Introduction
Gastrointestinal stromal tumors (GISTs) are the most
common type of primary gastrointestinal mesenchymal tumor,
accounting for 2% of all gastrointestinal tumors. GISTs are
typically identified via immunohistochemical staining for CD117 in
the neoplastic cell membrane, which is generally positive, and in
neoplastic cytoplasm or nucleus, which is diffusely positive, and
exhibit a phenotype similar to that of interstitial cell of Cajal.
The majority of GIST cases exhibit an activated c-Kit gene
(KIT) or PDGFRA mutations, which obtain conductivity
activation independently of each other (1,2). In the
absence of ligand binding, c-Kit or PDGFRA proteins maintain
a sustained tyrosine kinase activity, which activates downstream
signaling pathways (1–3).
With regard to GIST, previous studies investigating
the associations among the mutation rate, mutation types and
biological behavior in KIT or PDGFRA are not in
agreement (4–7). Imatinib is a targeted drug therapy for
limiting the pathogenesis of the GIST, and has been widely adopted
for the treatment of GISTs in recent years. In vitro and
in vivo experiments have clinically demonstrated that the
efficacy of imatinib against GIST depends considerably on the
presence or absence of genetic mutations, and on the mutation sites
and types (8,9). Therefore, investigations into the
mutations associated with GIST have high clinical significance.
In the present study, polymerase chain reaction
(PCR) amplification and sequencing methods were used to detect the
presence of gene mutations and analyze GIST-associated gene
mutations in patients from North China. The aim of the current
study was to provide the basis of the molecular pathology
underlying GIST to allow for targeted therapy with imatinib.
Materials and methods
Specimen collection
Test specimens were collected from the General
Military Hospital of Beijing PLA (Beijing, China), the Affiliated
Zhongshan Hospital of Dalian University (Dalian, China) and the
People's Hospital of Weifang (Weifang, China) between 2007 and 2013
for the pathological diagnosis of GIST. Every specimen was reviewed
by three pathologists, who collected the relevant clinical data.
All protocols were approved by the Human Clinical and Research
Ethics Committees of the General Military Hospital of Beijing PLA
(Beijing, China), the Affiliated Zhongshan Hospital of Dalian
University (Dalian, China) and the Peoples Hospital of Weifang
(Weifang, China). Written informed consent was obtained from all
patients.
Reagents and instruments
A DNA extraction kit was purchased from Qiagen
(Hilden, Germany) and a nucleic acid-protein concentration
measuring instrument was purchased from Shanghai Chong Meng
Biotechnology Co., Ltd. (B-500; Shanghai, China). CD117 (clone
YR145), S-100 (clone 4C4.9) and DOG-1 (clone SP31) antibodies were
purchased from Fuzhou Maixin Biotechnology Development Co., Ltd.
(Fuzhou, China), while smooth muscle actin (SMA; clone IA4) and
desmin (clone ZC18) antibodies were purchased from Beijing
Zhongshan Golden Bridge Biotechnology Co., Ltd. (Beijing, China).
Primers synthesis was performed by Shanghai Handsome Biotech Co.,
Ltd. (Shanghai, China), and the sequences are shown in Table I. An ABI 9700 PCR Cycler was
purchased from Applied Biosystems Life Technologies (Foster City,
CA, USA).
 | Table I.Primers for each exon of KIT
and PDGFRA. |
Table I.
Primers for each exon of KIT
and PDGFRA.
| Exon | Primer
sequences | Fragment length
(bp) |
|---|
| 9 (KIT) | Forward:
TCCTAGAGTAAGCCAGGGCTT | 261 |
|
| Reverse:
TGGTAGACAGAGCCTAAACATCC |
|
| 11
(KIT) | Forward:
CCAGAGTGCTCTAATGACTG | 225 |
|
| Reverse:
TGACATGGAAAGCCCCTGTT |
|
| 13
(KIT) | Forward:
GCTTGACATCAGTTTGCCAG | 193 |
|
| Reverse:
AAAGGCAGCTTGGACACGCCTTTA |
|
| 17
(KIT) | Forward:
TACAAGTTAAAATGAATTTAAATGGT | 228 |
|
| Reverse:
AAGTTGAAACTAAAAATCCTTTGC |
|
| 12
(PDGFRA) | Forward:
TCCAGTCACTGTGCTGCTTC | 260 |
|
| Reverse:
GCAAGGGAAAAGGGAGTCTT |
|
| 18
(PDGFRA) | Forward:
ACCATGGATCAGCCAGTCTT | 250 |
|
| Reverse:
TGAAGGAGGATGAGCCTGACC |
|
Immunohistochemistry
All specimens (4-µm serial sections) were embedded
in paraffin and incubated at 60°C for 90 min. Using the EnVision
two-step method, the experiments were performed in strict
accordance with the kit instructions. Tissue sections were
deparaffinized, rehydrated and incubated with 3%
H2O2 in methanol for 15 min at room
temperature to eliminate endogenous peroxidase activity. The
antigen was retrieved at 95°C for 20 min by placing the slides in
0.01 M sodium citrate buffer (pH 6.0). Slides were then incubated
with primary antibody at 4°C overnight. After incubation at room
temperature for 30 min with biotinylated secondary antibody, slides
were incubated with streptavidin-peroxidase complex at room
temperature for 30 min. Then slides were immunostained with 3,
3-diaminobenzidine, and counterstained with Mayer's hematoxylin
(10).
GIST diagnostic criteria
Among the 93 specimens collected, 85 cases were
morphologically consistent with a diagnosis of GIST, since CD117
immunohistochemistry revealed strongly positive diffusion in the
cellular membranes. Among the remaining eight CD117-negative cases,
three cases were found to be positive for DOG-1. Although the
remaining five cases were CD117-negative, due to the negativity for
SMA, desmin and S-100, and following the exclusion of neurogenic
tumors and of smooth muscle origin, the cases were included in the
study since they were morphologically consistent with a GIST.
Primer design
KIT and PDGFRA gene sequences were
obtained from the GenBank database (http://www.ncbi.nlm.nih.gov) under the accession
numbers of NC_000004 and NG_009250, respectively. Primers were
designed using OLIGO Primer analysis software (Molecular Biology
Insights, Inc., Colorado Springs, CO, USA). Attention was paid to
prevent the formation of complementary sequences over three base
pairs (bp) or between primers. The primer fragment size ranged
between 190 and 270 bp, while the length of the primer ranged
between 17 and 25 bp. The GC content of the primers varied between
40 and 70%, and the melting temperatures of the forward and reverse
primers were maintained as similar as possible. The primer products
were checked with BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi), with all the
primer products meeting the requirements. Primer synthesis was
conducted by the Shanghai Handsome Biotech Co., Ltd.
DNA extraction
DNA was extracted from the various tissue specimens
using the QIAamp DNA FFPE Tissue kit, according to the
manufacturer's instructions. For each sample, five sections of
paraffin-embedded tissues of 10-µm thickness were used for DNA
extraction. Following isolation, the purity and concentration of
the DNA were estimated. DNA concentration was determined using a
NanoDrop spectrophotometer, purchased from (Thermo Fisher
Scientific, Waltham, MA, USA). A 1-ml sample of water was used as a
blank control to zero the spectrophotometer. The aborbance of 5-µl
samples dissolved in 1 ml water was measured at 280 and 260 nm. A
ratio of >1.8 was considered to indicate that the DNA purity and
concentration was qualified.
PCR amplification
PCR assays were conducted in a 50-µl reaction
system, which contained 25 µl 2X Taq PCR Master Mix, 2 µl DNA
template, 2 µl (10 pmol) each primer and 19 µl sterile deionized
water. The reaction conditions were as follows: Initial
denaturation at 94°C for 5 min, followed by 45 cycles of annealing
at 94°C for 30 sec, 56°C for 45 sec and 72°C for 1 min, and a final
extension at 72°C for 5 min, after which the samples were cooled to
4°C for 5 min. Sterile deionized water was used instead of the
template as the negative control. The purified PCR products were
sequenced by Beijing Jin Wei Zhi Biotech Co., Ltd. (Beijing,
China). Following the identification of the mutations, specimens
carrying mutations were reverse-sequenced for confirmation.
Statistical analysis
Data were analyzed using SPSS software, version 19.0
(IBM SPSS, Armonk, NY, USA). Results were analyzed using
χ2 and Fisher's exact tests, with a test level of
α=0.05. The P-value was set to bilateral distribution, and
P<0.05 was considered to indicate a statistically significant
difference.
Results
Clinical and pathological features of
GIST
Among the 93 patients pathologically confirmed to
have a GIST, 53 were male and 40 were female (male/female ratio,
1.33:1). The age range of the study population was 23–83 years
(median age, 57 years), and the tumor diameter ranged between 5 and
45 cm (median diameter, 8.7 cm). The tumors were distributed as
follows: Gastric origin, 76.34% (71/93); duodenum, 8.60% (8/93);
ileum, 1.08% (1/93); colon, 1.08% (1/93); rectum, 5.38% (5/93);
esophagus, 5.38% (5/93); pleural origin, 1.08% (1/93); and
intrauterine origin, 1.08% (1/93). Based on the study by Fletcher
et al (11), the risk of GIST
was divided into three groups. Firstly, patients were classified
with a very low risk of invasion if the tumor diameter was <5 cm
and they had a mitotic index of <5/50 high-power fields (HPF;
44.09%, 41/93 samples). Secondly, patients were classified into the
moderate risk invasion group if their tumor diameter ranged between
5 and 10 cm and had a mitotic index of <5/50 HPF, or if a tumor
with a diameter of <5 cm was accompanied by a mitotic index of
6–10/50 HPF (18.28%, 17/93 samples). Finally, patients were
classified with a high risk of invasion if they had a tumor
diameter of >5 cm and a mitotic index of >5/50 HPF, a tumor
diameter of >10 cm, or a mitotic index of >10/50 HPF (37.63%,
35/93 samples). The CD117-positive cases accounted for 91.40%
(85/93) of the study population, with the ages ranging between 23
and 83 years (median age, 58 years). The CD117-negative cases
accounted for 8.60% (8/93) of the study population, with the ages
ranging between 46 and 63 years (median age, 53 years). Of the
eight CD117-negative cases, three were male and one was female, and
two cases were of pleural origin, one case was of gastric origin,
and one case was an esophageal tumor. These eight cases of GIST
were followed-up and found to be malignant (Table II).
| Table II.Mutations in KIT and
PDGFRA and the associations with the clinical and
pathological features of GIST. |
Table II.
Mutations in KIT and
PDGFRA and the associations with the clinical and
pathological features of GIST.
|
| KIT (n) |
| PDGFRA
(n) |
|
|---|
|
|
|
|
|
|
|---|
| Parameter | Mutant | No mutant | P-value | Mutant | No mutant | P-value |
|---|
| Gender |
|
| 0.490 |
|
| 0.820 |
|
Male | 38 | 15 |
| 3 | 50 |
|
|
Female | 26 | 14 |
| 1 | 39 |
|
| Age (years) |
|
| 0.260 |
|
| 1.000 |
|
≥59 | 23 | 14 |
| 2 | 35 |
|
|
<59 | 41 | 15 |
| 2 | 54 |
|
| Diameter (cm) |
|
| 0.446 |
|
| 0.683 |
|
<5 | 23 | 8 |
| 1 | 30 |
|
|
5–10 | 36 | 20 |
| 3 | 53 |
|
|
>10 | 5 | 1 |
| 0 | 6 |
|
| Position |
|
| 0.040 |
|
| 0.002 |
|
Stomach | 53 | 18 |
| 0 | 71 |
|
|
Intestinal | 9 | 6 |
| 2 | 13 |
|
| Outside
the gastrointestinal tract | 2 | 5 |
| 2 | 5 |
|
| Mitotic index
(HPF) |
|
| 0.071 |
|
| 0.144 |
|
<5/50 | 23 | 6 |
| 3 | 26 |
|
|
6–10/50 | 33 | 14 |
| 1 | 46 |
|
|
>10/50 | 8 | 9 |
| 0 | 17 |
|
| Risk
classification |
|
| 0.057 |
|
| 0.086 |
| Very
low/low degree | 23 | 18 |
| 0 | 41 |
|
|
Moderate | 14 | 3 |
| 1 | 16 |
|
|
High | 27 | 8 |
| 3 | 32 |
|
KIT mutation analysis
Among the 93 GIST cases, KIT mutations were
detected in 64 cases (68.82%), of which all were CD117-positive. Of
the KIT mutations, 87.50% (56/64) were located on exon 11,
6.25% (4/64) were located on exon 9, 4.69% (3/64) were located on
exon 13, which included a concomitant mutation on exon 11, and
1.56% (1/64) were located on exon 17, which included a concomitant
mutation of exon 11. In the CD117-positive cases, the mutation
ratio of KIT reached 75.29% (64/85). Among the mutations on
exon 11, 91.07% (51/56) were heterozygous and only 8.93% (5/56)
were homozygous. As shown in Fig. 1,
the mutation types identified on exon 11 included deletion
mutations (55.36%, 31/56), point mutations (26.79%, 15/56),
insertion mutations (tandem repeats; 14.29%, 8/56) and point
mutations in the deletion mutation (3.57%, 2/56). The majority of
the mutations (62.50%, 35/56) were located at the ‘hot’ zone,
involving codons 550–560, at the 5′-end. The most common form was
the WK (Trp-Lys) deletion mutation at the 5′-end of codons 557–558
(12.50%, 7/56), followed by the V (Val) deletion mutation at codon
559, and the V575A point mutation (7.14%, 4/56). In 14.29% (8/56)
of the GIST cases, 2–14 amino acids were inserted into exon 11,
including five cases with 3′-end tandem repeats of internal tandem
duplications (ITDs) and three cases with 5′-end ITDs. Among the
five cases with the 3′-end ITDs, two were female and three were
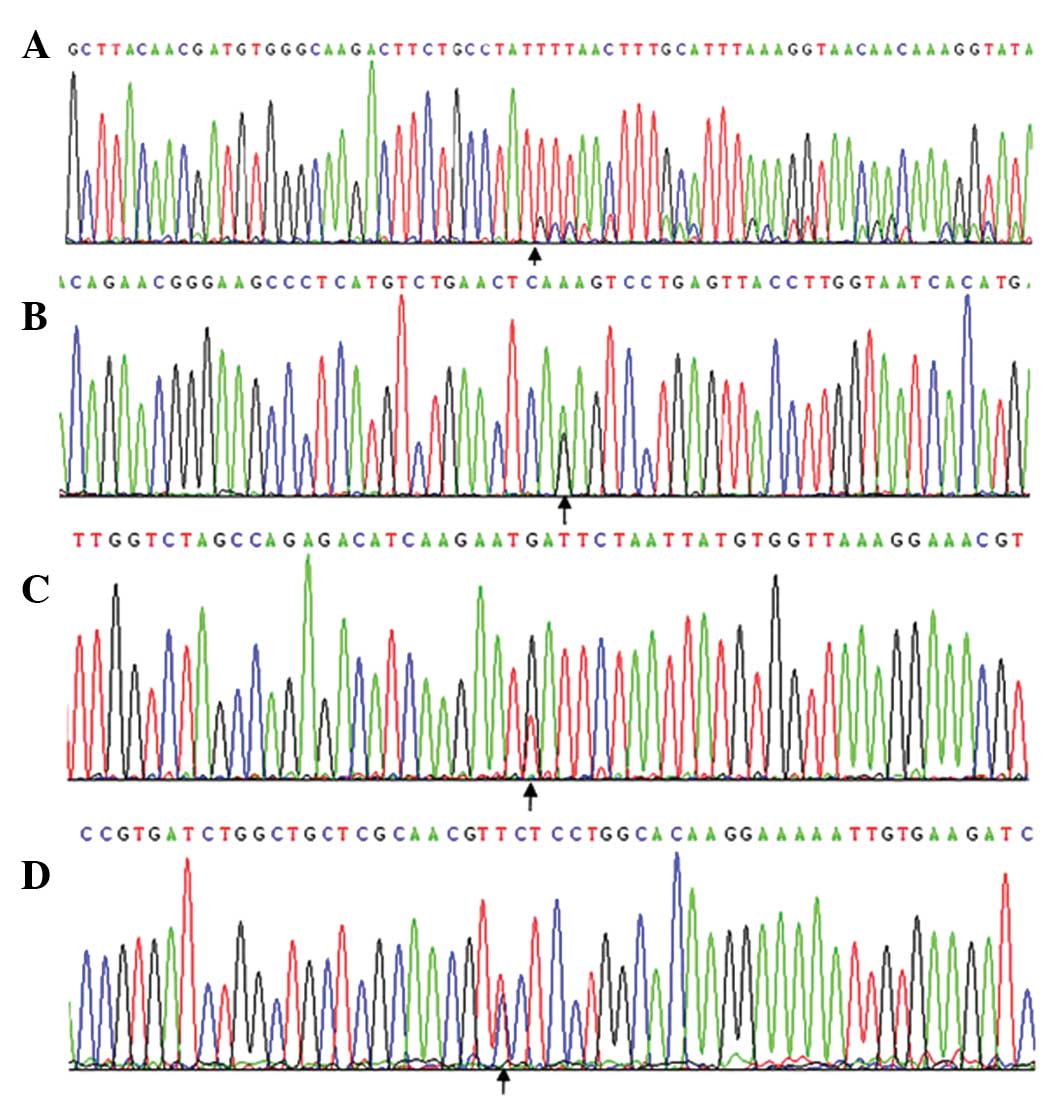
male. In 6.25% (4/64) of the GIST cases, KIT mutations were
detected in exon 9, which corresponded to codons 502 and 503
(Ala-Tyr) with six ITDs, of which three were heterozygous mutations
(Fig. 2A). Three cases originated in
the duodenum and one case originated in the stomach. In 4.69%
(3/64) of the GIST cases, mutations were detected in exon 13, which
comprised a K642R point mutation (Fig.
2B). Only in 1.56% (1/64) of the GIST cases was a mutation
detected in exon 17, and this mutation coexisted with a double
deletion mutation on exon 11 at codons 551–554 (KPMY,
Lys-Pro-Met-Tyr) and with a D820Y point mutation (Fig. 2C).
PDGFRA mutation analysis
Among the 29 GIST cases without a KIT
mutation, a mutation in PDGFRA was detected in three cases (3.23%,
3/93; 10.34%, 3/29). Only one GIST patient with a mutation in
PDGFRA on exon 18, which corresponded to a Val 824 internal
GTC>GTT base point mutation, also had a mutation in exon 11 of
KIT, which corresponded to a L576P point mutation (Fig. 2D). The remaining three cases also
exhibited mutations on exon 18, all of which corresponded to a
D842V point mutation. These three patients with a mutation in
PDGFRA were CD117-negative.
Discussion
The c-Kit proto-oncogene is located on the
long arm of the fourth human chromosome, within zones 2 and 3
(4q12-13). The product of KIT is a type III tyrosine kinase
growth factor receptor, which is a 145-kDa transmembrane
glycoprotein. The KIT gene contains 21 exons, and activation
of the gene depends on ligand binding with stem cell factor, which
enables the phosphorylation of substrate proteins. Subsequently,
certain signal transduction pathways are activated, which stimulate
important cellular functions, such as proliferation and apoptosis.
Mutations in KIT that cause autophosphorylation without the
presence of the ligand lead to uncontrolled cell proliferation,
which eventually induces tumor development (12). Furthermore, Hirota et al
(12) indicated that a
gain-of-function mutation within KIT may be one of the core
events underlying tumor development, with the localization of the
intracellular membrane-proximal domain of the c-Kit protein
(mutation on exon 11) being the most common mutation. Due to the
differences in the tissues used for DNA extraction, the
experimental methods employed and the cases constituting each study
group, previous studies have reported a wide range of mutation
rates, varying between 21 and 92% (9,13–15).
Three quantitative studies conducted using a large study population
from other countries revealed that the mutation rate of exon 11 in
GISTs was 69.32% (296/427), 72.36% (233/322) and 51.50% (103/200)
(15–17), respectively, which is consistent with
the results of the present study (60.21%, 56/93).
Analysis of the clinicopathological features of GIST
in the present study revealed that mutations in KIT were the
only significant variable (P=0.040), while gender, age, tumor
diameter, mitotic index and risk classification exhibited no
statistically significant differences (P>0.05). The primary
location of the KIT mutations was at the 5′-end of exon 11,
with point mutations and in-frame deletions being the most relevant
events, whereas the second most common location was at the 3′-end
of the same exon, in which ITDs were the predominant events. In
accordance with the results of the present study, previous studies
have demonstrated that the latter type of mutation occurs primarily
in the stomach and is more common in female patients (18,19). In
the current study, only four cases were identified to have a
mutation in exon 9 of KIT, which corresponded to the
extracellular domain of c-Kit. This mutation rate was
similar to that reported by Lasota et al who used a larger
cohort of samples (3.00%, 6/200) (17), but slightly lower compared with the
7.2–10.84% reported by Corless et al (16), Antonescu et al (18) and Wozniak et al (15). Mutations on exon 17 of KIT,
which correspond to the phosphotransferase domain of c-Kit,
have been rarely observed. In the present study, one case was found
to have an exon 17 mutation that coexisted with a D820Y point
mutation on exon 11, which was newly identified. Although the
identification of genetic mutations is an important tool, analysis
of CD117-negative GIST mutations is very difficult, and the results
reported in the literature are not consistent (20,21).
PDGFRA and KIT are located on adjacent
positions on the fourth human chromosome and share high amino acid
sequence homology (1). Heinrich
et al (1) found that in cases
of GIST without KIT mutations, PDGFRA mutations were
more frequent, which may be one of the mechanisms underlying GIST
development. The PDGFRA gene, whose signal transduction
pathway is similar to that of KIT, contains 23 exons and has
an epithelial cell-based phenotype. Mutations in PDGFRA can
generate autophosphorylation, which triggers a signal transduction
cascade that enables uncontrolled and disordered cell growth
(1). Analysis of the
clinicopathological factors of GIST in the present study indicated
that mutations in KIT were the only significant events
(P=0.002), while gender, age, tumor diameter, mitotic index and
risk classification exhibited no statistically significant
differences (P>0.05). In the present study, among the 93 samples
studied, mutations in PDGFRA accounted for 5.30% (4/93) of
the sample population, with one case coexisting with a mutation on
exon 11 of KIT. The remaining three cases (10.34%, 3/29)
were represented by a D842V point mutation on exon 18, associated
with a lack of KIT expression, which was observed in 37.50%
(3/8) of the CD117-negative cases. Tu et al (22) reported the case of a renal transplant
recipient with a GIST, in which a Val824 internal GTC>GTT base
point mutation was identified in PDGFRA with no KIT
mutations present. This was the second report of this type of
mutation, which coexisted with the L576P point mutation on exon
11.
Imatinib, which exerts a selective inhibition on
BCR-ABL, PDGFRA and KIT, is a small molecular
agent with tyrosine kinase activity whose clinical treatment for
GIST is very effective (23).
However, the location and nature of the mutations can affect the
response of GIST to imatinib (24).
A previous study found that GIST patients with a mutation on exon
11 of KIT exhibited a good response to imatinib, while the
response of patients with a mutation on exon 9 to Gleevec was
slightly worse (25). A number of
in vitro and clinical studies have indicated that for cases
with mutations in the kinase locus, such as mutations in exon 17 of
KIT or in exon 18 of PDGFRA, treatment with the
inhibitor should have been terminated (26). However, additional studies have
demonstrated that 30% of the mutations in PDGFRA are
sensitive to imatinib, indicating that attention should be focused
on CD117-negative GIST cases, and that the corresponding genetic
mutations should be tested against imatinib (27).
In conclusion, to the best of our knowledge, the
present study is the first to investigate the associations between
mutations in the KIT and PDGFRA genes with GIST in a
cohort of patients from North China. The results of the present
study demonstrated that the most frequent mutation occurred on the
two ends of exon 11 in KIT. In addition, mutations in
PDGFRA were primarily observed in CD117-negative GIST
patients. Notably, one case was identified to have a syngeneic
double mutant on exon 17 of PDGFRA and exon 11 of
KIT, which to the best of our knowledge, is the only case to
have been detected worldwide. To date, only one case of an
allogeneic double mutant has been reported in China, which involved
a Val824 internal GTC>GTT base point mutation on exon 18 of
PDGFRA. In the present study, gene mutation types were
identified using PCR amplification and sequencing method, and
associated gene mutations were analyzed to determine the
development process, prognosis and impact of imatinib-targeted
therapy in patients with GIST, indicating that it may provide a
molecular basis for evaluating pathologies.
References
|
1
|
Heinrich MC, Corless CL, Duensing A,
McGreevey L, Chen CJ, Joseph N, Singer S, Griffith DJ, Haley A,
Town A, et al: PDGFRA activating mutations in gastrointestinal
stromal tumors. Science. 299:708–710. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nedeljkovic SS, Wasan A and Jamison RN:
Assessment of efficacy of long-term opioid therapy in pain patients
with substance abuse potential. Clin J Pain. 18 (4 Suppl):S39–S51.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Joensuu H and Kindblom LG:
Gastrointestinal stromal tumors - a review. Acta Orthop Scand
Suppl. 75:62–71. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lasota J, Jasinski M, Sarlomo-Rikala M and
Miettinen M: Mutations in exon 11 of c-kit occur preferentially in
malignant versus benign gastrointestinal stromal tumors and do not
occur in leiomyomas or leiomyosarcomas. Am J Pathol. 154:53–60.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim TW, Lee H, Kang YK, Choe MS, Ryu MH,
Chang HM, Kim JS, Yook JH, Kim BS and Lee JS: Prognostic
significance of c-kit mutation in localized gastrointestinal
stromal tumors. Clin Cancer Res. 10:3076–3081. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Corless CL, McGreevey L, Haley A, Town A
and Heinrich MC: KIT mutations are common in incidental
gastrointestinal stromal tumors one centimeter or less in size. Am
J Pathol. 160:1567–1572. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
He HY, Fang WG, Zhong HH, Li Y, Zheng J,
Du J, Heng WJ and Wu BQ: Status and clinical implication of c-kit
and PDGFRA mutations in 165 cases of gastrointestinal stromal tumor
(GIST). Zhonghua Bing Li Xue Za Zhi. 35:262–266. 2006.(In Chinese).
PubMed/NCBI
|
|
8
|
Debiec-Rychter M, Dumez H, Judson I, Wasag
B, Verweij J, Brown M, Dimitrijevic S, Sciot R, Stul M, Vranck H,
Scurr M, Hagemeijer A, van Glabbeke M and van Oosterom ATEORTC Soft
Tissue and Bone Sarcoma Group: Use of c-KIT/PDGFRA mutational
analysis to predict the clinical response to imatinib in patients
with advanced gastrointestinal stromal tumours entered on phase I
and II studies of the EORTC soft tissue and bone sarcoma group. Eur
J Cancer. 40:689–695. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Miettinen M and Lasota J: Gastrointestinal
stromal tumors: Review on morphology, molecular pathology,
prognosis and differential diagnosis. Arch Pathol Lab Med.
130:1466–1478. 2006.PubMed/NCBI
|
|
10
|
Piao J and Liu S, Xu Y, Wang C, Lin Z, Qin
Y and Liu S: Ezrin protein overexpression predicts the poor
prognosis of pancreatic ductal adenocarcinomas. Exp Mol Pathol.
98:1–6. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fletcher CD, Berman JJ, Corless C,
Gorstein F, Lasota J, Longley BJ, Miettinen M, O'Leary TJ, Remotti
H, Rubin BP, et al: Diagnosis of gastrointestinal stromal tumors: A
consensus approach. Hum Pathol. 33:459–465. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hirota S, Isozaki K, Moriyama Y, Hashimoto
K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M,
et al: Gain-of-function mutations of c-kit in human
gastrointestinal stromal tumors. Science. 279:577–580. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hou YY and Zhu XZ: Gastrointestinal
stromal tumor molecular biology. Cancer Res Clin. 18:507–512.
2006.
|
|
14
|
Battochio A, Mohammed S, Winthrop D,
Lefresne S, Mulder K, Chu Q, O'Hara C and Lai R: Detection of c-KIT
and PDGFRA gene mutations in gastrointestinal stromal tumors:
Comparison of DHPLC and DNA sequencing methods using a single
population-based cohort. Am J Clin Pathol. 133:149–155. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wozniak A, Rutkowski P, Piskorz A,
Ciwoniuk M, Osuch C, Bylina E, Sygut J, Chosia M, Rys J, Urbanczyk
K, et al Polish Clinical GIST Registry: Prognostic value of
KIT/PDGFRA mutations in gastrointestinal stromal tumours (GIST):
Polish Clinical GIST Registry Experience. Ann Oncol. 23:353–360.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Corless CL, Fletcher JA and Heinrich MC:
Biology of gastrointestinal stromal tumors. J Clin Oncol.
22:3813–3825. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lasota J, Wozniak A, Sarlomo-Rikala M, Rys
J, Kordek R, Nassar A, Sobin LH and Miettinen M: Mutations in exons
9 and 13 of KIT gene are rare events in gastrointestinal stromal
tumors. A study of 200 cases. Am J Pathol. 157:1091–1095. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Antonescu CR, Sommer G, Sarran L,
Tschernyavsky SJ, Riedel E, Woodruff JM, Robson M, Maki R, Brennan
MF, Ladanyi M, et al: Association of KIT exon 9 mutations with
nongastric primary site and aggressive behavior: KIT mutation
analysis and clinical correlates of 120 gastrointestinal stromal
tumors. Clin Cancer Res. 9:3329–3337. 2003.PubMed/NCBI
|
|
19
|
Lasota J, Dansonka-Mieszkowska A, Stachura
T, Schneider-Stock R, Kallajoki M, Steigen SE, Sarlomo-Rikala M,
Boltze C, Kordek R, Roessner A, et al: Gastrointestinal stromal
tumors with internal tandem duplications in 3′ end of KIT
juxtamembrane domain occur predominantly in stomach and generally
seem to have a favorable course. Mod Pathol. 16:1257–1264. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Medeiros F, Corless CL, Duensing A,
Hornick JL, Oliveira AM, Heinrich MC, Fletcher JA and Fletcher CD:
KIT-negative gastrointestinal stromal tumors: Proof of concept and
therapeutic implications. Am J Surg Pathol. 28:889–894. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tzen CY and Mau BL: Analysis of
CD117-negative gastrointestinal stromal tumors. World J
Gastroenterol. 11:1052–1055. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tu H, Li Q, Cai J, Chen Z, Yang H, Jiang
H, Mao Y, Shou Z and Chen J: Extragastrointestinal stromal tumor in
a kidney transplant recipient. Clin Exp Nephrol. 16:350–353. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li J: Molecular target therapy for
gastrointestinal stromal tumor. Zhong Hua Xiao Hua Wai Ke Za Zhi.
12:253–256. 2013.(In Chinese).
|
|
24
|
Nickl NJ: Gastrointestinal stromal tumors:
New progress, new questions. Curr Opin Gastroenterol. 20:482–487.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen H: Mutation of c-kit and PDGFRA Genes
in Gastrointestinal Stromal Tumor. Zhong Guo Sheng Wu Hua Xue Yu
Fen Zi Sheng Wu Xue Bao. 12:697–701. 2009.(In Chinese).
|
|
26
|
He H, Xiang Y, Li Y, Zhong G, Wu B and
Zheng J: c-kit and PDGFRA mutat ions in 60 cases of
gastrointestinal stromal tumors (GISTs). Beijing Da Xue Xue Bao.
37:320–324. 2005.(In Chinese). PubMed/NCBI
|
|
27
|
Kim SY, Janeway K and Pappo A: Pediatric
and wild-type gastrointestinal stromal tumor: New therapeutic
approaches. Curr Opin Oncol. 22:347–350. 2010. View Article : Google Scholar : PubMed/NCBI
|