Introduction
Bullous pemphigoid (BP) is a chronic autoimmune
disorder caused by antibodies targeting BP180, a type XVII
collagen, HD1 (BPAG1), a 230-kDa glycoprotein located on chromosome
6p11-6p12 and HD4 (BPAG2), a 180-kDa hemidesmosome glycoprotein
located on chromosome 10q 24.3. The intracellular hemidesmosome
plaque contains the 230 kDa BP antigen, a non-collagenous protein
of the plakin family that serves as an autoantigen in BP located in
basal keratinocytes, resulting in the formation of a subepithelial
blisters. The 180-kDa BP antigen, a transmembrane collagenous
protein known as type XVII collagen, interacts with α6β4 integrin
and extends from the intracellular compartment of basal cells to
the extracellular space, thus stabilizing the association of basal
keratinocytes to the underlying basement membrane (1).
The term ‘pemphigoid’ is derived from a Greek word
(pemphix) meaning ‘pustule’, and ‘oid’ means ‘to resemble’. The
lesions of BP occur in the oral cavity following the onset of
pruritic papules in the extremities (2). The diagnosis of such lesions is more
challenging for the oral physician. The lesions of bullous
pemphigoid occur as blisters on the oral mucous membranes, which
can rupture easily by the frictional forces of the teeth during
chewing (2). The ulcers are
usually multiple and exhibit chronicity due to the autoimmune
destruction of the hemidesmosome proteins, anti-BP 180 and BP 230,
targeting the basement membrane zone, resulting in the formation of
subepithelial blisters (2). The
pruritic rashes on the skin develop before the occurrence of the
oral lesions (2). Patients who are
known to be hypertensive and are already under anti-hypertensive
medications, such as amlodipine and prazosin develop BP (2). Patients who are already known
diabetics and are being administered dipeptidyl peptidase IV drugs,
such as vildagliptin, sitagliptin, linagliptin, alogliptin,
anagliptin and teneligliptin also develop BP. Penicillin,
cephalosporins, sulfonamides and antifungals also increase the risk
of developing BP lesions (2).
Preexisting lichen planus and psoriasis are also associated with
the risk of developing BP (2). The
occurrence of BP must not be overlooked, and a complete drug
history and clinical examination must be sought after by an oral
physician.
The present study describes the case of a
38-year-old male patient with BP who was a known hypertensive and
was thus being treated with amlodipine. In addition, following a
database search, the findings of various studies on BP are
discussed, in an aim to shed further light on this pathology.
Case report
A 38-year-old male patient reported to the
Department of Oral Medicine and Radiology, Vinayaka Mission's
Sankarachariyar Dental College, Vinayaka Mission's Research
Foundation (Deemed to be University) (Salem, Tamil Nadu, India)
with a chief complaint of soreness while eating foods. An analysis
of his medical history revealed that he was a known hypertensive
and under amlodipine medication 10 mg once daily for 2 years. The
patient complained of similar soreness in his oral cavity, for
which he was already treated with intravenous methylprednisolone
(500 mg in 5% dextrose) administered over a period of 6 h.
Cyclophosphamide (500 mg in 250 ml of 5% dextrose) was
intravenously infused slowly for 3 h. The patient developed high
blood pressure (170/110 mm Hg) during the intravenous
methylprednisolone therapy for which amlodipine 5 mg tablets were
prescribed twice daily. He was also prescribed prazosin (an α1
blocker) tablets at 75 mg once daily, and propranolol (40 mg) once
daily for 2 weeks. The vital signs of the patient were monitored
during the drug therapy. His medical history also revealed the
occurrence of pruritic rashes on the extensor aspect of his left
extremity on his left forearm and also on the right and left side
of his abdomen 2 years prior. For pruritic skin lesions, he was
prescribed levocetirizine (5 mg) tablets once daily for 2 weeks.
Hydroxyzine tablets (25 mg) were also prescribed once daily for 2
weeks. Upon an extraoral examination of his skin, multiple discrete
papules were observed on the extensor aspect of his left forearm
and also over the skin of his right and left abdomen (Fig. 1).
Following the occurrence of pruritic papules on his
left forearm, he developed a blister on the right buccal mucosa,
which ruptured simultaneously and was superimposed with a white
lesion (Fig. 2A). An intraoral
examination of his left buccal mucosa revealed multiple tiny ulcers
near the palatal aspect of the gingiva in relation to the
interdental papilla of the tooth no. 23 and 24 region, and another
ulcer on the left buccal mucosa (Fig.
2B). An examination of the floor of the mouth revealed a tissue
tag formed as a result of an attempt to heal the ruptured bulla
(Fig. 2C). The intraoral
examination also revealed desquamation involving the marginal
gingiva in relation to mandibular anterior teeth region (Fig. 2D) and marginal gingiva in relation
to the right maxillary posterior tooth region (Fig. 2E).
The intraoral examination also revealed a ruptured
blister surrounded by a white lesion near the mucobuccal vestibule
and an ulcer on the buccal aspect of the attached gingiva in
relation to the region of tooth 46 (Fig. 3).
The biopsied tissue from the perilesional site of
the oral mucosa was placed in Michel medium (transport medium) at
room temperature and stored. Biopsy specimens were washed for 30
min in phosphate-buffered saline (PBS) at pH 7.2. At -20˚C,
embedding material (Jung tissue freezing medium, Leica Microsystems
GmbH) was snap-frozen and 3-4-µ-thick sections were produced using
a cryostat. A minimum of three portions were washed three times for
10 min with PBS and dried in air. A drop of FITC-regent-labeled
antihuman IgG, IgM, IgA, complement C3, and fibrin was utilized.
The slides were then incubated at 37˚C for 30 min in a humid
environment and washed three times for 10 min with PBS to remove
the conjugate. The slides were air-dried and mounted with glycerol
that had been buffered at (pH 8). A reading was performed utilizing
a fluorescence microscope (EVOS M5000 fluorescence Microscope,
Thermo Fischer Scientific, Inc.).
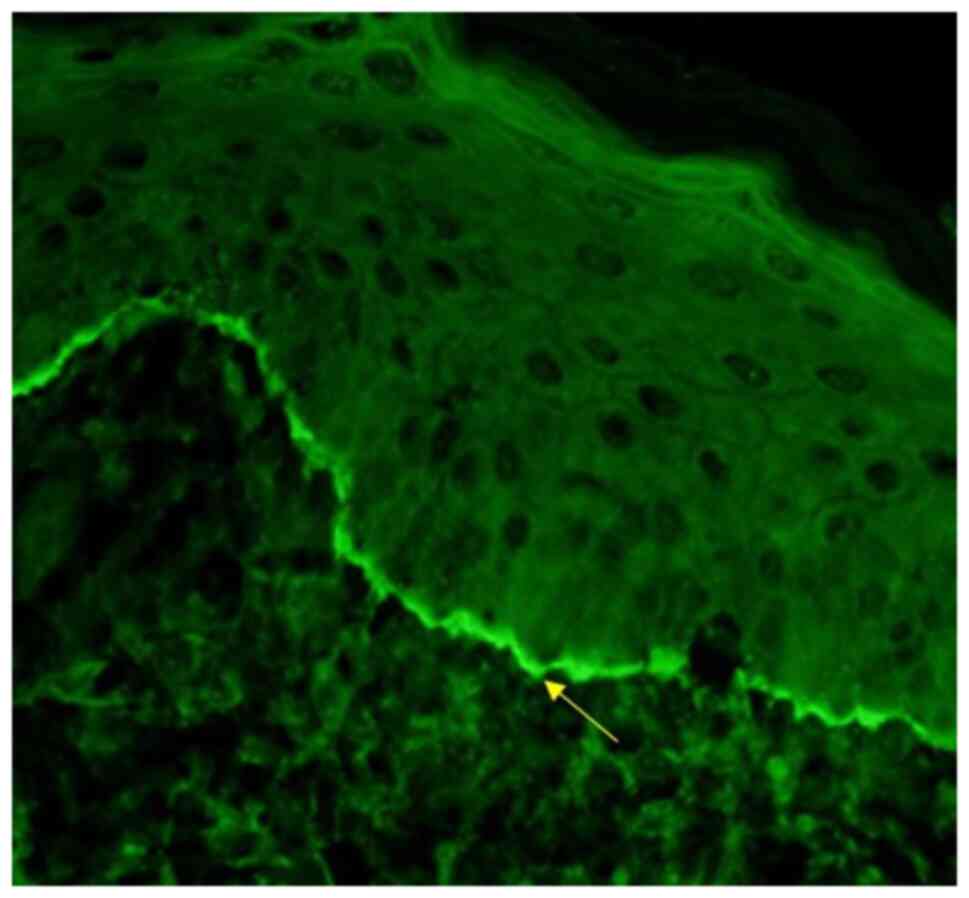
The direct immunofluorescence analysis from the
intact, unaffected site of his skin using the salt-split technique
revealed the deposition of IgG and complemented C3 only along the
epidermal side of the basal layer of the epithelium (Fig. 4). The titres for BP180 antibodies
using ELISA (using the MBL Bion DSG 1 & DSG 3 ELISA TEST.
SYSTEM) were 105.7 U/ml.
The patient again developed blisters and ulcers on
the oral mucosa and gingiva after 2 years of terminating the
prescribed methylprednisolone (12.5 mg) medication, which indicated
the recurrent nature of the lesion. The patient was prescribed
mycophenolate mofetil tablets 500 mg twice daily for 2 weeks as he
is a known hypertensive. The patient was advised to undergo
periodic follow-up sessions and evaluation.
Discussion
History of BP
BP was first differentiated from pemphigus in 1953
by Walter Lever. He described the histopathological hallmarks of
pemphigus and the intraepidermal split formation and loss of cell
adherence between keratinocytes (acantholysis). By contrast, he
coined the term ‘pemphigoid’ for conditions where a sub-epidermal
split formation was typically present (2). A decade later, Jordan et al
(3) demonstrated that in BP,
tissue-bound and serum autoantibodies against proteins of the
dermal-epidermal junction (DEJ) were present. Further milestones in
the understanding of BP included the immunochemical
characterization of the hemidesmosome target proteins BP180 (type
XVII collagen, also termed BPAG2) and BP230 (BPAG1-e), the cloning
of their genes and the demonstration that autoantibodies against
BP180 are pathogenic (3).
Pathogenesis of BP
The pathogenic importance of humoral and cellular
autoimmunity against BP180 has been demonstrated. Fcγ receptor III
and IV mediate tissue destruction in a Novel Adult Mouse Model of
Bullous Pemphigoid. More specifically, complement activation at the
DEJ and the activation of mast cells appears to be crucial for
attracting neutrophils and macrophages at the DEJ. The subsequent
release of reactive oxygen species and various proteases then
induces dermal-epidermal splitting. Targeting mast cells,
neutrophils, complement activation and the cytokine network may
open novel therapeutic avenues for this disease (4).
Autoantibodies
In almost all patients with BP, autoantibodies bind
to BP180 (type XVII collagen and BPAG2). The extracellular portion
of the 16th non-collagenous domain (NC16A) located directly
adjacent to the cellular membrane is the immunodominant region in
BP and is recognized by autoantibodies in 75-90% of patients. The
importance of anti-BP180 NC16A reactivity is further highlighted by
the observation that serum levels of BP180 NC16A-specific IgG
antibodies are associated with disease activity in patients with
BP. IgG4 and IgG1 are the major IgG subclasses of anti-BP180 NC16A
antibodies. The majority of patients also have increased levels of
IgG antibodies against epitopes outside the NC16A domain, while
initial reactivity appears to target NC16A. IgG reactivity with
C-terminal epitopes appears to be associated with mucosal
involvement and more severe skin disease, whereas the intracellular
domain is preferentially targeted at an early clinical stage. The
majority of patients with BP develop, apart from IgG, IgA and IgE
anti-BP180 reactivity. IgE anti-BP180 NC16A antibodies are
associated with a severe form of BP, a longer duration for
remission, and the requirement for more intensive therapies
(4).
BP230 (also known as BPAG1-e and BPAG1) is
recognized by 50-70% of BP sera. As regards BP180, B-cell epitopes
are not equally distributed on the molecule, but preferentially
localize to the globular C-terminal domain of BP230. In addition to
IgG reactivity, IgE antibodies against BP230 are detected in the
majority of BP sera (4).
Cellular immune response
In contrast to the humoral immune response, the
cellular immune response is less widely studied in human BP. T- and
B-cell reactivity against the NH2-terminal portion of the BP180
ectodomain is associated with severe BP, while the central part is
more frequently recognized in patients with limited disease. By
contrast, combined T- and B-cell response against the COOH- and
NH2-terminal globular domains of BP230 are found in <50% of
cases. The response to the BP180 ectodomain is restricted to the
DQβ1*0301 allele. Autoreactive T-cells in patients with BP produce
a Th1/Th2 mixed cytokine profile. While the number of circulating
CD4+CD25+FOXP3+ regulatory
T-cells, natural killer T-cells, and natural killer cells are
normal, γδT cell numbers are reduced in patients with BP. The
number of peripheral follicular helper T-cells, a T-cell subset
known to be pivotal for B-cell activation, is higher in patients
with active disease compared to healthy volunteers and patients
with BP in remission and is associated with serum levels of
anti-BP180 antibodies (5).
Cytokines and chemokines
Elevated levels of IL-1β, IL-2, IL-4, IL-5, IL-6,
IL-8, IL-10, IL-15, IL-16, IL-17, IL-21, eotaxin, monocyte
chemotactic protein 4 (MCP-4), TNF-α and CCL-18 occur in the sera
and blister fluids of patients with BP. Serum levels of TNF-α,
IL-6, IL-8, IL-15, IL-21 and CCL18 are associated with the extent
of BP skin lesions, pointing to a pathological relevance of these
mediators. Furthermore, the assumption that Th2-type cytokines are
essential in human BP is supported by the increased frequency of
cutaneous lymphocyte-associated antigen-positive IL-4- and
IL-13-producing cells in the peripheral blood. More recently, the
potential role of Th17 cells in BP has been highlighted (6).
Functionally relevant pathogenic
mechanisms
When cultured normal human keratinocytes were
treated with anti-BP180 IgG, a signal-transducing event leading to
a dose- and time-dependent release of IL-6 and IL-8 was observed.
In the same model, the internalization and creased expression of
BP180 and weakening of keratinocyte attachment in response to
anti-BP180 IgG were observed. Recently, the release of IL-6 and
IL-8 and reduction of the number of hemidesmosomes were also
observed following incubation with anti-BP180 IgE. Using
cryosections of normal human skin, BP180 NC16A-specific IgG induced
a dermal-epidermal separation when co-incubated with leukocytes
from healthy volunteers. This effect was mediated by the Fc portion
of autoantibodies and the Fcγ receptors IIA and IIIA on human
neutrophils, resulting in the release of matrix metalloproteinase-9
and neutrophil elastase. Both enzymes were found in blister fluid
and lesional biopsies from patients with BP and were capable of
degrading BP antigen 180(7).
Passive transfer of rabbit IgG raised against the
murine homolog of the human BP180 NC16A domain into neonatal
wild-type mice produced clinical, histopathological and
immunopathological alterations similar to those observed in
patients with BP. In this model, blister formation was dependent on
complement activation, the degranulation of mast cells, the
recruitment of macrophages and neutrophils, and the release of
various proteases, including the plasminogen/plasmin system, mast
cell proteinase 4, matrix metalloproteinase 9, α1-proteinase
inhibitor and neutrophil elastase. More specifically, in the early
stages of blistering, matrix metalloproteinase nine is mainly
activated by plasmin, which is formed by the activation of
plasminogen by tissue plasminogen activator and/or urokinase
plasminogen activator. Plasmin and the mast cell-specific serine
protease four can activate matrix metalloproteinase nine, which
then inactivates α1-proteinase inhibitor, the physiological
inhibitor of neutrophil elastase. The unrestrained activity of
neutrophil elastase is responsible for the degradation of the DEJ
structural proteins, including BP180(8). A recent passive transfer model in
adult mice highlighted the importance of FcγR IIB, FcγR III and
FcγR IV (9). Amongst these models,
three distinct lines of transgenic mice that expressed human BP180
in murine skin elegantly replicated essential features of human BP
(10). In one of these models, the
complement dependency of experimental BP was questioned when the
passive transfer of F(ab)2 fragments of human BP led to skin
frangibility in Col17-humanized mice (11). Subsequently, two ‘active’ mouse
models were developed that do not depend on the transfer of
anti-BP180 antibodies: Wild-type mice were immunized with
recombinant murine NC15A, and in the second model,
Rag-2-/-/COL17m-/-,h+ mice
(immunoexpression human BP180) received splenocytes from wild-type
mice that had been immunized by grafting of
COL17m-/-,h+ mouse skin and subsequently
developed anti-BP180 antibodies and a blistering phenotype. In the
latter model, the importance of NC16A-reactive CD4+
T-cells has corroborated previous in vitro studies with
human cells that showed a restriction of NC16Areactive.
CD4+ T cells to the HLA-DQB1*301 allele. In vivo
evidence for the pathogenic role of IgE autoantibodies was provided
by clinical observations and two additional mouse models. IgE
Bullous pemphigoid 180 NC16A-specific antibodies were associated
with more severe forms of human BP, associated with the extent of
skin lesions. Individual corticosteroid-resistant BP patients
responded well to omalizumab, a humanized monoclonal antibody that
inhibits IgE binding to its high-affinity receptor (12).
In contrast to BP180, the pathogenic relevance of
autoantibodies against BP230 remains elusive: Two animal models
investigating the pathogenic relevance of antibodies to BP230 have
been reported; however, blisters were not or were not consistently
seen (13). Studies on the
association of serum anti-BP230 autoantibodies with the disease
have been contradictory, with the majority finding no association
between serum anti-BP230 levels and disease activity (13). However, in BP230-/-
mice, in addition to mild skin fragility, neurological defects with
sensory neuron degeneration were developed, and thus highlighting
the association between BP and neurological disorders; autoimmunity
to BP230 may contribute not only to the skin phenotype in patients
with BP, but also to the extracutaneous features (12).
The etiopathogenesis of bullous pemphigoid occurs in
various steps that act concurrently without being dependent on each
other. These include the following:
Complement activation. Complement activation
occurs after the interaction of antibodies targeting hemidesmosomal
glycoprotein antigens, namely BP180 and BP230(14).
Release of protease enzymes from neutrophils and
eosinophils. Proteases enzymes are released from neutrophils
and eosinophils independent of complement activation, and are
recruited to the sites of inflammatory reaction in BP (14).
Micropinocytosis of anti-BP180 from
keratinocytes. Anti-BP180 IgG may induce BP180 internalization
from the keratinocyte cell membrane through micropinocytosis
(1).
TNF-related weak inducer of apoptosis
(TWEAK). TWEAK, a member of the TNF superfamily, and
TWEAK/fibroblast growth factor-inducible 14 (Fn14) interaction aids
in inducing blister formation in BP (15).
Eosinophils. Eosinophilic cationic protein,
the major basic protein from eosinophils, activates the cytokines,
IL-5, -4 and -13 that trigger immunological reactions in BP.
Eosinophils have also been demonstrated to be the main source of
tissue factor, an initiator of blood coagulation that favors
coagulability of blood, leading to pulmonary embolism (15).
Intercellular adhesion molecule 1 (ICAM-1) on
activated leukocytes. The expression of ICAM-1 on keratinocytes
is induced in basal and lower suprabasal layers by IFN-γ and TNF-α,
which are products released by lymphocytes infiltrating inflamed
skin. Activated lymphocyte IFN-γ induces the keratinocyte
expression of ICAM-1 and HLA-DR, promoting inflammatory and
allergic epidermal responses (4,16).
Chymase from mast cells. Chymase is a 30-kDa
monomeric protease stored in the same secretory granules as
tryptase in the MCTC subset of mast cells. Chymase contributes to
the splitting of the dermal-epidermal junction in BP (17). BP is a chronic autoimmune disorder
that is caused by the production of autoantibodies targeted against
BP180 and BP230, the hemidesmosomal proteins that help to connect
the basement membrane and underlying connective tissue. The binding
of antibodies to the basement membrane initiates the complement
cascade with recruitment of neutrophils and eosinophils to the area
(17).
Predisposing risk factors for BP
Several triggering or predisposing factors for BP
include trauma, burns, skin grafting, radiotherapy and UV radiation
including sunlight, UVA1, psoralen and UVA (PUVA) and photodynamic
therapy. Furthermore, most frequently against influenza, BP occurs
following vaccination for COVID-19 and influenza (18,19).
Numerous case reports have described the triggering of BP by drugs,
most frequently frusemide, least likely with spironolactone,
phenothiazines with aliphatic side-chains and loop diuretics
(20-22).
The use of these drugs should thus be carefully evaluated. Various
groups of drugs are more commonly implicated in the pathogenesis of
BP and these are presented in Table
I.
 | Table IDrugs that have a high association
with Bullous pemphigoid. |
Table I
Drugs that have a high association
with Bullous pemphigoid.
| Drug group | Drug |
|---|
| Dipeptidyl
peptidase IV Inhibitors | Linagliptin,
vidagliptin, alogliptin, anagliptin, teneligliptin |
| Check point
inhibitors: Programmed cell death protein-1 (PD-1) and the
programmed death ligand-1 (PD-L1) | Pembrolizumab,
nivolumab and durvalumab |
| Antineoplastic
drug, kinase inhibitors | Everolimus |
|
Immunosupressants | Tacrolimus |
| Diuretics | Thiazide diuretics,
hydrochlorthiazide, chlorthalidone, spironolactone, frusemide |
| Antitubercular
drug | Rifampicin |
| Antifungal
drugs | Terbinafine,
griseofulvin |
| Non-steroidal
anti-inflammatory drugs | Ibuprofen, aspirin,
phenacetin (paracetamol) |
|
Antibiotics-Penicillin Group | Amoxicillin,
ampicillin |
| Cephalosporins | Cephalexin |
|
Fluoroquinolones | Ciprofloxacin |
| Sulfonamides | Sulfasalazine |
| Statins | Rosuvastatin |
Lichen planus, psoriasis, multiple sclerosis,
Alzheimer's disease, pre-existing coagulopathies, such as elevated
D-dimer levels and the overexpression of tissue factors can result
in increased risk for thromboembolic events, such as pulmonary
embolism (2). COVID-19 mRNA
vaccines also pose a risk for development of BO (2). Hematological malignancies, such as
Hodgkin's lymphoma, non-follicular lymphoma, mature T/NK-cell
lymphoma, non-Hodgkin's lymphoma, myeloid leukemia, and other types
of cancer, such as gastric, renal, bladder, colorectal, prostate,
laryngeal, non-small cell lung and breast cancers predispose to BP
(22).
Clinical features
A prodromal non-bullous phase usually precedes the
development of tense generalized blisters. This prodromal phase may
last for several weeks or even months. At this stage, a clinical
diagnosis is difficult. Pruritus, from mild to intractable, is
typical and may even occur without skin lesions. In the prodromal
phase, excoriated papules, eczematous, or urticarial lesions,
hemorrhagic crusts and excoriations prevail. The bullous stage is
characterized by intense pruritus accompanied by widespread tense
blisters and vesicles on apparently normal or erythematous skin
(23).
Frequently, partly hemorrhagic crusts and urticated
and infiltrated erythematous plaques with an occasionally annular
or figurate pattern are present for several centimeters and contain
clear sometimes hemorrhagic exudates; Nikolsky's sign is negative.
Pruritus, which may be incapacitating, is almost constantly
present. Blisters are typically symmetrically distributed and may
persist for several days. Following mechanical irritation, erosions
and yellowish or hemorrhagic crusts develop. Predilection sites
involve the flexural aspects of the limbs and abdomen. In the
intertriginous areas, vegetating plaques may occur and oral lesions
develop in 10-20% of cases. The mucosae of the eyes, nose, pharynx,
esophagus, and anogenital areas are rarely affected. Without severe
superinfection, all lesions heal without scarring. Erythema may
persist at the sites of previous blisters for many weeks or months.
Milia formation only rarely occurs (24).
Clinical variants of BP
Cutaneous manifestations of BP can be highly
polymorphic. This notion has led to the description of several
clinical variants. In all of these, direct immunofluorescence
microscopy of a perilesional biopsy reveals linear deposits of IgG
and/or C3 at the DEJ. At present, fine specificities of serum
autoantibodies were not shown to differ from the classic form.
Several clinical variants of BP have been described with a variety
of different denominations, such as dyshidrosiform, prurigo
nodularis-like, prurigo-like, erythrodermic, ecthyma
gangrenosum-like, intertrigo-like, papular, eczematous,
lymphomatoid papulosis-like, vegetating, vesicular and toxic
epidermolysis-like pemphigoid. Some forms, such as prurigo-like,
papular, eczematous, vesicular and erythrodermic pemphigoid may
later develop into tense blisters and transform into the classical
type (24). 20% of patients with
BP present with the non-classical form at the time of diagnosis
(24).
Localized BP
In some patients, the disease is limited to certain
body parts, most frequently the lower extremities and notably, the
pretibial area. In addition, other regions such as the flexures,
palms, soles, genital area and the umbilicus have been described,
as well as around stomata and hemodialysis fistulae. Localized
lesions may remain localized or develop into classical BP (24).
Childhood BP
Two peaks of incidences of BP in childhood have been
reported, in the first year of life (infantile BP) and around at
the age of 8 years (24). Multiple
cases with a close association with preceding vaccinations have
been reported, most of these in infants. Due to the high rate of
vaccinations in this age group, a causative relation is difficult
to confirm. In infants, the distribution of the lesions is often
acral, in particular palmar and plantar. In older children, the
involvement of the genital region occurs in almost half of the
cases. No immunopathological differences between BP in childhood
and in adults have been reported (24). Autoantibodies mainly target the
NC16A domain of BP180. Generally, infants and children with BP have
a good prognosis with remissions within weeks to a few months under
therapy. For treatment, systemic corticosteroids are usually
combined with dapsone or sulfapyridine. The blisters may obtain a
diameter; pruritus may be an initial early symptom of BP. Patients
with BP may experience one episode or recurrent bouts of pruritus,
which initially present as macules and papules (24). BP is self-limiting, but can last
for months to years without therapy. This is followed by the
development of multiple bullae on the skin, which eventually heal
without scarring. The oral lesions of bullae are prone to rupture
as a result of constant low-grade trauma by the teeth due to
masticatory forces to which they are subjected, resulting in
multiple shallow ulcers with distinct margins. Oral involvement in
BP occurs in 15-20% of patients. The oral mucosal involvement in BP
is less severe than skin involvement, forms more slowly over a
period of time, is less painful, and the disease severity is
expressed only when there is a loss of immunological tolerance. The
gingival lesions consist of inflammation and desquamation with
irregular areas of ulcer caused by the rupture of blisters
(24). The various literature
reviews on BP are presented in Table
II (7,25-43).
| Table IILiterature reviews on Bullous
pemphigoid. |
Table II
Literature reviews on Bullous
pemphigoid.
| Author | Year of
publication | Study
conclusion | (Refs.) |
|---|
| Cohen | 2021 | Bullous pemphigoid
is characterized by localized or widespread Pruritic tense
subepidermal blisters which usually develop in flexural areas that
are proximally located, such as the axilla and groin | (25) |
| Deotto et
al | 2022 | Bullous pemphigoid
related to aging | (26) |
| Niebel et
al | 2022 | Bullous pemphigoid
occurs in patients receiving immune checkpoint inhibitors for the
treatment of melanoma, non-small cell lung cancer and
cholangiocarcinoma (pembrolizumab, nivolumab, durvalumab,
atezolizumab, | (27) |
| Tsiogka et
al | 2021 | Bullous pemphigoid
was reported in patients treated with pemrolizumab, durvalumab,
nivolimumab, anti-programmed cell death protein 1 and
anti-programmed cell death ligand 1 therapy used in the treatment
of patients with melanoma and non-small cell lung carcinoma | (28) |
| Zheng et
al | 2021 | Bullous pemphigoid
was reported in a 62-year-old woman induced by apatinib mesylate
prescribed for treatment of malignant breast cancer | (29) |
| Pruessmann et
al | 2021 | The
immunomodulator, galectin-9, a chemoattractant from eosinophils was
increased in blood and perilesional skin of patients with bullous
pemphigoid | (30) |
| Kridin et
al | 2021 | Patients with
bullous pemphigoid experience increased COVID-19-associated
mortality and need to be monitored closely | (31) |
| Kim et
al | 2021 | Bullous pemphigoid
occurred after 3 days on the thighs and buttocks of a 75-year-old
male, who underwent total knee arthroplasty for knee pain after
surgical placement of stryker posterior stabilized prosthesis | (32) |
| Liu et
al | 2020 | There exists a
strong association of bullous pemphigoid with use of dipeptidyl
peptidase IV inhibitors, aldosterone antagonists, and
anticholinergics | (33) |
| Genovese et
al | 2019 | There exists an
increased risk of thromboembolic events such as pulmonary embolism
in bullous pemphigoid patients | (7) |
| Tasanen et
al | 2019 | Bullous pemphigoid
in patients receiving dipeptidyl peptidase IV inhibitors | (34) |
| Kridin and
Bergman | 2018 | Reported bullous
pemphigoid in diabetics receiving dipeptidyl peptidase IV
inhibitors | (35) |
| Hoffer et
al | 2018 | Bullous pemphigoid
was reported in a 77-year-old male, who was diagnosed with
parkinsonism and was prescribed amantadine drug 100 mg three times
daily after 3 weeks of drug therapy | (36) |
| Hoffmann et
al | 2018 | Bullous pemphigoid
was reported in an 81-year-old patient who received adalimumab
therapy for ulcerative colitis | (37) |
| Jang et
al | 2018 | Bullous pemphigoid
was reported in a 78-year-old male with hepatitis C virus
infection | (38) |
| Flamm et
al | 2017 | Gabapentin-induced
bullous pemphigoid pruritic lesions on the skin of arm, leg and
torso were reported in a 87-year-old male who was treated for
diabetic neuropathy after 3 weeks of gabapentin treatment. The
lesions involved the torso, but no facial skin and oral involvement
were reported | (39) |
| Mendonça et
al | 2016 | Reported occurrence
of three cases of bullous pemphigoid, one with linagliptin and two
cases with vildagliptin | (40) |
| Williams et
al | 2013 | Mucous membrane
pemphigoid was noted in a 78-year-old male, who was a known
hypertensive and was under amlodipine medication. The skin
reactions stopped after 6 weeks of terminating treatment with the
anti-hypertensive drug, amlodipine | (41) |
| Park et
al | 2011 | Amlodipine is
associated with bullous pemphigoid | (42) |
| Monteagudo et
al | 2008 | Bullous pemphigoid
was reported after 30 days of treatment with amlodipine, a calcium
channel blocker in a 70-year-old woman with known history of
diabetes, hypertension and hypercholesterolemia | (43) |
Differential diagnosis
Bullous lichen planus, pemphigus, epidermolysis
bullosa dystrophica, adverse drug reactions, chronic urticaria,
erythema multiforme and BP are all associated with some form of
skin blisters or sores. Bullous lichen planus caused by
T-cell-mediated immune disorder is characterized by the presence of
fine radiating purple-colored striae known as Wickham's striae in
areas of preexisting lesions of lichen planus. The activation of
CD8+ cells plays a pivotal role in bullous lichen
planus. This is achieved either directly by an antigen binding to
the major histocompatibility complex (MHC) class I on lesional
keratinocytes or through activation from CD4+ cells. In
addition, there are increased numbers of CD4+ and
Langerhans cells. CD4+ cells are activated by antigens
associated with MHC class II molecules, present in Langerhans cells
and keratinocytes. These CD4+ T-cells activate
CD8+ cells through receptor interaction and by the
concurrent action of IL-2 and IFN-γ. IFN-γ, in turn, induces the
production of TNF-α from keratinocytes, as well as an increase in
class II MHC, resulting in increased interaction with helper
T-cells. Furthermore, it induces the production of VCAM-1 and
ICAM-1 by keratinocytes and dendritic/Langerhans cells, which
facilitates lymphocyte adhesion to keratinocytes and results in
keratinocyte apoptosis. Histologically, such lesions are confirmed
by the presence of Civatte bodies, which represents apoptotic or
dyskeratotic keratinocytes, and Max Joseph spaces, which represent
the vacuolar degeneration of the basal layer that results in
separate areas between the epidermis and dermis, ‘saw-tooth
appearance’ caused by acanthosis and an increased granular cell
layer (44).
Multiple mechanisms have been suggested to explain
this apoptotic process: i) The expression of Fas-ligand on T-cell
surface binds to FAS on keratinocytes; ii) TNF-α secretion by
T-cells binds to the TNF-α receptor on keratinocytes; iii) the
release of cytotoxic molecules, such as perforin and granzymes B
that function together to trigger cell lysis (45).
Pemphigus results in large irregular areas of ulcers
caused by flaccid or easily rupturable thin-walled bullae with
extensive labial involvement, which is not observed in BP. Adverse
drug reactions are an undesirable and a usually unanticipated
response, independent of the intended therapeutic purpose of the
medication, which may be either immunologic (i.e., drug allergy) or
non-immunologic (i.e., drug intolerance) which accounts for 90% of
the adverse drug reactions (46).
Chronic urticaria is most often idiopathic; however,
50% of cases have an autoimmune basis, characterized by triple
response erythema caused by vasodilatation, increased vascular
permeability (wheal) and axon reflex (flare) produced by the
stimulation of cutaneous sensory nerve endings, with antidromic
conduction of the impulse and release of the neurokinin substance
P. Substance P is a vasodilator that causes the release of
histamine and other mediators from cutaneous mast cells, thus
augmenting the urticarial lesions (47).
Erythema multiforme is an acute, self-limited,
inflammatory mucocutaneous disorder that manifests on the skin,
oral mucosa and genitalia. Erythema multiforme is caused by
infection with the herpes simplex virus (HSV), cytomegalovirus,
retrovirus, influenza, pneumococci, hepatitis C virus and
Rotavirus. Characteristically, lesions of erythema multiforme
involve hemorrhagic crusting occurring on the vermilion border of
the lip and target, or Bull's eye lesions occurring on the palms
and soles, which is not observed in patients with BP (48).
Mucous membrane pemphigoid involves conjunctiva,
resulting in scarring and adhesions between palpebral and bulbar
conjunctiva termed symblepharon, inwardly placed eyelashes
(entropion), inwardly positioned eyelashes injuring the cornea
(trichiasis) and resulting in corneal scarring and blindness. No
such scarring occurs in BP. The various diagnostic methods for BP
are presented in (Table III).
The methods for the management of BP are described in Table IV. Plasmapheresis and intravenous
immunoglobulin also prove beneficial in recalcitrant cases of BP
(48).
| Table IIIDiagnostic tests used for bullous
pemphigoid. |
Table III
Diagnostic tests used for bullous
pemphigoid.
| Diagnostic
tests | Detected antigens
and antibodies |
|---|
| Direct
immunofluorescence-salt-split skin technique-perilesional site from
uninvolved skin transported in Mitchell's medium | Gold standard for
diagnosis of bullous pemphigoid. Reveals deposition of IgG and C3
bound in a linear band to the basement membrane |
|
Histopathology-hematoxylin and eosin
stain | Inflammatory
infiltrate rich in eosinophils and neutrophils |
| Enzyme-linked
immunosorbent assay (ELISA) | To detect
antibodies to the NC16A domain of BP180, also known as BPAG2 |
| Table IVMedical management of bullous
pemphigoid. |
Table IV
Medical management of bullous
pemphigoid.
| Medication | Dosage |
|---|
| Prednisolone
(Wysolone) tablets | 0.5-1 mg/kg body
weight |
| Pulse therapy:
Methylprednisolone (Zempred) tablets | 16 mg twice daily
for two weeks, 10 mg for the third week, and 4 mg for the fourth
week |
| Mycophenolate
mofetil (CellCept) tablets | 500 mg twice daily
for two weeks |
| Azathioprine
(Imuran) tablets | 0.5-2 mg/kg/day
once daily for two weeks |
| Doxycycline (Adoxa)
tablets | 100 mg twice daily
for 1 week |
| Tab. Dapsone
(Acnesone) | 100 mg per day for
1 week |
| Rituximab
(Truxima) | 1 g on day 1 and
14th day, 375 mg intravenously per week for 4 weeks |
| Omalizumab
(Xolair) | 300 mg
subcutaneously every 2-4 weeks |
| Dupilumab
(Dupixent) | 300 mg
subcutaneously every 2-4 weeks |
Management guidelines for BP
The management guidelines proposed by the French
reference centers for autoimmune bullous diseases, the British
Association of Dermatologists, the German Dermatological Society,
and the European Academy of Dermatology and Venereology for
treating BP are presented in Table
V (49).
| Table VGuidelines from the French reference
centres for autoimmune bullous diseases, the British Association of
Dermatologists, the German Dermatological Society, and the European
Academy of Dermatology and Venereology for the treatment of bullous
pemphigoid. |
Table V
Guidelines from the French reference
centres for autoimmune bullous diseases, the British Association of
Dermatologists, the German Dermatological Society, and the European
Academy of Dermatology and Venereology for the treatment of bullous
pemphigoid.
| Severity of bullous
pemphigoid | First-line
drugs | Second-line
drugs |
|---|
| Localized or
mild | Potent topical
steroids, e.g., 0.1% flucinonide (Vanos), 0.05% diflorosane
(Psorcon), 0.025% triamcinolone acetonide (Kenalog in orabase),
0.05% clobetasol (Temovate) | - |
| Moderate | Very potent topical
corticosteroids on the whole-body surface 2 mg/day | Very potent topical
corticosteroids on the whole body surface 2 mg/day plus (in
alphabetical order): Azathioprine 2.5 mg/kg/day [with normal
thiopurine methyl transferase (TPMT) activity] or Dapsone 1.0-1.5
mg/kg/day or doxycycline 200 mg/day ± nicotinamide 2 g/day or
methotrexate 10-20 mg/week or mycophenolates (mofetil 2 g/day,
gastro-resistant mycophenolic acid (Myfortic®) 1.44
g/day) or prednisolone 0.5 mg/kg/day tapering, with or without
azathioprine, dapsone, doxycycline, methotrexate, mycophenolate
mofetil (CellCept) |
| Severe or extensive
bullous pemphigoid | Very potent topical
corticosteroids on the whole-body surface 2 mg/day plus
azathioprine, dapsone, doxycycline, methotrexate, mycophenolates
(see earlier) or very potent topical corticosteroids on the
whole-body surface 1 mg/day plus prednisolone 0.5 mg/kg/day
tapering, with or without azathioprine, dapsone, doxycycline,
methotrexate, mycophenolates | In case of
insufficient response treat with oral prednisolone, increase dose
to 0.75 mg/kg/day and, if still insufficient, to mg/kg/day plus
immunosuppressants rituximab 375 mg/m2 1x/week for 4
consecutive weeks or 2x1 g in and interval of 2-3 weeks or
intravenous immunoglobulin (IVIG) 2 g/kg every 4 weeks for 3-6
months followed by prolonged intervals of up to 6 weeks |
Measurement tools for the assessment
of the treatment response to BP: BP disease area index (BPDAI)
Total BPDAI activity and total BPDAI damage are the
two scores that the BPDAI tool computes. The three subcomponents of
cutaneous blisters/erosions, cutaneous urticaria/erythema and
mucosal blisters/erosions add to the total BPDAI activity score.
The arithmetic sum of the elements evaluated regionally for damage
brought on by more permanent features, such as post-inflammatory
hyperpigmentation, scarring and others comprise the overall BPDAI
damage score. BPDAI quantifies the lesion numbers and size
thresholds. Based on the areas affected, lesions are rated. To more
clearly distinguish the clinical response in BP, the BPDAI provides
extra weight for the limbs and other skin regions most commonly
affected by BP, while placing less focus on the scalp and face. The
values for BPDAI total activity can range from 0 to 360 (with a
maximum of 240 for total skin activity and 120 for mucosal
activity), and for BPDAI damage, they can range from 0 to 12.
Higher scores indicate more disease activity or damage.
Additionally, BPDAI features a unique subjective measurement termed
BPDAI-pruritus (50).
BPDAI-pruritis. A significant BP symptom that
may indicate the beginning or relapse of the condition is pruritus.
This severity is assessed using BPDAI-pruritus, a different
subjective component of the BPDAI. A visual analog scale is used to
rate the severity of pruritus, with 0 denoting no itching and 10
denoting the most intense itching. To indicate the degree of
itching today, last week, and last month, the patient marks an ‘x’,
yielding a total score of 30. Pruritus is deduced from the severity
of excoriations, which is also graded on a 30-point scale in cases
where the patient could not complete the grading correctly
(51).
Autoimmune bullous skin disorder intensity score
(ABSIS). For the measurement of disease activity in patients
with blistering autoimmune illnesses, such as BP, the ABSIS
(51). tool is frequently
utilized. The palm of the patient's hand is set as 1% of the total
body surface area when using ABSIS's Rule of Nines and Rule of
Palms techniques. The weighting factors are 1.5 (erosive, exudative
lesions, blisters), 1 (erosive, dry lesions) and 0.5 (erosive, dry
lesions) and are added to the estimated proportion of body surface
area involved (re-epithelialized lesions). Higher scores indicate
more severe disease, with a total score from 0 to 206. In addition,
it has sections for mucosal extent, skin involvement, and
patient-reported mucosal discomfort, with a maximum score of 150
for skin involvement and an 11 for mucosal stretch (top score of
45) (51).
Physician Global Assessment (PGA). PGA is a
10-point Likert scale that ranges from 0 to 10, with 10
representing the best possible skin condition to the worst. The
disease activity can be rated based on a general overall perception
using this scoring method. The primary clinician also classifies
patients as having mild, moderate, or severe illness (52).
Autoimmune bullous quality of life (ABQOL).
The quality of life (QOL) unique to autoimmune bullous illness is
assessed using the ABQOL, which has been found to have good
validity and reliability. This 17-item questionnaire covers the
physical toll of the disease, psychological impacts and effects on
daily functioning. Every question carries a point value from 0 to
3, with higher scores indicating lower QOL. The ABQOL score cap is
51(53).
Treatment of autoimmune bullous quality of life
(TABQOL). The risk of medical consequences and QOL
degradation from the side-effects of autoimmune blistering diseases
treatment are high. The TABQOL questionnaire is used to assess the
burden related to autoimmune bullous disease-specific treatment
effects. This 17-item questionnaire is similar to the ABQOL in that
higher scores indicate a lower QOL due to treatment effects
(54).
Eosinophil cationic protein (ECP). The
severity of BP can be assessed using ECP, a chemoattractant protein
released from eosinophils. A decrease in ECP concentrations of at
least 12.8 ng/ml have a positive predictive value of 81% for
remission, demonstrating that ECP serum fluctuation may be a
valuable biomarker for identifying BP patients who are at risk of
relapse (55).
In conclusion, BP is a chronic autoimmune disorder
caused by antibodies targeting against BP180, a type XVII collagen,
HD1 (BPAG1), a 230-kDa glycoprotein located on chromosome 6p11-6p12
and HD4 (BPAG2), a 180-kDa hemidesmosome glycoprotein located on
chromosome 10q 24.3. BP is a chronic autoimmune disorder caused by
antibodies targeting BP proteins BP180 and 230, resulting in
blisters and multiple ulcers in the oral cavity. BP can increase
the risk of thromboembolic events, such as pulmonary embolism.
Patients with pre-existing hematological malignancies, such as
leukemia, Hodgkin's lymphoma, mantle-cell lymphoma, and colorectal,
breast, gastric and lung cancer are more prone to bullous
pemphigoid lesions. The occurrence of BP in a patient must be
thoroughly evaluated in such types of cancer, and treatment must be
initiated on time. Such ulcers caused by BP exhibit chronicity and
occur in multiple sites of the oral cavity. The treatment of this
type of BP is challenging for the oral physician.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
KRM was involved in the literature collection of the
articles to be included in the literature review, as well as in the
conception of the study. SMF was involved in the intellectual
content of the study and in the study design. SPGS was involved in
the conception of the study and in the literature collection of the
articles to be included in the literature review. RPT was involved
in the drafting of the manuscript and in the collection of the
immunofluorescence images. KRM and RPT confirm the authenticity of
all the raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
The patient provided informed consent to participate
in the present study.
Patient consent for publication
Patient consent was obtained for the publication of
his clinical images.
Competing interests
The authors declare they do not have no competing
interests.
References
|
1
|
Baigrie D and Nookala V: Bullous
pemphigoid. StatPearls (Internet). StatPearls Publishing, Treasure
Island, FL, 2022.
|
|
2
|
LEVER WF: Pemphigus. Medicine (Baltimore).
32:1–123. 1953.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Jordan RE, Day NK, Sams WM Jr and Good RA:
The complement system in bullous pemphigoid. I. Complement and
component levels in sera and blister fluids. J Clin Invest.
52:1207–1214. 1973.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Cole C, Vinay K, Borradori L and Amber KT:
Insights into the pathogenesis of bullous pemphigoid: The role of
complement-independent mechanisms. Front Immunol.
13(912876)2022.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Sadik CD and Schmidt E: Resolution in
bullous pemphigoid. Semin Immunopathol. 41:645–654. 2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Tabatabaei-Panah PS, Moravvej H, Aghaei S,
Akbari M, Rajabi S, Kia A, Ebrahimi E, Sadaf Z, Atoon A, Behravesh
N, et al: TH17/IL23 cytokine gene polymorphisms in bullous
pemphigoid. Mol Genet Genomic Med. 8(e1519)2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Genovese G, Di Zenzo G, Cozzani E, Berti
E, Cugno M and Marzano AV: New insights into the pathogenesis of
bullous pemphigoid: 2019 update. Front Immunol.
10(1506)2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Kasperkiewicz M, Zillikens D and Schmidt
E: Pemphigoid diseases: Pathogenesis, diagnosis, and treatment.
Autoimmunity. 45:55–70. 2012.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Verbeek JS, Hirose S and Nishimura H: The
complex association of FcγRIIb With autoimmune susceptibility.
Front Immunol. 10(2061)2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Bournazos S and Ravetch JV: Fcγ receptor
pathways during active and passive immunization. Immunol Rev.
268:88–103. 2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Ujiie H, Shibaki A, Nishie W, Sawamura D,
Wang G, Tateishi Y, Li Q, Moriuchi R, Qiao H, Nakamura H, et al: A
novel active mouse model for bullous pemphigoid targeting humanized
pathogenic antigen. J Immunol. 184:2166–2174. 2010.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Franziska SK, Beckmann T, Nimmerjahn F,
Ishiko A, Collin M, Köhl J, Goletz S, Zillikens D, Ludwig R and
Schmidt E: Fcγ receptors III and IV mediate tissue destruction in a
novel adult mouse model of bullous pemphigoid. Am J Pathol.
184:2185–2196. 2014.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Heimbach L, Li N, Diaz A and Liu Z:
Experimental animal models of bullous pemphigoid. G Ital Dermatol
Venereol. 144:423–431. 2009.PubMed/NCBI
|
|
14
|
Hiroyasu S, Turner CT, Richardson KC and
Granville DJ: Proteases in pemphigoid diseases. Front Immunol.
10(1454)2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Liu Y, Peng L, Li L, Liu C, Hu X, Xiao S
and Xia Y: TWEAK/Fn14 activation contributes to the pathogenesis of
bullous pemphigoid. J Invest Dermatol. 137:1512–1522.
2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Karashima T, Hachisuka H, Okubo K and
Sasai Y: Epidermal keratinocytes of bullous pemphigoid express
intercellular adhesion molecule-1 (ICAM-1). J Dermatol. 19:82–86.
1992.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Kaminska R, Naukkarinen A, Glinski W,
Horsmanheimo M and Harvima IT: Mast cells in developing
subepidermal bullous diseases: Emphasis on tryptase, chymase and
protease inhibitors. Acta Derm Venereol. 79:351–355.
1999.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Maronese CA, Caproni M, Moltrasio C,
Genovese G, Vezzoli P, Sena P, Previtali G, Cozzani E, Gasparini G,
Parodi A, et al: Bullous pemphigoid associated with COVID-19
vaccines: An Italian multicentre study. Front Med (Lausanne).
9(841506)2022.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Aronson JK: Influenza vaccine. In:
Meyler's Side Effects of Drugs: The International Encyclopedia of
Adverse Drug Reactions and Interactions. Vol 7. 16th edition.
Elsevier, pp98-106, 2016.
|
|
20
|
Modeste AB, Cordel N, Courville P, Gilbert
D, Lauret P and Joly P: Bullous pemphigoid induced by
spironolactone. Ann Dermatol Venereol. 129:56–58. 2002.PubMed/NCBI(In French).
|
|
21
|
Warner C, Kwak Y, Glover MHB and Davis LS:
Bullous pemphigoid induced by hydrochlorothiazide therapy. J Drug
Dermatol. 13:360–362. 2014.PubMed/NCBI
|
|
22
|
Verheyden MJ, Bilgic A and Murrell DF: A
systematic review of drug-induced pemphigoid. Acta Derm Venereol.
100(adv00224)2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Wofford J, Patel M, Readinger A and Menter
A: Widespread dermal ulcerations and bullae. Proc (Bayl Univ Med
Cent). 25:155–158. 2012.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Griffiths C, Barker J, Bleiker T, Chalmers
R and Creamer D: Immunobullous diseases. In: Rook's Textbook of
Dermatology. Vol 2. 9th edition. Wiley Blackwell Publishers, part
4, chapter 50, 2016.
|
|
25
|
Cohen PR: Dyshidrosiform bullous
pemphigoid. Medicina (Kaunas). 57(398)2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Deotto ML, Spiller A, Sernicola A and
Alaibac M: Bullous pemphigoid: An immune disorder related to aging
(Review). Exp Ther Med. 23(50)2022.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Niebel D, Wilsmann-Theis D, Bieber T,
Berneburg M and Wenzel J: Braegelmann C: Bullous pemphigoid in
patients receiving immune-checkpoint inhibitors and psoriatic
patients-focus on clinical and histopathological variation.
Dermatopathology (Basel). 9:60–81. 2022.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Tsiogka A, Bauer JW and Patsatsi A:
Bullous pemphigoid associated with anti-programmed cell death
protein 1 and anti-programmed cell death ligand 1 therapy: A review
of the literature. Acta Derm Venereol. 101(adv00377)2021.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Zheng Q, Ma Y, Shen F, Wang Q, Song X,
Jiang W and Xie S: Case of bullous pemphigoid induced by apatinib
mesylate. Br J Clin Pharmacol. 87:2158–2159. 2021.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Pruessmann J, Pruessmann W, Holtsche MM,
Linnemann B, Hammers CM, van Beek N, Zillikens D, Schmidt E and
Sadik CD: Immunomodulator galectin-9 is increased in blood and skin
of patients with bullous pemphigoid. Acta Derm Venereol.
101(adv00419)2021.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Kridin K, Schonmann Y, Weinstein O,
Schmidt E, Ludwig RJ and Cohen AD: The risk of COVID-19 in patients
with bullous pemphigoid and pemphigus: A population-based cohort
study. J Am Acad Dermatol. 85:79–87. 2021.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Kim YB, Choi HS, Cho HK and Seo GW:
Diagnosis and treatment of bullous pemphigoid that developed twice
after total knee replacement arthroplasty: A case report. BMC
Musculoskelet Disord. 22(118)2021.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Liu SD, Chen WT and Chi CC: Association
between medication use and bullous pemphigoid: A systematic review
and meta-analysis. JAMA Dermatol. 156:891–900. 2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Tasanen K, Varpuluoma O and Nishie W:
Dipeptidyl peptidase-4 inhibitor-associated bullous pemphigoid.
Front Immunol. 10(1238)2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Kridin K and Bergman R: Association of
bullous pemphigoid with dipeptidyl-peptidase 4 inhibitors in
patients with diabetes: Estimating the risk of the new agents and
characterizing the patients. JAMA Dermatol. 154:1152–1158.
2018.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Hoffer S, Hategan A and Bourgeois JA: .
Amantadine-associated bullous pemphigoid. J Clin Psychopharmacol.
38:394–395. 2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Hoffmann S, Berneburg M and Schreml S:
Bullous pemphigoid associated with adalimumab therapy in a patient
with ulcerative colitis. Case Rep Dermatol. 10:145–148.
2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Jang H, Jin YJ, Yoon CH, Kim CW and Kim L:
Bullous pemphigoid associated with chronic hepatitis C virus
infection in a hepatitis B virus endemic area: A case report.
Medicine (Baltimore). 97(e0377)2018.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Flamm A, Sachdev S and Dufresne F:
Gabapentin-induced bullous pemphigoid. J Am Osteopath Assoc.
117:191–193. 2017.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Mendonça FM, Martín-Gutierrez FJ,
Ríos-Martín JJ and Camacho-Martinez F: Three cases of bullous
pemphigoid associated with dipeptidyl peptidase-4 inhibitors-one
due to linagliptin. Dermatology. 232:249–253. 2016.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Williams G, Goodwin R and Hughes D:
Amlodipine as a cause of mucous membrane pemphigoid: First report
of amlodipine as a causative agent in MMP. Eye (Lond).
27(1425)2013.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Park KY, Kim BJ and Kim MN:
Amlodipine-associated bullous pemphigoid with erythema
multiforme-like clinical features. Int J Dermatol. 50:637–639.
2011.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Monteagudo B, Heras C, Bouza P, Almagro M,
Álvarez JC and Cacharrón JM: Bullous pemphigoid after treatment
with amlodipine. Med Cutan Iber Lat Am. 36:308–311. 2008.
|
|
44
|
Mukhopadhyay AK: Two eponyms in the
histopathology of lichen planus: Creation and confusion. Indian J
Dermatol Venereol Leprol. 88:270–273. 2022.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Yang M, Wu H, Zhao M, Chang C and Lu Q:
The pathogenesis of bullous skin diseases. J Transl Autoimmun.
2(100014)2019.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Mozafari N, Ganji R and Toossi P: A rare
new presentation of pemphigus vulgaris. Clin Case Rep.
10(e5979)2022.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Criado PR, Criado RF, Maruta CW and Reis
VM: Chronic urticaria in adults: State-of-the-art in the new
millennium. An Bras Dermatol. 90:74–89. 2015.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Kaur S and Handa S: Erythema multiforme
following vaccination in an infant. Indian J Dermatol Venereol
Leprol. 74:251–253. 2008.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Feliciani C, Joly P, Jonkman MF, Zambruno
G, Zillikens D, Ioannides D, Kowalewski C, Jedlickova H, Kárpáti S,
Marinovic B, et al: Management of bullous pemphigoid: The european
dermatology forum consensus in collaboration with the european
academy of dermatology and venereology. Br J Dermatol. 172:867–877.
2015.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Clapé A, Muller C, Gatouillat G, Le Jan S,
Barbe C, Pham BN, Antonicelli F and Bernard P: Mucosal involvement
in bullous pemphigoid is mostly associated with disease severity
and to absence of Anti-BP230 autoantibody. Front Immunol.
9(479)2018.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Wijayanti A, Zhao CY, Boettiger D, Chiang
YZ, Ishii N, Hashimoto T and Murrell DF: The reliability, validity
and responsiveness of two disease scores (BPDAI and ABSIS) for
bullous pemphigoid: Which one to use? Acta Derm Venereol. 97:24–31.
2017.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Pratasava V, Sahni VN, Suresh A, Huang S,
Are A, Hsu S and Motaparthi K: Bullous pemphigoid and other
pemphigoid dermatoses. Medicina (Kaunas). 57(1061)2021.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Sebaratnam DF, Okawa J, Payne A, Murrell
DF and Werth VP: Reliability of the autoimmune bullous disease
quality of life (ABQOL) questionnaire in the USA. Qual Life Res.
24:2257–2260. 2015.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Kouris A, Platsidaki E, Christodoulou C,
Armyra K, Korkoliakou P, Stefanaki C, Tsatovidou R, Rigopoulos D
and Kontochristopoulos G: Quality of life, depression, anxiety and
loneliness in patients with bullous pemphigoid. A case control
study. An Bras Dermatol. 91:601–603. 2016.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Giusti D, Gatouillat G, Le Jan S, Plée J,
Bernard P, Antonicelli F and Pham BN: Eosinophil cationic protein
(ECP), a predictive marker of bullous pemphigoid severity and
outcome. Sci Rep. 7(4833)2017.PubMed/NCBI View Article : Google Scholar
|