograft model
A total of 2×106 HSC-2 and SCC-9 cells
treated with vehicle control or siRNA-PTTG1 were randomly divided
and injected into independent groups of 5 BALB/c-nu/nu male nude
mice (total n=20; mean weight, 16±1 g; ORIENT BIO, Inc.) aged 5-6
weeks, respectively. The mice were housed in a pathogen-free animal
facility with a 12 h light/dark cycle at 20–24°C with 45–55%
humidity and allowed free access to food and water. All animals
were examined for tumor formation and growth every 3 days. To
produce OSCC xenograft models, 100 µl of the tumor resuspended in
Dulbecco's Phosphate Buffered Saline (D-PBS) was injected
subcutaneously into the flank. After ~3 weeks, when the tumor size
approached 102 mm3, and the tumor volume was
calculated by V=(L × W2)/2 where V=volume
(mm3); L=the largest dimension (mm); W=perpendicular
diameter (mm), the mice were anesthetized via inhalation of
isoflurane (IFRAN LIQ., Hana Pharm Co., Ltd.) with an induction
concentration of 4–5% and a maintenance concentration of 2–3%, and
the tumors xenografts form each mouse were removed. The humane
endpoint of the experiment was reached when any of the following
criteria applies: The tumor's largest diameter reaches <20 mm,
the tumor weight exceeds 10% of the animals' body weight, the body
weight losses >20% of animals' body weight or the animal stops
eating and is deemed to be in poor health. After the experiment was
completed, all mice were sedated with a
CO2/O2 mixture (70/30%). Once the mice lost
consciousness, they were euthanized with 100% CO2.
Euthanasia was performed in a chamber for 5 min, and death was
confirmed by monitoring breathing, heart beating and pupil dilation
for an additional 5 min. Nude mouse tumors were explanted for
protein and RNA extraction, embedded in paraffin blocks, and
frozen. The present study was approved (approval no.
GWNUIRB-2020-36-1) by the Committee for Ethical Review of Research
of Gangneung-Wonju National University (Gangneung-si, Korea) and
conducted from December 2020 to January 2023.
Terminal
deoxynucleotidyl-transferase-mediated dUTP nick end labeling
(TUNEL) assay
To observe cell death in vivo, a TUNEL assay
was performed using the in situ Cell Death Detection kit
(cat. no. 12156792910; Roche Diagnostics) following the
manufacturer's protocol. Briefly, frozen tissues were
cryo-sectioned at a thickness of 7 µm using a cryostat microtome
(Leica Microsystems, Inc.). The sections were fixed on slides with
4% paraformaldehyde for 20 min at RT and then washed thrice with
PBS. The slides were then permeabilized with 0.1% sodium citrate
for 2 min on ice and washed thrice with PBS. The samples were added
to the TUNEL reaction mixture, incubated at 37°C, without a
CO2 incubator, for 1 h, and washed thrice with PBS. For
quantification, at least seven images were obtained, and the
samples were analyzed using a fluorescence microscope (BX53;
Olympus Corporation) at ×40 magnification. The experiments were
performed in triplicate.
2,5-diphenyl-2H-tetrazolium bromide
(MTT) assay
The MTT assay was performed to analyze the effect of
doxorubicin (DOX) on DNA damage mediated by the expression of PTTG1
and p21 in OSCC cell lines. Briefly, OSCC cells were seeded in a
96-well plate and cultured at 37°C in a 5% CO2
humidified incubator. After 24 h, the cells were treated with 0.05,
0.5, 5, 50 and 100 µM of DOX (cat. no. D4193; Tokyo Chemical
Industry Co., Ltd.) for 24 h and 200 µl DMSO (cat. no. 276855;
Sigma-Aldrich; Merck KGaA) for 30 min. Absorbance at 570 nm was
then measured using and enzyme-linked immunosorbent assay (ELISA)
microplate reader (Molecular Devices, LLC). All experiments were
repeated at least three times.
Statistical analyses
All assays were performed in triplicate, and the
data are presented as the mean ± standard error (SE) of the mean.
Differences in test variables between the control and experimental
groups were analyzed using an unpaired Student's t-test or one-way
ANOVA, followed by Dunnett's post hoc test. *P<0.05 was
considered to indicate a statistically significant difference.
Statistical analyses were performed using SPSS (version 20.0; IBM
Corp.) and GraphPad Prism 9 (GraphPad Software; Dotmatics).
Results
PTTG1 is overexpressed in OSCC
tissues
To investigate the expression and potential role of
PTTG1 in OSCC, IHC was performed on 32 pairs of OSCC and adjacent
healthy tissue samples. PTTG1 expression was significantly higher
in OSCC tissues than in healthy tissues (Fig. 1A). Samples from patients with OSCC
were further examined using RT-qPCR and western blot analysis which
confirmed that PTTG1 was significantly overexpressed in tumors
compared with that in healthy tissues (Fig. 1B and C; protein fold change: 2.10,
2.14, 2.10 and 2.00, respectively, compared with non-tumor
tissues). In addition, PTTG1 was expressed in HSC-2 and SCC-9, two
OSCC cell lines. Interestingly, the expression of the anti-oncogene
p21, which is associated with several key characteristics of
cellular senescence (i.e., a modified transcriptome, DNA damage,
and SASP) (30,31) exhibited an opposite pattern in HSC-2
and SCC-9 cell lines in contrast to that of PTTG1 (Fig. 1D and E; protein fold change: 1.44
and 0.56, respectively, between HSC-2 and SCC-9 cells). The results
of the independent analyses indicated that PTTG1 is an oncogene
highly expressed in both OSCC tissues and cells.
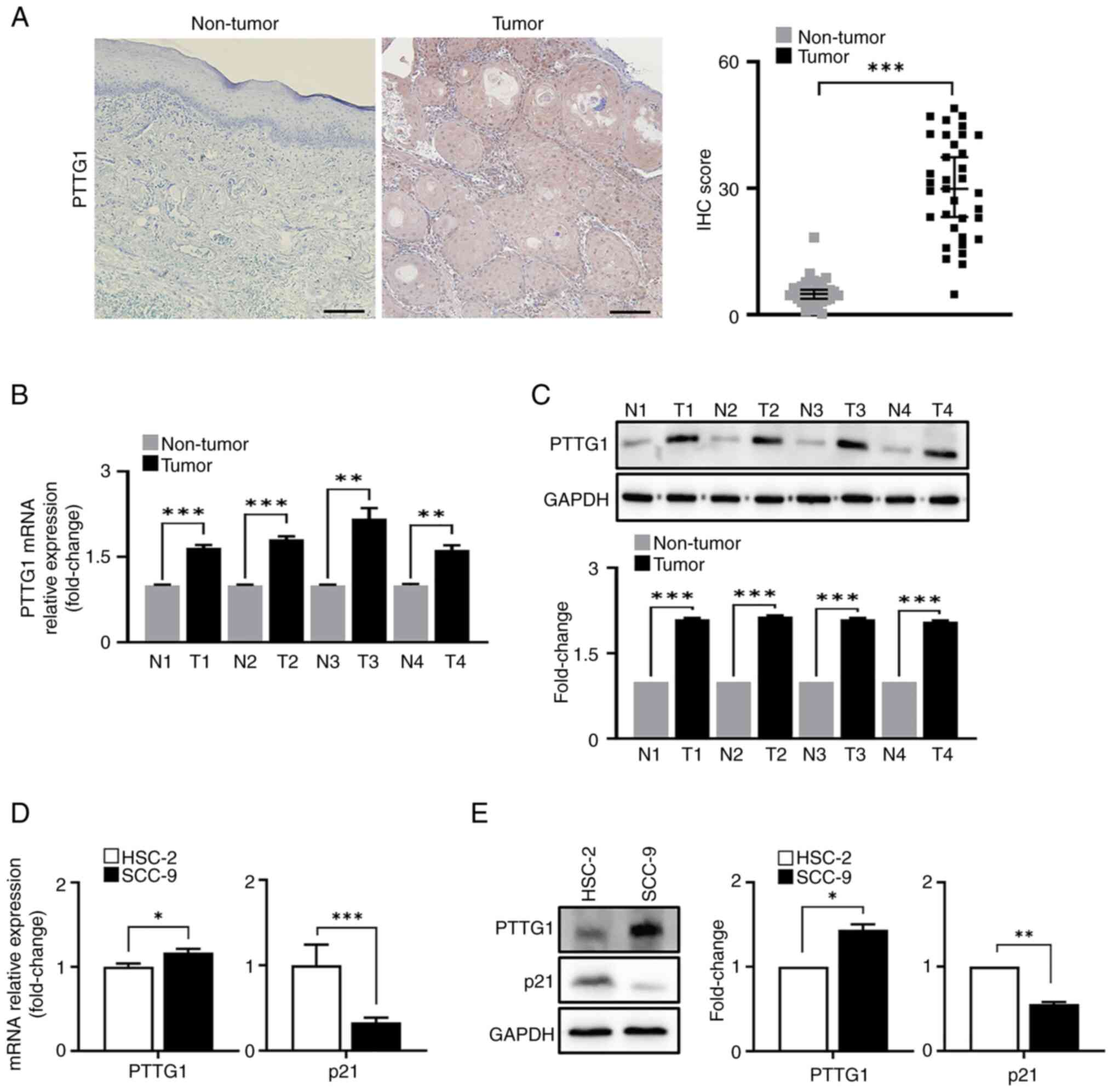 | Figure 1.PTTG1 expression in OSCC tissues and
cells. (A) Representative images (left) and quantification (right)
of PTTG1 immunohistochemistry staining in healthy (n=32) and OSCC
tissues (n=32). Scale bar, 100 µm; original magnification, ×20. (B)
PTTG1 expression was analyzed by RT-qPCR for the pooled OSCC tissue
samples. *P<0.05, **P<0.01 and ***P<0.001 vs. non-tumor.
(C) Protein expression of PTTG1 in the pooled OSCC samples revealed
by western blotting. The upper panel shows the membranes stained
with antibodies, with GAPDH as an internal control, and the lower
panel shows the relative quantification of protein expression. (D)
mRNA and (E) protein expression (left) and fold change (right) of
PTTG1 and p21 were analyzed by RT-qPCR and western blotting in OSCC
cell lines. *P<0.05, **P<0.01 and ***P<0.001 vs. HSC-2
cell lines using Student's t-test. All experiments were performed
in triplicate. PTTG1, pituitary tumor-transforming gene 1; OSCC,
oral squamous cell carcinoma; RT-qPCR, reverse transcription
quantitative PCR; N, non-tumor; T, tumor. |
PTTG1 depletion in OSCC cells
suppresses cell proliferation and promotes apoptosis via cell cycle
arrest
To evaluate the effect of PTTG1 on OSCC cell
proliferation, the expression of PTTG1 was inhibited using siRNA.
The mRNA and protein expression of PTTG1 was significantly
downregulated in HSC-2, SCC-9 and YD-10B cells transfected with
siRNA-PTTG1 compared with levels in those transfected with the
vehicle control (Figs. 2A and
S1A). Furthermore, the expression
of PCNA, a DNA replication marker (32), was significantly reduced in both
OSCC cell lines transfected with siRNA-PTTG1 compared with levels
in those transfected with the vehicle control (Figs. 2B and S1B). The percentage of DNA synthesis was
significantly suppressed in HSC-2 and SCC-9 cells transfected with
siRNA-PTTG1, leading to a decrease in cell viability, compared with
that in cells transfected with the vehicle control (Fig. 2C and D). Additionally, cyclins D1,
B1 and E1, which are responsible for cell proliferation, cell
division and DNA replication, respectively, were significantly
downregulated in both OSCC cell lines transfected with siRNA-PTTG1
compared with levels in those transfected with the vehicle control
(Figs. 2E and S1C). Aberrant changes in the cell cycle
in cancer cells lead to an imbalance between cell survival and
apoptosis (33). In the present
study, the expression ratio of apoptotic markers (including c-Cas-7
and c-PARP) was significantly increased in both OSCC cell lines
transfected with siRNA-PTTG1 compared with levels in those
transfected with the vehicle control (Figs. 2F and S1D). These findings suggested that PTTG1
depletion in OSCC cells restricts cell proliferation and promotes
cell death through cell cycle arrest.
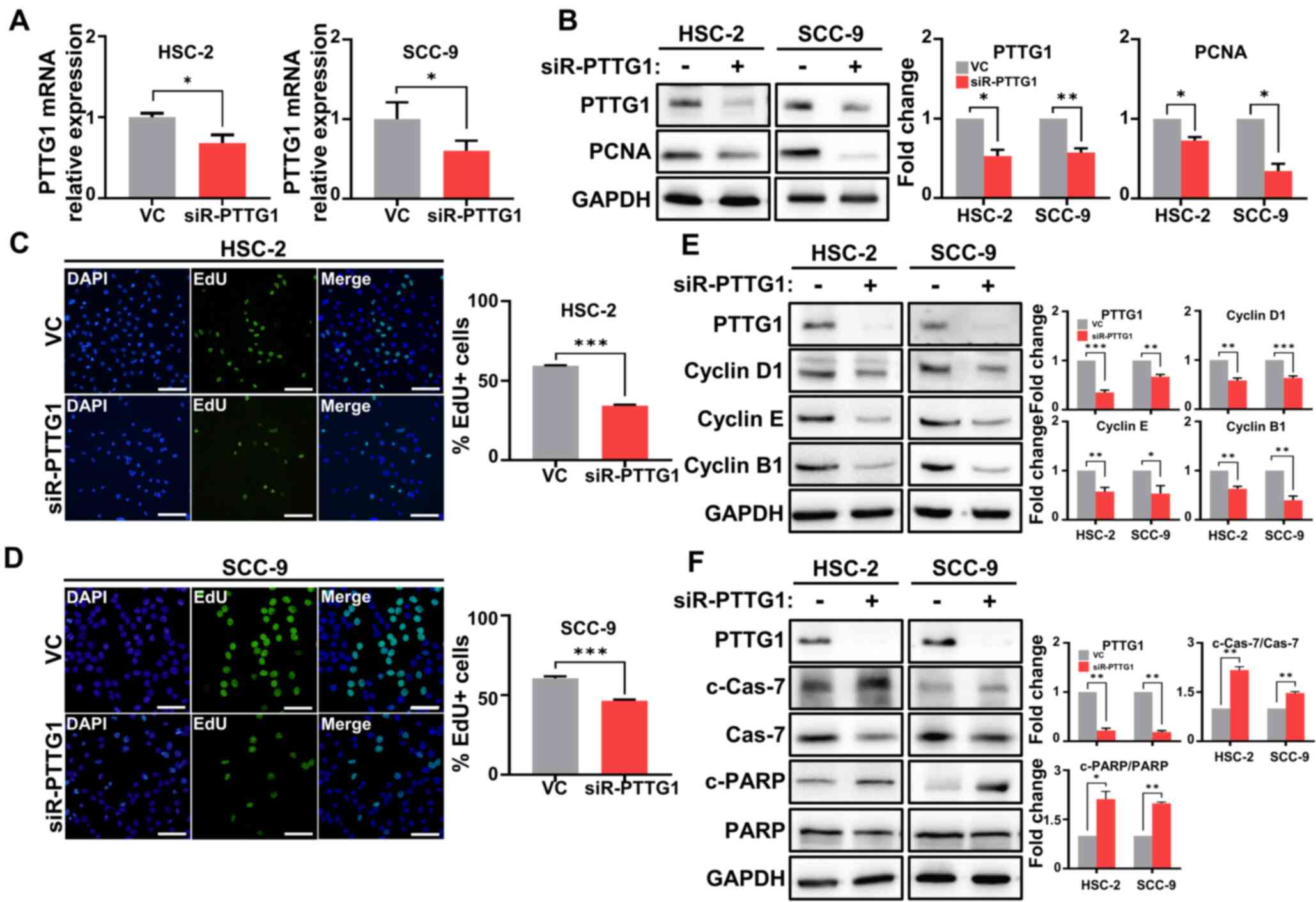 | Figure 2.PTTG1 expression in relation to cell
proliferation, cell cycle and apoptosis in OSCC cells. (A) PTTG1
expression was analyzed by reverse transcription quantitative PCR
in OSCC cells. (B) Protein expression of PTTG1 and PCNA in OSCC
cell lines revealed by western blotting. The left panel shows the
membranes stained with antibodies, with GAPDH as an internal
control, and the right panel shows the relative quantification of
protein expression. (C and D) Representative images of cell
proliferation ability (left) and quantification (right) of PTTG1
using the EdU assay in the (C) HSC-2 and (D) SCC-9 cell lines
(scale bars, 100 µm and 50 µm, respectively; original
magnification, ×40). The percentages of HSC-2 and SCC-9 cells
treated with VC or siR-PTTG1 were determined by EdU incorporation
(green) and DAPI (blue). (E) Protein expression (left) and the fold
change (right) of cell cycle markers, including cyclins D1, E, and
B1 in OSCC cells. (F) Protein expression (left) and fold change
(right) related to apoptosis markers, including Cas-7, c-Cas-7, and
c-PARP in OSCC cells. *P<0.05, **P<0.01 and ***P<0.001 vs.
VC using Student's t-test. All experiments were performed in
triplicate. PTTG1, pituitary tumor-transforming gene 1; OSCC, oral
squamous cell carcinoma; PCNA, proliferating cell nuclear antigen;
VC, vehicle control; siR-PTTG1, small interfering RNA-PTTG1; EdU,
5-ethynyl-2′-deoxyuridine; Cas-7, Caspase-7; c-, cleaved-; PARP,
poly (ADP-ribose) polymerase. |
PTTG1 depletion in OSCC cells promotes
cellular senescence via p21
p21 is a cyclin-dependent kinase inhibitor and a
senescence regulator in senescent cells, where its activation
maintains stable cell cycle arrest (25). To determine whether cell cycle
arrest caused by PTTG1 results in cellular senescence, alterations
in senescent phenotypes were evaluated following PTTG1 knockdown.
The mRNA and protein expression of p21 was significantly increased
in both cell lines transfected with siRNA-PTTG1 compared with
levels in those transfected with the vehicle control (Fig. 3A and B). Moreover, intracellular
SA-β-gal significantly accumulated in both OSCC cell lines
transfected with siRNA-PTTG1 compared with that in those
transfected with vehicle control (Fig.
3C). To understand the mechanism underlying apoptosis induction
in senescent cells following PTTG1 knockdown and to test whether an
increased DNA damage response (DDR) causes this effect after
knockdown, the phosphorylation levels of ATM and ATR, which are
involved in the DDR, were evaluated. It was found that PTTG1
knockdown in the two OSCC cell lines led to a substantial increase
in the phosphorylation of these proteins (Fig. 3D). Furthermore, there was a
significant increase in the number of γH2AX foci in the nucleus of
PTTG1 knockdown cells compared with that in those transfected with
the vehicle control, where only a few γH2AX foci were observed
(Fig. 3E and F). In the YD-10B cell
line, it was observed that γH2AX expression and SA-β-gal
accumulation increased compared with the vehicle control after
PTTG1 knockdown (Fig. S2).
Furthermore, in the HSC-2 and SCC-9 cell lines, PTTG1 knockdown
resulted in decreased p53 expression and increased p-Rb expression
compared to the vehicle control (Fig.
S3). These findings suggested that loss of PTTG1 promotes DNA
damage in a p21-dependent manner, followed by cellular senescence
regardless of cell types.
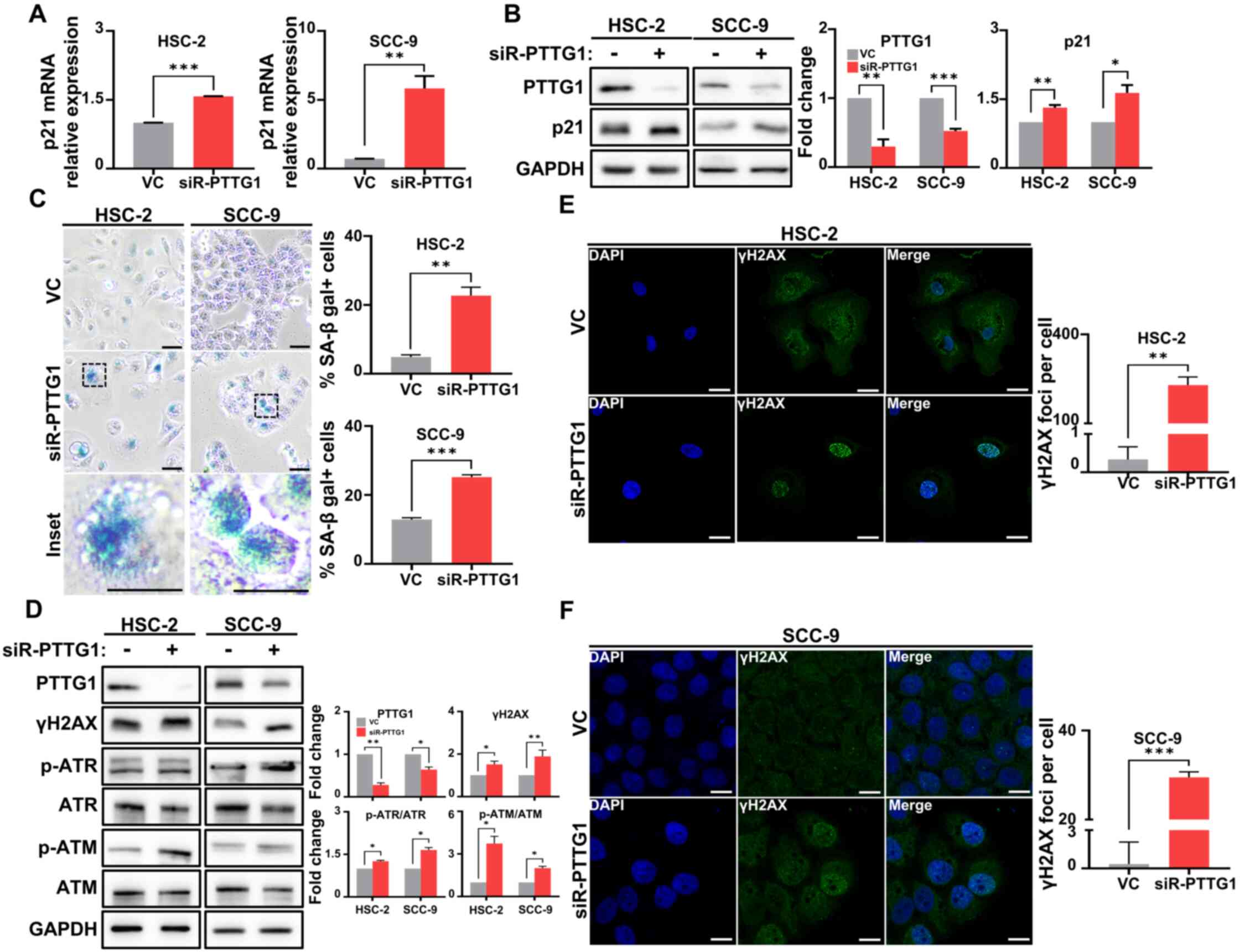 | Figure 3.The effect of PTTG1 on cellular
senescence and DNA damage in OSCC. (A) mRNA expression of p21 was
analyzed by reverse transcription quantitative PCR in OSCC cells.
(B) Protein expression (left) and quantification (right) of PTTG1
and p21 were analyzed by western blotting in OSCC cells. (C)
Representative images (left) and numbers (right) of cellular
senescence in OSCC cells detected by senescence-associated
β-galactosidase staining (blue). The percentages of HSC-2 and SCC-9
were determined by β-galactosidase incorporation (scale bars, 50 µm
and 200 µm, respectively; original magnification, ×40). (D) Protein
expression (left) and fold change (right) related to DNA damage,
including γH2AX, p-ATR and p-ATM in OSCC cells. GAPDH was used as
an internal control. (E) Representative images (left) and numbers
(right) of chromosomal damage detected by γH2AX staining in HSC-2
cells (scale bar, 50 µm; original magnification, ×63). (F)
Representative images (left) and numbers (right) of chromosomal
damage detected by γH2AX staining in SCC-9 cells (scale bar, 250
µm; original magnification, ×63). *P<0.05, **P<0.01 and
***P<0.001 vs. VC using Student's t-test. All experiments were
performed in triplicate. PTTG1, pituitary tumor-transforming gene
1; OSCC, oral squamous cell carcinoma; γH2AX, phosphorylated
histone H2AX; ATR, ataxia telangiectasia and Rad3-related protein;
p-, phosphorylated; ATM, ataxia telangiectasia mutant; siR-PTTG1,
small interfering RNA-PTTG1; SA-β gal, senescence-associated
beta-galactosidase; VC, vehicle control. |
PTTG1 depletion in OSCC cells promotes
apoptosis via DNA damage in vivo
To verify the effect of PTTG1 on tumor progression,
6-week-old nude mice were subcutaneously injected with
2×106 vehicle control-treated or PTTG1-knockdown HSC-2
or SCC-9 cells. Tumor growth, weight, DNA damage and apoptosis were
analyzed to determine the effect of PTTG1 on tumor progression
(Fig. 4A). Compared with that in
the vehicle control-treated group, a steady reduction in tumor size
was observed in the groups injected with PTTG1-knockdown HSC-2 and
SCC-9 cell lines starting from 15 days after injection (Fig. 4B). The largest tumor observed in the
SCC-9 VC group had a maximum diameter of 14.06 mm and a volume of
12.6 mm (V=1116 mm3). Moreover, the groups injected with
the PTTG1-knockdown cell lines showed a statistically significant
decrease in tumor weight 3 weeks after cell injection compared with
that in the vehicle control group (Fig.
4C). Immunoblotting results revealed that PTTG1 knockdown
significantly reduced the expression of PCNA, a cell proliferation
marker, and elevated the expression of p21 (senescence), c-Cas-7
(apoptosis), markers of γH2AX p-ATR and p-ATM (DNA damage),
compared with that in the vehicle control-treated group (Fig. 4D). To examine whether the reduction
in tumor growth by PTTG1 knockdown was associated with DNA
damage-dependent cancer cell apoptosis, a TUNEL assay was performed
to measure DNA breaks in cells undergoing apoptosis (34). In the vehicle control group, the
nuclei of cells present in the tumor tissues exhibited a negligible
number of DNA breaks. Conversely, tumor tissues injected with
PTTG1-knockdown HSC-2 and SCC-9 cells displayed a significant
increase in DNA breaks, similar to the effects observed with
DNA-damaging chemicals in vitro (Figs. 4E, F and S4). Thus, these findings suggested that
PTTG1 expression plays a crucial role in regulating tumor growth
and apoptosis and that these effects are mediated by DNA
damage.
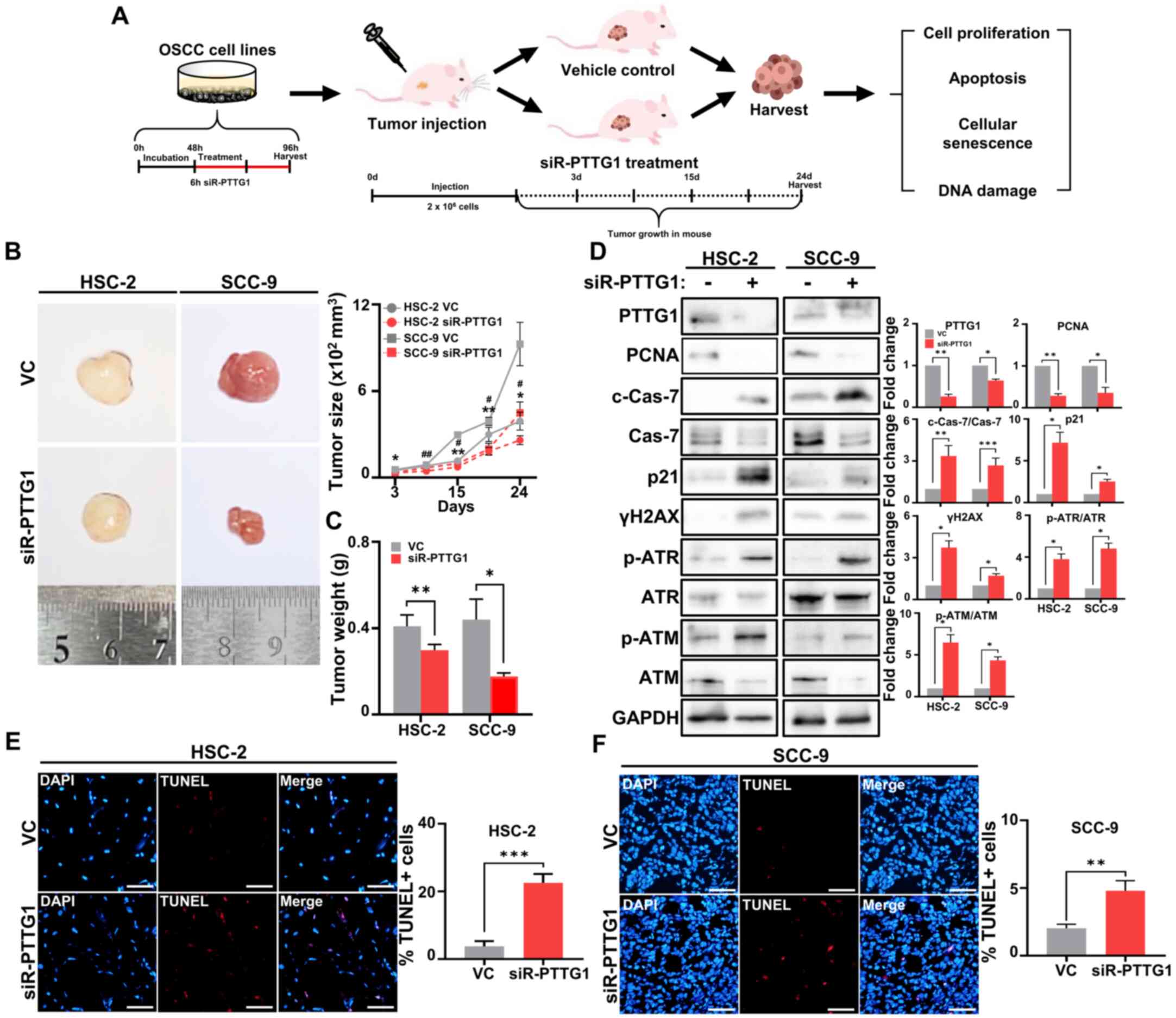 | Figure 4.The effect of PTTG1 downregulation on
tumor growth in vivo. (A) A schematic diagram of
transfection in vivo. (B) Representative images of tumor
sizes in vivo. The tumor size of each group was depicted
graphically in vivo. (scale bar, 500 mm). (C) Tumor weights
of each group in vivo. (D) Protein expression (left) and
fold change (right) related to cell proliferation, apoptosis,
cellular senescence and DNA damage, including PCNA, c-Cas-7, p21,
γH2AX and p-ATM in OSCC cell lines. GAPDH was used as an internal
control. (E and F) Representative images (left) and numbers (right)
of apoptotic DNA damage analyzed by TUNEL assay (red) and DAPI
(blue) in (E) HSC-2 and (F) SCC-9 cells (scale bar, 50 µm; original
magnification, ×20). *P<0.05, **P<0.01, ***P<0.001,
#P<0.05 and ##P<0.01 vs. VC using
Student's t-test. All experiments were performed in triplicate.
PTTG1, pituitary tumor-transforming gene 1; PCNA, proliferating
cell nuclear antigen; c-Cas-7, cleaved Caspase-7; γH2AX,
phosphorylated histone H2AX; ATM, ataxia telangiectasia mutant; p-,
phosphorylated; Cas-7, Caspase-7; ATR, ataxia telangiectasia and
Rad3-related protein; ATM, ataxia telangiectasia mutant; OSCC, oral
squamous cell carcinoma; TUNEL, terminal deoxynucleotidyl
transferase-mediated dUTP nick end labeling; VC, vehicle
control. |
Discussion
Numerous studies have investigated the role of PTTG1
in promoting the malignant progression of cancer through various
mechanisms (35–37). These studies emphasize the crucial
significance of PTTG1 in tumor progression and further support the
present and related previous studies, which identified a role for
PTTG1 in OSCC growth and metastasis. It was previously demonstrated
by the authors that suppressing the expression of PTTG1 led to a
significant reduction in the migration and invasion abilities of
the OSCC cell lines YD-10B and YD-15. However, the differential
effects of PTTG1 on the growth of YD-10B and YD-15 cells are
attributed to the expression of p53 (38), which was not expressed in YD-10B
cells but was strongly expressed in YD-15 cells. p53 is a potent
tumor suppressor gene, and mutations in this gene have been
observed in most cancers. These mutations disrupt the function of
antitumor genes and contribute to cancer progression facilitated by
oncogenes (39). In the YD-10B cell
line, independent of p53 expression, PTTG1 knockdown significantly
inhibited PCNA expression compared with that in the vehicle control
(Fig. S1A and B, P<0.05).
Furthermore, PTTG1 knockdown resulted in inhibition of proteins
involved in the cell cycle or induction of apoptotic markers
(Fig. S1C and D). Interestingly,
consistent with the findings observed in YD-10B, PTTG1 knockdown
induced DNA damage (Fig. S2A) and
cellular senescence (Fig. S2B),
which is in contrast with observations in the corresponding vehicle
control. Therefore, in the present study, the HSC-2 and SCC-9 cell
lines were utilized, both of which harbor p53 mutations, to
comprehensively investigate the role of PTTG1 in OSCC progression
(40,41).
In the present study, PTTG1 was observed
overexpression in patients with OSCC and it was revealed that PTTG1
negatively regulates the expression of p21, leading to stable cell
cycle arrest. The suppression of PTTG1 expression in OSCC cells
resulted in irreversible DNA damage and sustained senescence.
However, the molecular mechanisms underlying the regulation of the
p21-dependent pathway by PTTG1 require further investigation.
Moreover, whether the DDR pathway is activated in OSCC following
DNA damage induced by PTTG1 inhibition requires further
investigation to provide a comprehensive understanding of the
molecular mechanisms involved. Finally, the present in vivo
results demonstrated that inhibiting PTTG1 expression induces
apoptosis in OSCC cells. Therefore, the current study provided
novel evidence supporting the significant role of PTTG1 in OSCC
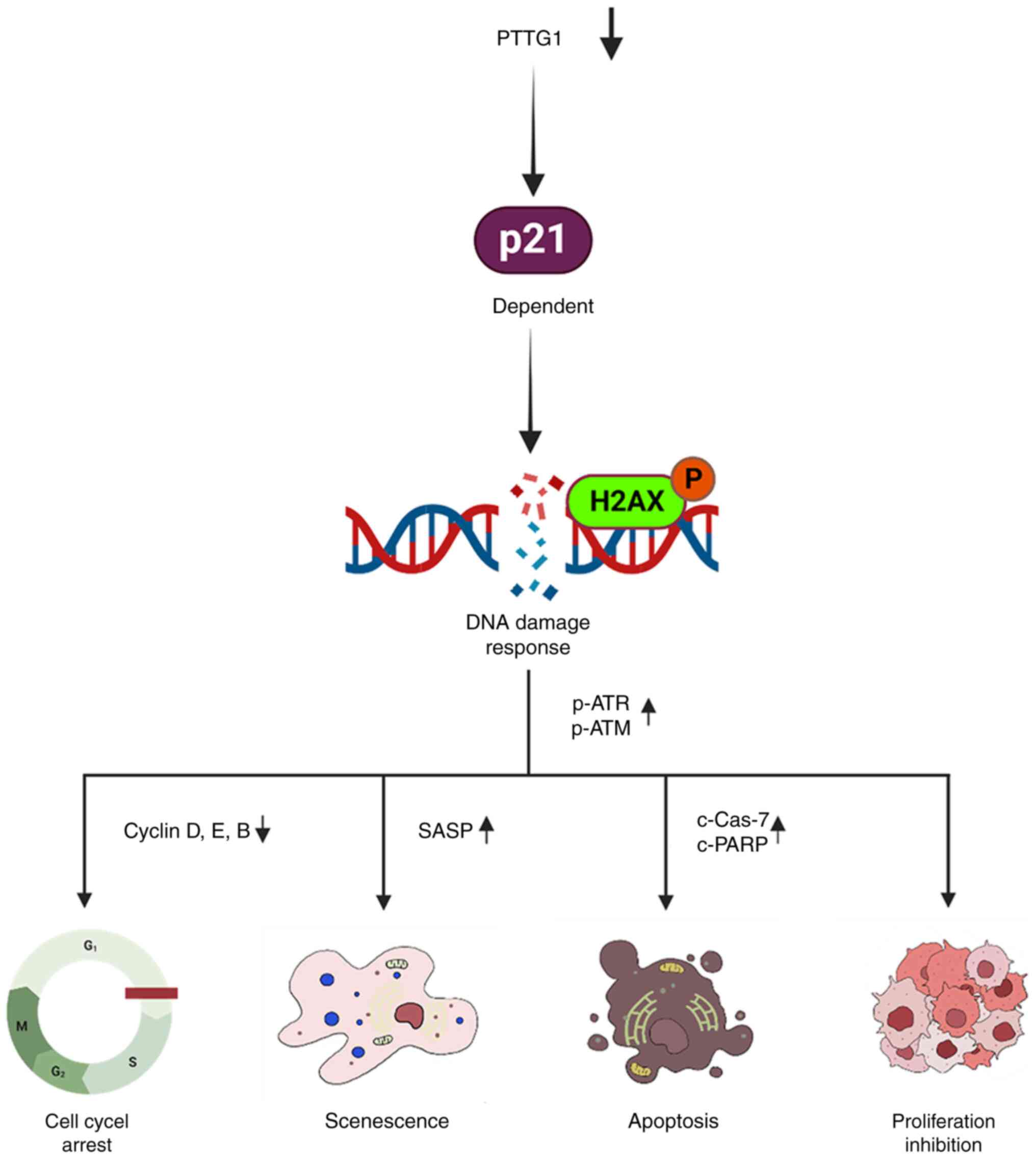
proliferation and metastasis through cell cycle arrest, DNA damage
and cell death mediated by p21 (Fig.
5).
DNA damage refers to the disruption or breakage of
DNA strands due to the generation of abnormal nucleotides or
nucleotide fragments. This can be induced by various intrinsic or
extrinsic factors, including ionizing radiation, reactive oxygen
species and drugs, in normal cells (42). Persistent DNA damage triggers the
activation of cellular processes, such as apoptosis or senescence,
which prevent the replication of a damaged genome (43). In contrast to normal cells, cancer
cells frequently arise because of replication stress induced by
oncogenes. Additionally, DDR mechanisms, which continuously repair
DNA damage to prevent senescence and apoptosis, prolong the
survival of cancer cells (44).
Therefore, most antitumor therapeutic strategies aim to inhibit the
growth of cancer cells, induce apoptosis, and impose continuous and
significant DNA damage to fundamentally block the DDR mechanism
(45). Following DNA damage stress
and DNA double-strand breaks, ATM and ATR undergo excessive
phosphorylation, which leads to phosphorylation of the histone
variant H2AX at the Ser139 residue, resulting in the
formation of p-H2AX (γH2AX) (46).
γH2AX plays a crucial role in inducing cell apoptosis in cancer
cells (47). Therefore, p53 may
play a central role in determining the spontaneous fate of cells
following DNA damage. Recently, Engeland et al (48) demonstrated that p53, during
activation by DNA damage, upregulates p21 expression and induces
cell cycle arrest via Rb-E2F complex formation. However, in the
present study, it was found that PTTG1 downregulation led to the
activation of p21 expression and the downregulation of p53
expression. PTTG1-induced senescence and apoptosis in OSCC cells
were regulated independently of p53 and Rb (Figs. 3B and S3). Similar results regarding cancer
apoptosis through p53-independent induction of p21 have been
reported by other research groups in studies on prostate and liver
cancers (49,50). These findings further support the
observation of p53-independent senescence and apoptosis in OSCC
cells. In addition, DOX was used to investigate changes in the
expression of PTTG1 and p21 following anticancer drug treatment.
DOX is known to cause DNA double-strand breaks and is currently
used as a clinical anticancer treatment (51,52).
Therefore, to investigate the appropriate concentration of DOX,
IC50 values in HSC-2 (2.7 µM) and SCC-9 cells (27 µM)
were analyzed MTT assays (Fig. S4A and
B). The present results identified that mRNA expression of
PTTG1 was significantly decreased after DOX treatment in both cell
lines (Fig. S4C). Conversely, the
mRNA expression of p21 was significantly increased in both cell
lines (Fig. S4D). In addition, it
was found that DOX treatment in the two OSCC cell lines led to a
substantial increase in the number of γH2AX foci in the nucleus
compared with that in control (Fig.
S4E and F). These results suggested that the p21-dependent
expression of PTTG1 may elucidate the mechanism of anticancer
treatment. However, the current results also have certain
limitations. First, it is needed to analyze the expression of PTTG1
in OSCC patients who underwent chemotherapy and radiation therapy.
Second, it is needed to identify the mechanisms that induce
senescence and apoptosis in OSCC though p21-only silencing in both
in vitro and in vivo experiments. Lastly, the role of
p21-dependent PTTG1 after chemotherapy in in vitro
experiments needs to be investigated in future studies.
Taken together, the findings of the present study
support the proposed role of PTTG1 as an oncogene and demonstrated
that inducing DNA damage through PTTG1 downregulation suppresses
OSCC growth and metastasis by promoting senescence and apoptosis in
a p53-independent and p21-dependent manner. These novel insights
contribute to understanding of the mechanisms underlying OSCC
development and metastasis and may open avenues for the development
of diagnostic and therapeutic strategies for OSCC.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Basic Science Research
Program through a National Research Foundation of Korea grant
funded by the Korean Government (MSIT) (grant no. 2020R1C1C1004007)
and the Ministry of Education, Science, and Technology (grant no.
2019R1I1A33A01057886).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
SP and JC designed the study and wrote the
manuscript. SP and SK performed the experiments. MYK and SSL
collected clinical specimens and wrote the manuscript. SP and SK
revised the paper. SP, SK and JC confirm the authenticity of all
the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Committee for
Ethical Review of Research of Gangneung-Wonju National University
(approval no. GWNUIRB-2020-26-1; Gangneung-si, Korea). Written
informed consent was provided by all patients before surgery.
Animal studies were approved by the Gangneung-Wonju National
University Ethics Committee on Animal Experiments (approval no.
GWNUIRB-2020-36-1; Gangneung-si, Korea).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
OSCC
|
oral squamous cell carcinoma
|
|
PTTG1
|
pituitary tumor-transforming gene
1
|
|
SA-β-gal
|
senescence-associated
beta-galactosidase
|
|
c-
|
cleaved-
|
|
Cas7
|
Caspase-7
|
|
PARP
|
poly (ADP-ribose) polymerase
|
|
PCNA
|
proliferating cell nuclear antigen
|
|
GAPDH
|
glyceraldehyde 3-phosphate
dehydrogenase
|
|
γH2AX
|
phosphorylated histone H2AX
|
|
ATM
|
ataxia telangiectasia mutant
|
|
ATR
|
ataxia telangiectasia and Rad3-related
protein
|
|
siRNA
|
small interfering RNA
|
|
IHC
|
immunohistochemistry
|
|
TUNEL
|
terminal
deoxynucleotidyl-transferase-mediated dUTP nick end labeling
|
|
RT-qPCR
|
reverse transcription-quantitative
polymerase chain reaction
|
|
EdU
|
5-Ethynyl-2′-deoxyuridine
|
|
RT
|
room temperature
|
References
|
1
|
No authors listed: Oral cancer-the fight
must go on against all odds…. Evid Based Dent. 23:4–5. 2022.
View Article : Google Scholar
|
|
2
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Johnson DE, Burtness B, Leemans CR, Lui
VWY, Bauman JE and Grandis JR: Head and neck squamous cell
carcinoma. Nat Rev Dis Primers. 6:922020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kuo TJ, Jean YH, Shih PC, Cheng SY, Kuo
HM, Lee YT, Lai YC, Tseng CC, Chen WF and Wen ZH: Stellettin
B-induced oral cancer cell death via endoplasmic reticulum
stress-mitochondrial apoptotic and autophagic signaling pathway.
Int J Mol Sci. 23:88132022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kruijtzer CM, Beijnen JH and Schellens JH:
Improvement of oral drug treatment by temporary inhibition of drug
transporters and/or cytochrome P450 in the gastrointestinal tract
and liver: An overview. Oncologist. 7:516–530. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dobler C, Jost T, Hecht M, Fietkau R and
Distel L: Senescence induction by combined ionizing radiation and
DNA damage response inhibitors in head and neck squamous cell
carcinoma cells. Cells. 9:20122020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pei L and Melmed S: Isolation and
characterization of a pituitary tumor-transforming gene (PTTG). Mol
Endocrinol. 11:433–441. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jallepalli PV, Waizenegger IC, Bunz F,
Langer S, Speicher MR, Peters JM, Kinzler KW, Vogelstein B and
Lengauer C: Securin is required for chromosomal stability in human
cells. Cell. 105:445–457. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vlotides G, Eigler T and Melmed S:
Pituitary tumor-transforming gene: Physiology and implications for
tumorigenesis. Endocr Rev. 28:165–186. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Teveroni E, Di Nicuolo F, Bianchetti G,
Epstein AL, Grande G, Maulucci G, De Spirito M, Pontecorvi A,
Milardi D and Mancini F: Nuclear localization of PTTG1 promotes
migration and invasion of seminoma tumor through activation of
MMP-2. Cancers (Basel). 13:2122021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang Z, Yu R and Melmed S: Mice lacking
pituitary tumor transforming gene show testicular and splenic
hypoplasia, thymic hyperplasia, thrombocytopenia, aberrant cell
cycle progression, and premature centromere division. Mol
Endocrinol. 15:1870–1879. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lai Y, Xin D, Bai J, Mao Z and Na Y: The
important anti-apoptotic role and its regulation mechanism of PTTG1
in UV-induced apoptosis. J Biochem Mol Biol. 40:966–972.
2007.PubMed/NCBI
|
|
13
|
Kim DS, Franklyn JA, Smith VE, Stratford
AL, Pemberton HN, Warfield A, Watkinson JC, Ishmail T, Wakelam MJ
and McCabe CJ: Securin induces genetic instability in colorectal
cancer by inhibiting double-stranded DNA repair activity.
Carcinogenesis. 28:749–759. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Read ML, Fong JC, Modasia B, Fletcher A,
Imruetaicharoenchoke W, Thompson RJ, Nieto H, Reynolds JJ, Bacon A,
Mallick U, et al: Elevated PTTG and PBF predicts poor patient
outcome and modulates DNA damage response genes in thyroid cancer.
Oncogene. 36:5296–5308. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pei L: Identification of c-myc as a
down-stream target for pituitary tumor-transforming gene. J Biol
Chem. 276:8484–8491. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bernal JA, Luna R, Espina A, Lázaro I,
Ramos-Morales F, Romero F, Arias C, Silva A, Tortolero M and
Pintor-Toro JA: Human securin interacts with p53 and modulates
p53-mediated transcriptional activity and apoptosis. Nat Genet.
32:306–311. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liao LJ, Hsu YH, Yu CH, Chiang CP, Jhan
JR, Chang LC, Lin JJ and Lou PJ: Association of pituitary tumor
transforming gene expression with early oral tumorigenesis and
malignant progression of precancerous lesions. Head Neck.
33:719–726. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang E, Liu S, Xu Z, Huang S, Tan X, Sun
C and Lu L: Pituitary tumor-transforming gene 1 (PTTG1) is
overexpressed in oral squamous cell carcinoma (OSCC) and promotes
migration, invasion and epithelial-mesenchymal transition (EMT) in
SCC15 cells. Tumour Biol. 35:8801–8811. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Piskorz WM and Cechowska-Pasko M:
Senescence of tumor cells in anticancer therapy-beneficial and
detrimental effects. Int J Mol Sci. 23:110822022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kuilman T, Michaloglou C, Mooi WJ and
Peeper DS: The essence of senescence. Genes Dev. 24:2463–2479.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Campisi J and d'Adda di Fagagna F:
Cellular senescence: When bad things happen to good cells. Nat Rev
Mol Cell Biol. 8:729–740. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Childs BG, Baker DJ, Kirkland JL, Campisi
J and van Deursen JM: Senescence and apoptosis: Dueling or
complementary cell fates? EMBO Rep. 15:1139–1153. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gorgoulis V, Adams PD, Alimonti A, Bennett
DC, Bischof O, Bishop C, Campisi J, Collado M, Evangelou K,
Ferbeyre G, et al: Cellular senescence: Defining a path forward.
Cell. 179:813–827. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hernandez-Segura A, Nehme J and Demaria M:
Hallmarks of cellular senescence. Trends Cell Biol. 28:436–453.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Herranz N and Gil J: Mechanisms and
functions of cellular senescence. J Clin Invest. 128:1238–1246.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
HAYFLICK L: THE LIMITED IN VITRO LIFETIME
OF HUMAN DIPLOID CELL STRAINS. Exp Cell Res. 37:614–636. 1965.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen YC, Hsieh HH, Chang HC, Wang HC, Lin
WJ and Lin JJ: CDC25B induces cellular senescence and correlates
with tumor suppression in a p53-dependent manner. J Biol Chem.
296:1005642021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jung SH, Hwang HJ, Kang D, Park HA, Lee
HC, Jeong D, Lee K, Park HJ, Ko YG and Lee JS: mTOR kinase leads to
PTEN-loss-induced cellular senescence by phosphorylating p53.
Oncogene. 38:1639–1650. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jeoung DI, Tang B and Sonenberg M:
Induction of tumor suppressor p21 protein by kinase inhibitors in
MCF-7 cells. Biochem Biophys Res Commun. 214:361–366. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Englund DA, Jolliffe A, Aversa Z, Zhang X,
Sturmlechner I, Sakamoto AE, Zeidler JD, Warner GM, McNinch C,
White TA, et al: p21 induces a senescence program and skeletal
muscle dysfunction. Mol Metab. 67:1016522023. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bologna-Molina R, Mosqueda-Taylor A,
Molina-Frechero N, Mori-Estevez AD and Sánchez-Acuña G: Comparison
of the value of PCNA and Ki-67 as markers of cell proliferation in
ameloblastic tumors. Med Oral Patol Oral Cir Bucal. 18:e174–e179.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Loftus LV, Amend SR and Pienta KJ:
Interplay between cell death and cell proliferation reveals new
strategies for cancer therapy. Int J Mol Sci. 23:47232022.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mirzayans R and Murray D: Do TUNEL and
other apoptosis assays detect cell death in preclinical studies?
Int J Mol Sci. 21:90902020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Meng C, Zou Y, Hong W, Bao C and Jia X:
Estrogen-regulated PTTG1 promotes breast cancer progression by
regulating cyclin kinase expression. Mol Med. 26:332020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang G, Xiog Y, Wang G, Li W, Tang T, Sun
J and Li J: miR-374c-5p regulates PTTG1 and inhibits cell growth
and metastasis in hepatocellular carcinoma by regulating
epithelial-mesenchymal transition. Mol Med Rep. 25:1482022.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang H, He Z, Qiu L, Wei J, Gong X, Xian
M, Chen Z, Cui Y, Fu S, Zhang Z, et al: PRR11 promotes cell
proliferation by regulating PTTG1 through interacting with E2F1
transcription factor in pan-cancer. Front Mol Biosci. 9:8773202022.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee SS, Choi JH, Lim SM, Kim GJ, Lee SK
and Jeon YK: Alteration of pituitary tumor transforming gene 1 by
microRNA-186 and 655 regulates invasion ability of human oral
squamous cell carcinoma. Int J Mol Sci. 22:10212021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hu J, Cao J, Topatana W, Juengpanich S, Li
S, Zhang B, Shen J, Cai L, Cai X and Chen M: Targeting mutant p53
for cancer therapy: Direct and indirect strategies. J Hematol
Oncol. 14:1572021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nakagaki T, Tamura M, Kobashi K, Koyama R,
Fukushima H, Ohashi T, Idogawa M, Ogi K, Hiratsuka H, Tokino T and
Sasaki Y: Profiling cancer-related gene mutations in oral squamous
cell carcinoma from Japanese patients by targeted amplicon
sequencing. Oncotarget. 8:59113–59122. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hauser U, Balz V, Carey TE, Grénman R, Van
Lierop A, Scheckenbach K and Bier H: Reliable detection of p53
aberrations in squamous cell carcinomas of the head and neck
requires transcript analysis of the entire coding region. Head
Neck. 24:868–873. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Alhmoud JF, Woolley JF, Al Moustafa AE and
Malki MI: DNA damage/repair management in cancers. Cancers (Basel).
12:10502020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yousefzadeh M, Henpita C, Vyas R,
Soto-Palma C, Robbins P and Niedernhofer L: DNA damage-how and why
we age? Elife. 10:e628522021. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Trenner A and Sartori AA: Harnessing DNA
double-strand break repair for cancer treatment. Front Oncol.
9:13882019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Abuetabh Y, Wu HH, Chai C, Al Yousef H,
Persad S, Sergi CM and Leng R: DNA damage response revisited: The
p53 family and its regulators provide endless cancer therapy
opportunities. Exp Mol Med. 54:1658–1669. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cimprich KA and Cortez D: ATR: An
essential regulator of genome integrity. Nat Rev Mol Cell Biol.
9:616–627. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bonner WM, Redon CE, Dickey JS, Nakamura
AJ, Sedelnikova OA, Solier S and Pommier Y: GammaH2AX and cancer.
Nat Rev Cancer. 8:957–967. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Engeland K: Cell cycle regulation:
p53-p21-RB signaling. Cell Death Differ. 29:946–960. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ho CJ, Lin RW, Zhu WH, Wen TK, Hu CJ, Lee
YL, Hung TI and Wang C: Transcription-independent and -dependent
p53-mediated apoptosis in response to genotoxic and non-genotoxic
stress. Cell Death Discov. 5:1312019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lee TK, Lau TC and Ng IO:
Doxorubicin-induced apoptosis and chemosensitivity in hepatoma cell
lines. Cancer Chemother Pharmacol. 49:78–86. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kciuk M, Gielecińska A, Mujwar S, Kołat D,
Kałuzińska-Kołat Ż, Celik I and Kontek R: Doxorubicin-an agent with
multiple mechanisms of anticancer activity. Cells. 12:6592023.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yun UJ, Lee JH, Shim J, Yoon K, Goh SH, Yi
EH, Ye SK, Lee JS, Lee H, Park J, et al: Anti-cancer effect of
doxorubicin is mediated by downregulation of HMG-Co A reductase via
inhibition of EGFR/Src pathway. Lab Invest. 99:1157–1172. 2019.
View Article : Google Scholar : PubMed/NCBI
|