Introduction
Gliomas account for ~50% of all brain tumors
(1,2). Among these, malignant gliomas are
extremely invasive and are rarely removable by en bloc resection.
Despite the use of aggressive therapies, including radical
resection, irradiation and chemotherapy, the median survival time
of the most malignant type of glioma, glioblastoma multiforme
(GBM), is ~1 year from the initial diagnosis (3,4).
Temozolomide
(8-carbamoyl-3-methylidazo(5,1-d)-1,2,3,5-terrazin-4(3H)-one; TMZ)
has emerged as a well-tolerated, orally administered alkylating
agent, delivering a methyl group to DNA purine bases
(O6-guanine; N7-guanine and
N3-adenine), and has the ability to cross the
blood-brain barrier and thereby treat primary and recurrent gliomas
(5,6). However, its relatively short half-life
(~1.8 h) and the presence of methylguanine-DNA methyltransferase
within the tumor result in a high recurrence rate following
TMZ-based monotherapy (7). Novel
strategies devised to enhance response and thwart resistance are,
therefore, the focus of clinical investigation, and are essential
for the future improvement of patient prognosis (8,9).
Previous studies have reported that glioma cells
respond to TMZ treatment with autophagy, referred as type II
programmed cell death (10–12). Notably, Kanzawa et al
(13) demonstrated that
3-methyladenine (3-MA) inhibits autophagy through the inhibition of
microtubule-associated protein 1 light chain 3 (LC3) localization
to the autophagosomal membrane, thereby attenuating glioblastoma
cell death. However, the role of autophagy is dependent on cellular
context; apart from a cytotoxic role during TMZ action, autophagy
induction following mild or moderate cellular stress (for example,
starvation) is a cytoprotective process that eliminates
stress-reactive cytoplasmic aggregates, macromolecules and
organelles in mammalian cells by the lysosomal system and, in turn,
supplies energy to the cells to maintain homeostasis through these
catabolic phenomena (14–16). Such paradoxical roles of autophagy
indicate that an autophagy activator may exert antitumor effects
when applied in combination with TMZ, causing increased
glioblastoma cell autophagy, whilst exerting no detrimental effects
or even being beneficial to normal tissue.
The hormonally active form of vitamin D (VD),
1,25-dihydroxycholecalciferol, has well-established actions on
autophagy and has low toxicity at low concentrations (<1,000
µg/day, causing conditions such as hypercalcemia only when
administered in excess) (17). The
present study thus aimed to investigate the potential of using VD
as a chemosensitizing agent in glioblastoma treatment. The
induction of autophagy by VD relies on an increase in cytoplasmic
Ca2+ concentration, which may result from VD
receptor-mediated changes to calcium-regulating protein expression.
An increase in cytoplasmic Ca2+ concentration activates
Ca2+/calmodulin-dependent kinase kinase-β, which is
followed by the activation of AMP-activated kinase (AMPK), a
well-known potent inducer of autophagy (18). AMPK activation induces autophagy via
the inhibition of mammalian target of rapamycin complex 1, the
major gatekeeper of mammalian autophagy, and the subsequent
activation of several autophagy-associated proteins (19,20).
The present study aimed to determine the anticancer
effect of VD and TMZ in the co-treatment of glioblastoma, and to
identify the underlying mechanism of action. Using the C6
glioblastoma cell line, the in vitro anticancer effects of
TMZ and VD were compared with those of TMZ alone through a cell
viability assay. In accordance with a previous study, which
demonstrated that 100 nM VD could trigger autophagy in breast tumor
cells without any signs of apoptotic morphology (21), a 100 nM concentration of VD was used
in the present study. Western blotting, flow cytometry,
transmission electron microscopy (TEM) and immunofluorescence (IF)
were also performed to identify whether autophagy enhancement was
the underlying mechanism of this anticancer effect. Finally, these
treatments were applied to rat orthotopic xenograft models to
determine their antitumor effects in vivo.
Materials and methods
Cell culture
The C6 rat glioblastoma cell line was purchased from
the American Type Culture Collection (ATCC, Manassas, VA, USA).
Cells were cultured under sterile conditions, at 37°C, in a humid
environment containing 5% CO2; culture medium consisted
of Dulbecco's modified Eagle's medium supplemented with 10% fetal
bovine serum, 4 mM glutamine, 100 U/ml penicillin and 100 mg/ml
streptomycin (all purchased from Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA). The cultures were regularly checked and
trypsinized when cells reached 85% confluence.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
In order to determine the half-maximal inhibitory
concentration (IC50 value) of TMZ, 5×103 C6
cells in single-cell suspensions were seeded into individual wells
of 96-well plates and incubated for 24 h at 37°C prior to treatment
with TMZ (0.1, 0.5, 1, 5 or 10 µM; Sigma-Aldrich, St. Louis, MO,
USA) diluted in dimethyl sulfoxide (DMSO; Gibco; Thermo Fisher
Scientific, Inc.) for 24 h (data not shown). Following
determination of the IC50 value of TMZ as 1 mM, cells
were treated for 24 h with DMSO alone, 1 mM TMZ, 100 nM VD (Tocris
Bioscience, Ellisville, MO, USA) or a combination of the two (1 mM
TMZ plus 100 nM VD). To reveal the role of autophagy on C6 cell
cytotoxicity, which may be induced by various treatments as
mentioned above, cells were treated with 1 mM 3-methyladenine
(3-MA; Sigma-Aldrich), an autophagy inhibitor, at 30 min
treatments. MTT solution (Thermo Fisher Scientific, Inc.) was added
to each well and the plate was incubated for 4 h at 37°C prior to
removing the culture medium. DMSO was then added for 30 min at room
temperature. Cell viability was determined using a
spectrophotometer by measuring the absorbance at 492 nm (Ultrospec
7000; Biochrom, Holliston, MA, USA). Viability for each group was
calculated as a percentage of that of the control group.
Clonogenic assay
Prior to plating, the cell culture medium was
removed and the cells were washed twice with phosphate-buffered
saline (PBS). Adherent cells were then trypsinized and counted. A
total of 5,000 cells were seeded into 60-mm tissue culture dishes
(Greiner Bio-One GmbH, Frickenhausen, Germany) containing culture
medium supplemented with DMSO, 1 mM TMZ, 100 nM VD or a combination
of 1 mM TMZ and 100 nM VD. Colonies were allowed to form for 72 h.
The cell culture medium was then removed and the cells were washed
twice with PBS. Colonies were fixed using 100% methanol for 30 min
at room temperature, and stained with Coomassie Blue (Thermo Fisher
Scientific, Inc.) for 15 min. The number of colonies containing ≥50
cells was counted. This experiment, and the following assays were
performed in triplicate.
Wound healing assay
Wound healing experiments were performed, as
previously described (22). Briefly,
the cells were seeded on 60-mm tissue culture dishes and grown to
confluence. The cells were treated with DMSO, TMZ, VD or a
combination of TMZ and VD, and scratched with a sterile 20-µl
pipette tip to create an artificial wound. Immediately and 24 h
after wounding, the wound healing process was imaged using an
inverted microscope with a 10X long-working-distance objective
(1X51; Olympus Corporation, Tokyo, Japan). Cell migration was
quantified by measuring the diameter of the wound at 0 and 24 h
using the image analyzing software, ImageJ (version 1.45; NIH,
Bethesda, MA, USA).
Hoechst 33258 staining
In order to evaluate levels of apoptosis,
1×106 cells/well were plated on coverslips (BD
Biosciences, Bedford, MA, USA) in a 12-well plate. The cells were
treated with DMSO, TMZ, VD or a combination of TMZ and VD and kept
in a CO2 incubator at 37°C for 24 h. Following
incubation and addition of fresh medium, the cells were incubated
with 5 µl Hoechst 33258 (Sigma-Aldrich) per well at 37°C for 10
min. The number of apoptotic cells was then assessed using an LSM
700 laser confocal microscope (Zeiss, Oberkochen, Germany).
Increased fluorescence with shrunken nuclei was indicative of
apoptotic cells, whereas weak fluorescence with normally-sized
nuclei was indicative of non-apoptotic cells. The number of
apoptotic cells was quantified by capturing images in random fields
and counting ≥200 cells from 4 random fields in each well.
IF staining
LC3 is a reliable marker for cells undergoing
autophagy (23). A total of
1×106 cells/well were seeded onto sterile glass
coverslips. After a 24 h treatment with DMSO, TMZ, VD or a
combination of TMZ and VD, cells were fixed using 4%
paraformaldehyde (Sigma-Aldrich), blocked with 3% normal goat serum
(Dako, Carpinteria, CA, USA) and incubated in 1% bovine serum
albumin (Sigma-Aldrich)/10% normal goat serum/0.3 M glycine in 0.1%
PBS-Tween for 1 h to permeabilize the plasma membrane and to block
non-specific protein-protein interactions. The cells were then
incubated with a rabbit polyclonal anti-LC3 antibody (1:500;
ab128025; Abcam, Cambridge, UK) for 2 h. Cells were washed twice
with PBS, incubated for an additional 30 min with fluorescein
isothiocyanate-conjugated anti-rabbit IgG antibody (1:1,000;
ab6717; Abcam), washed again with PBS and mounted onto slides with
4′,6-diamidino-2-phenylindole-conjugated mounting medium (Abcam).
Following image acquisition with the LSM-700 laser confocal
microscope, LC3 puncta in cells were quantified using 10
randomly-selected images from each drug treatment group.
Flow cytometry
Cells were analyzed for the presence of LC3 using a
FACSCanto II flow cytometer (BD Biosciences) according to the
manufacturer's protocol. Following 2 washes with PBS, cells were
fixed using 4% paraformaldehyde for 10 min at 37°C and
permeabilized with 0.25% Triton X-100 in PBS for 10 min. Cells were
stained with rabbit polyclonal anti-LC3 antibody (1:100; ab128025;
Abcam, Cambridge, UK) for 1–2 h at 4°C (1:100) and goat anti-rabbit
IgG antibody (1:200; ab6717; Abcam) for 1 h on ice. Following 2
additional washes with PBS, cells were fixed using 4%
paraformaldehyde and assayed immediately. Flow cytometry data were
collected using 10,000–30,000 cells and were analyzed using FlowJo
software (version 10.1; Tree Star, Ashland, OR, USA).
TEM
Cells were collected and fixed in 2%
paraformaldehyde and 0.1% glutaraldehyde in 0.1 M sodium cacodylate
for 2 h, post-fixed with 1% OsO4 for 1 h, PBS-washed and
stained for 1 h in 3% aqueous uranyl acetate. The cells were washed
again, dehydrated with graded alcohol and embedded in Epon-Araldite
resin (Canemco Inc., Gore, QC, Canada). Ultrathin sections were cut
using an ultramicrotome (Reichert, Inc., Depew, NY, USA),
counterstained with 0.3% lead citrate and examined using an HT7700
transmission electron microscope (Hitachi, Ltd., Tokyo, Japan).
Immunoblotting
C6 cells treated with vehicle, TMZ, VD or a
combination of TMZ and VD were lysed using a lysis buffer
containing 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 2 mM EDTA, 100 mM
NaF, 1 µg/ml leupeptin, 1 µg/ml antipain and 1 mM
phenylmethylsulfonyl fluoride. The protein content in the cell
lysates was determined using a BCA protein assay kit (Thermo Fisher
Scientific, Inc.). Immunoblotting was conducted by resolving 30–50
µg protein by 15% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis and electroblotting onto polyvinylidene difluoride
membranes for western blot analysis. Blots were probed with the
following 1:1,000-diluted primary rabbit antibodies: Anti-cleaved
caspase-3 (ab32042), β-actin (ab189073), LC3 (ab128025), beclin-1
(ab55878) and p62 (ab91526) overnight at 4°C, followed by
incubation with horseradish peroxidase-conjugated goat anti-rabbit
IgG (1:1,000; ab6717) secondary antibody for 1 h at room
temperature. The proteins were then visualized by an enhanced
chemiluminescence system (Thermo Fisher Scientific, Inc.) with
exposure to X-ray film. Finally, the blots were scanned and
densitometric analysis was performed using Scion Image (Beta 4.02
release) software (Scion Corporation, Torrance, CA, USA).
Experimental animals
Male Sprague-Dawley rats were obtained from the
Experimental Animal Center, Konyang University (Daejeon, South
Korea). A total of 24 male rats (age, 2-months; body weight,
250–300 g) were used. The animals were housed at 23°C and 60%
relative humidity, with a 12:12 h light:dark cycle and free access
to water and food. Animal handling and care conformed to the
Konyang University institutional guidelines, which comply with
international law and policy (as described in the NIH Guide for the
Care and Use of Laboratory Animals; NIH Publication No. 85-23,
1985, revised 1996). The study was approved by the ethics committee
of Konyang University, Daejeon, Korea). All experiments were
designed to minimize the number of animals used and the detriment
to the animals' mental and physical wellbeing.
Rat orthotopic xenograft model
To establish the rat glioblastoma model, the cell
implantation procedure was conducted based on the method developed
by Kobayashi et al (24).
Briefly, each animal was anesthetized (ketamine 40–90 mg/kg,
intraperitoneally and xylazine 5–10 mg/kg, subcutaneously; both
purchased from Daihan Pharmaceutical Co., Ltd., Seoul, Korea) and
immobilized on a stereotaxic unit (Stoelting Co., Wood Dale, IL,
USA). Following disinfection and incision of the skin with a
scalpel, a hole was drilled through the skull 2-mm lateral and 2-mm
anterior to the bregma, on the right side of the skull. A total of
1×106 C6 cells, resuspended in 10 µl saline solution,
were injected with a 25-gauge Hamilton syringe (Hamilton, Reno, NV,
USA) at a 3-mm depth from the dura, at a rate of 2 µl/min. A
waiting time of 2 min was implemented following injection to avoid
leakage. At post-operative day 7 (POD 7), animals were divided into
three groups (n=8 animals per group). The first group was treated
with 200 µl DMSO, the second group was treated intraorally (i.o.)
with 10 mg/kg/day TMZ, dissolved in 200 µl DMSO, and the third
group received a subcutaneous (s.c.) injection of 0.2 µg/kg/day of
VD dissolved in 200 µl of saline solution and also received TMZ as
described above. The treatment duration was 1 week. Magnetic
resonance imaging (MRI) was used to evaluate tumor size in
vivo. Prior to imaging, rats were anesthetized, using the
aforementioned anesthetics and doses, and placed in the imaging
chamber of a 7T/30 cm MRI scanner (Pharmascan 7T; Bruker BioSpin
GmbH, Karlsruhe, Germany). The parameters of the scan were as
follows: Field of view, 60 mm; slice thickness, 0.5 mm; multiple
echo times, 35.1 msec; and repetition time, 3,500 msec. Prior to
drug treatment, the confirmation of successful modeling was
conducted by T2-weighted MRI at POD 7. At the end of the 7-day
treatment period, on POD 14, tumors from each rat were imaged again
by MRI to compare the antitumor effects of the drug treatments. A
total of 20 MRI images per animal were obtained and tumor volume
was measured using 3D-Doctor Software (Able Software Corp,
Lexington, MA, USA) with a thresholding method (25).
Statistical analysis
The survival curves of the tumor-bearing rats were
estimated according to the Kaplan-Meier method, and the curves were
compared using a generalized Wilcoxon test. One-way analysis of
variance tests were performed to detect differences in effects
between the treatments. All data are presented as mean ± standard
error of the mean. Comparisons of the data between the groups were
performed with Student's t-tests using SPSS software (version IBM
Corp., Armonk, NY, USA). Differences with P-values <0.05 were
considered to be statistically significant. Each n-value refers to
the number of separate experiments conducted.
Results
Combination with VD enhances the
cytotoxicity of TMZ to a rat glioblastoma cell line in vitro
To examine whether co-treatment with TMZ and VD
inhibited the growth of C6 cells, a rat glioblastoma cell line,
in vitro experiments were performed. Cells were incubated in
a culture medium containing DMSO, VD alone, or TMZ with or without
VD, for 24 h. An MTT assay was then performed to compare the
cytotoxicity of each treatment. As demonstrated in Fig. 1A and B, treatment with VD alone did
not suppress cell growth, but TMZ alone or in combination with TMZ
significantly inhibited cell growth (P<0.001), compared with the
DMSO control. Notably, growth was inhibited to a greater extent in
cells treated with TMZ and VD when compared with the growth of
cells treated with TMZ alone (cell viability, 29.9±3.7 vs.
54.2±14.3%, respectively; P<0.001). These data were supported by
two additional experiments; TMZ treatment reduced the number of
colonies (Fig. 1C and D) and reduced
the wound healing (i.e., migratory) ability of the C6 cells
(Fig. 1E and F). TMZ and VD
co-treatment was more effective than TMZ alone in reducing colony
formation (2.6±2.2 vs. 5.9±0.8 colonies, respectively; P<0.01)
and inhibiting wound healing (wound distance, 0.56±0.05 vs.
0.28±0.03 mm; P<0.001). These data suggest that the combined use
of TMZ and VD may be an effective therapy for glioblastoma
treatment.
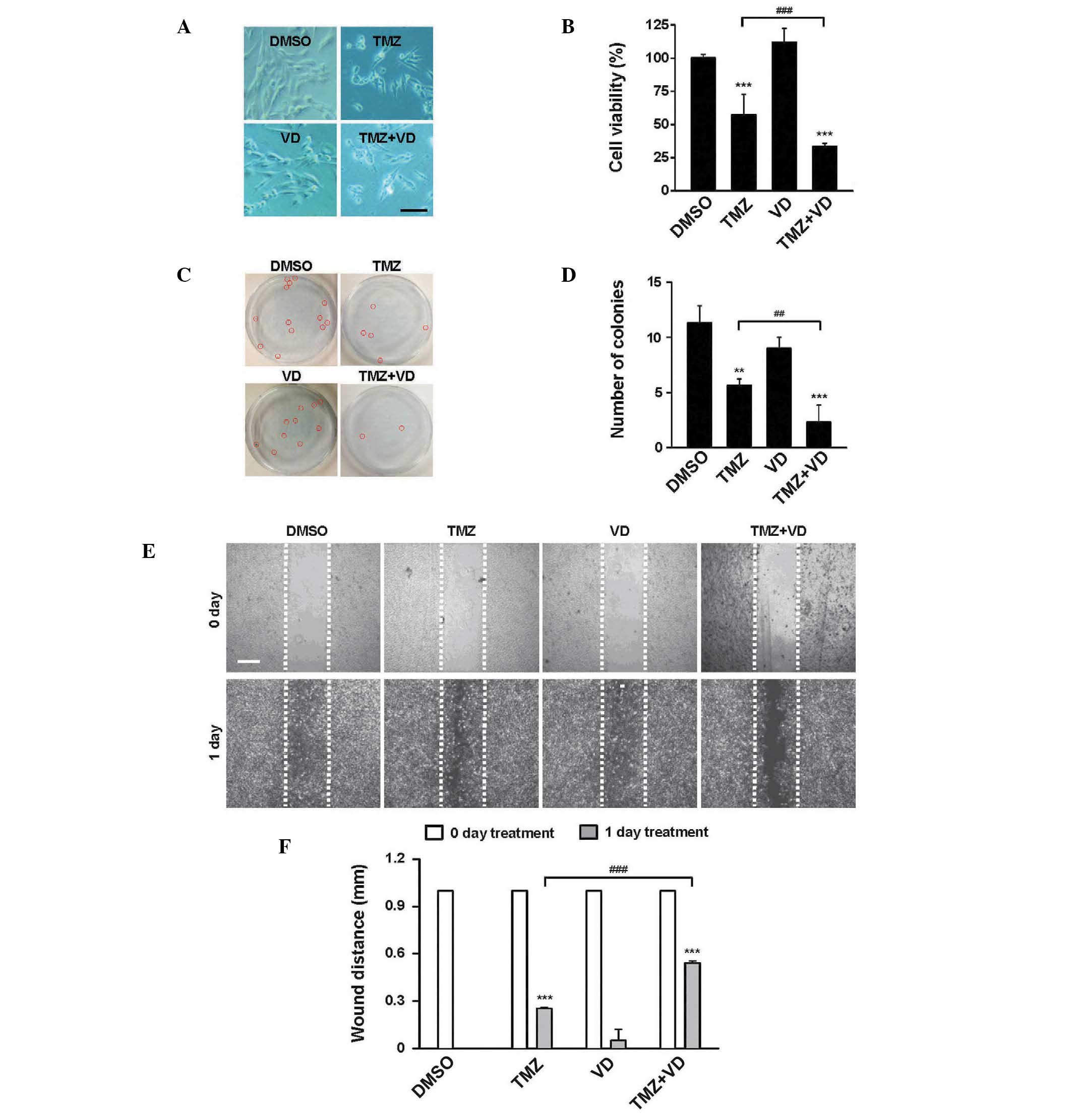 | Figure 1.In vitro effects of TMZ + VD
combination therapy in the C6 rat glioblastoma cell line. (A)
Representative microscopic images of C6 cells incubated with DMSO,
TMZ, VD or TMZ + VD for 24 h. Scale bar represents 100 µm. (B)
Graph of the treatment cytotoxicity, measured by MTT assay. (C)
Representative images of colonies formed by C6 cells incubated with
DMSO, TMZ, VD or TMZ + VD for 72 h., marked with red circles. (D)
Graph of the colony counts for each group. The number of colonies
containing ≥50 cells was selectively counted. (E) Representative
microscopic images of a wound healing assay using C6 cells treated
with DMSO, TMZ, VD or TMZ + VD for 24 h. Scale bar represents 500
µm. (F) Graph of the wound diameter remaining following treatment,
expressed as the mean ± standard error of the mean of ≥3
independent experiments. **P<0.01 and ***P<0.001 vs.
DMSO-treated cells; ##P<0.01 and
###P<0.001 vs. TMZ-treated cells. DMSO, dimethyl
sulfoxide; TMZ, temozolomide; VD, vitamin D; MTT,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide. |
TMZ and VD co-treatment enhances
autophagy in glioblastoma cells
TMZ and VD have separately been reported to induce
autophagy in numerous cell types (26,27), so
the present study examined the induction of autophagy by TMZ, VD or
a combination of TMZ and VD in C6 cells. After 24 h of treatment,
cells were collected and stained using IF (Fig. 2A and B) and examined by flow
cytometric analysis (Fig. 2C and D)
to detect the presence of LC3. LC3 is an established autophagy
marker due to its involvement in autophagosome membrane formation
(28,29). TEM observation (Fig. 2E) was performed to detect activation
of autophagy. All three drug treatments could activate autophagy in
C6 cells. In the cells treated with a combination of TMZ and VD
compared with the cells treated with TMZ alone, the estimated mean
number of LC3 puncta per cell (Fig. 2A
and B) and mean fluorescent intensity (Fig. 2C and D) were increased almost 2-fold
(22.1±3.6 vs. 11.9±0.2 puncta; P<0.01) and 1.5-fold
(1,210.3±62.5 vs. 820.2±34.4; P<0.001), respectively.
Ultrastructural observation using TEM (Fig. 2E) revealed the generation of small
autophagosomes, double-layered structures engulfing intracellular
organelles, in cells treated with TMZ or VD. However, the
autophagosome number and size increased in cells treated with TMZ
and VD compared with the cells treated with TMZ alone, also
confirmed by IF and TEM. The quantitative changes of
autophagy-related protein expression in C6 cells were also
measured. As demonstrated in Fig.
2F, TMZ treatment induced the conversion of LC3-I to LC3-II
(lipidated form), a reliable marker of autophagosome generation.
Furthermore, the expression levels of beclin-1 and p62, positive
and negative upstream regulators of LC3 recruitment, respectively,
were indicative of the induction of autophagy in TMZ-treated cells
(30). The alterations of the level
of autophagy-related protein expression were significantly
increased by the TMZ and VD treatment.
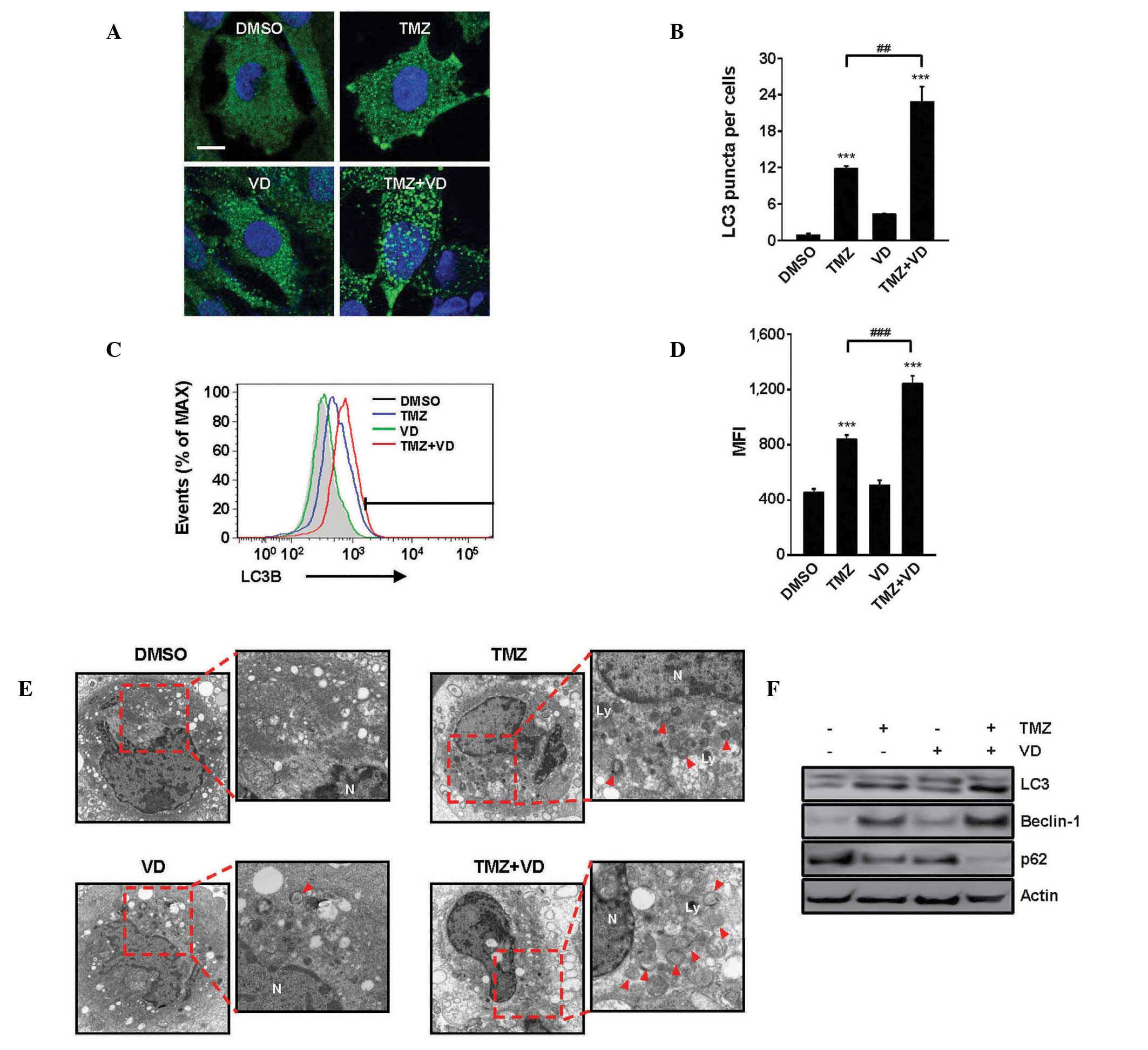 | Figure 2.Enhancement of autophagic activity in
C6 cells treated with TMZ and VD for 24 h. (A) Representative
immunofluorescence images demonstrating LC3 immunoreactivity in C6
cells treated with DMSO, TMZ, VD or TMZ + VD. Scale bar represents
10 µm. (B) Graph of LC3 puncta quantification, demonstrating the
average number of LC3 puncta per cell from 10 randomly selected
images. (C) Representative fluorescence-activated cell sorting
profiles for LC3 expression in C6 cells treated with DMSO, TMZ, VD
or TMZ + VD. (D) Graph of the MFI, expressed as the mean ± standard
error of the mean from ≥3 independent experiments. ***P<0.001
vs. DMSO-treated cells; ##P<0.01 and
###P<0.001 vs. TMZ-treated cells. (E) Autophagy in
transmission electron micrographs (x9,700 magnification) of C6
cells treated with DMSO, TMZ, VD or TMZ + VD. Red arrowheads
indicate autophagosomes. (F) Representative immunoblots
demonstrating LC3, beclin-1 and p62 expression in C6 cells treated
with the indicated drugs. Actin was used as a loading control.
DMSO, dimethyl sulfoxide; TMZ, temozolomide; VD, vitamin D; LC3,
microtubule-associated protein 1 light chain 3; MFI, mean
fluorescence intensity. |
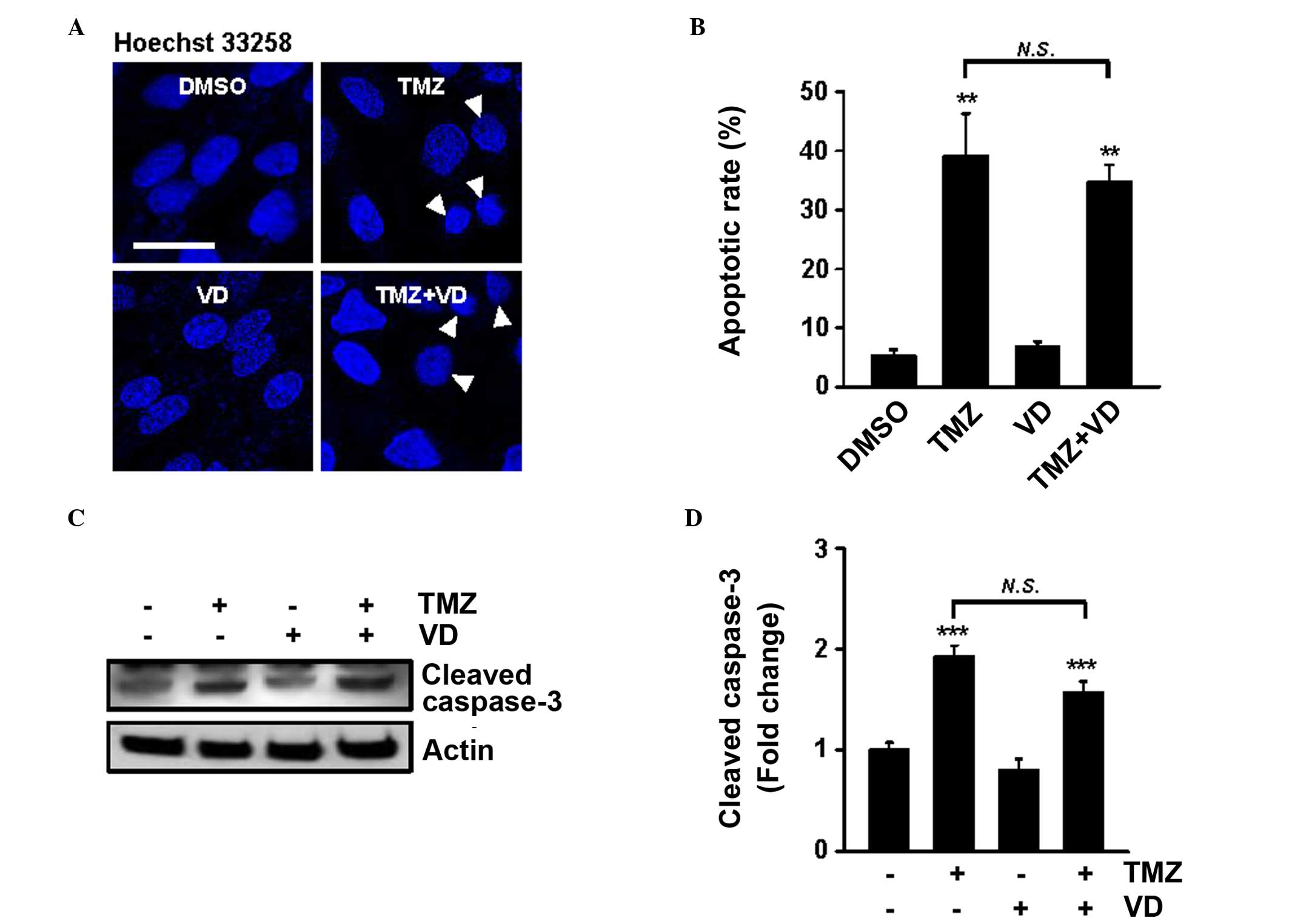
The effects of TMZ and VD co-treatment on apoptosis,
morphologically and quantitatively, were also examined (Fig. 3A and B). Following staining with
Hoechst 33258, small, bright, condensed nuclei occurred at a
significantly greater frequency in cells treated with TMZ alone or
TMZ and VD (39.1±8.3 and 35.0±3.4%, respectively), compared with
DMSO-treated cells (P<0.01). Furthermore, western blot analysis
to quantify cleaved (active-) caspase-3, the most important
effector of the apoptotic process, supported increased apoptosis
following these treatments (Fig. 3C and
D). Notably, no significant difference in apoptosis was
identified between the TMZ-treated and the TMZ plus VD-treated
cells. The present results suggest that TMZ and VD co-treatment
enhanced the autophagic process, but not apoptosis, in glioblastoma
cells.
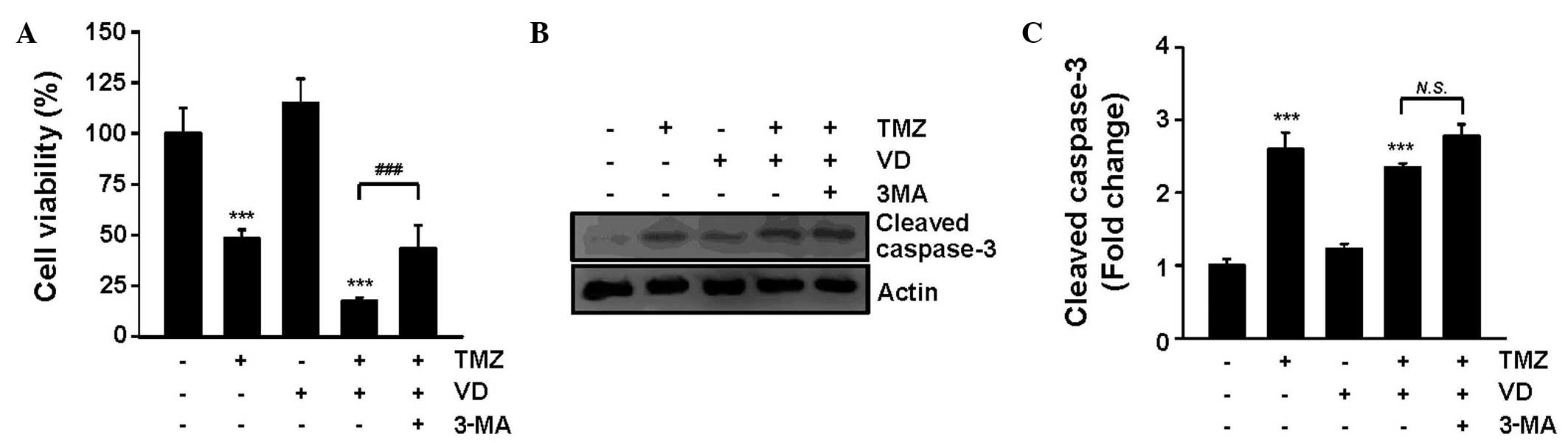
Involvement of cytotoxic autophagy in
the synergistic effect of VD and TMZ
The role of autophagy induced by the TMZ and VD
co-treatment on tumor suppression was elucidated in the current
study through use of the autophagy inhibitor 3-MA. As demonstrated
in Fig. 4A, 3-MA treatment
significantly attenuated the tumoricidal activity of the TMZ and VD
co-treatment (cell viability, 25.4±8.1 vs. 12.2±2.5% in TMZ and
VD-treated cells; P<0.001). Addition of 3-MA caused C6 cell
survival rates to be restored to those of C6 cells treated with TMZ
alone. The evaluation of caspase-3 expression levels using western
blot analysis revealed that 3-MA had no significant effect on
apoptosis (Fig. 4B and C). These
results indicate that the antitumor effect of the TMZ and VD
co-treatment is due, at least in part, to the enhancement of
autophagy, without affecting apoptosis.
Synergistic effect of VD in
combination of TMZ in the in vivo rat glioblastoma model
The growth dynamics of xenografted C6 tumors were
analyzed by MRI. At POD 7 in all tumor-bearing rats, the
glioblastoma presented as a hyperintensive tumor in the T2-weighted
images; no significant difference was observed in tumor size
between the experimental groups. In contrast to vehicle treatment,
the administration of TMZ alone and of TMZ plus VD for 1 week
significantly inhibited tumor growth (Fig. 5A), which reached 580±83 and 186±52
mm3 in rats treated with TMZ alone or TMZ plus VD,
respectively. These results suggest a synergism between TMZ and VD
(Fig. 5B). Tumor-bearing rats
treated with TMZ plus VD demonstrated a significantly prolonged
survival duration (Fig. 5C). The
median survival duration was 4 weeks in rats treated with TMZ only,
but ≥5 weeks for rats co-treated with TMZ and VD. These data
together indicate that treatment with a combination of TMZ and VD
enhanced TMZ therapeutic efficacy against glioblastoma in
vivo.
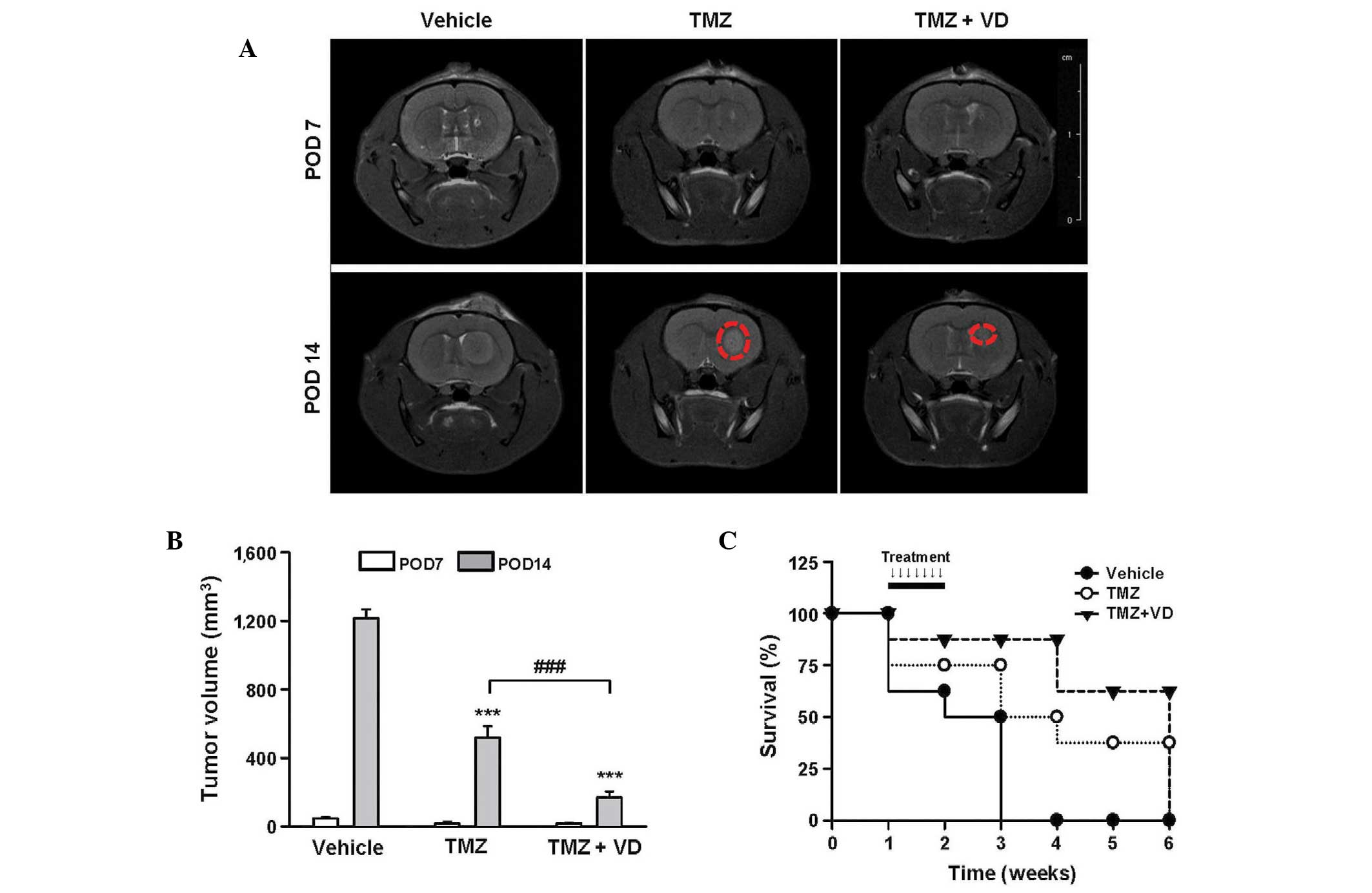 | Figure 5.In vivo effects of TMZ and VD
combination therapy on tumor size regression and prolonged survival
in orthotopic, xenografted tumor-bearing rats (n=8). (A)
Representative T2-weighted magnetic resonance images, used to
monitor brain tumor progression at POD 7 and 14 in rats treated for
7 days (from POD 8 to POD 14) with vehicle, TMZ (10 mg/kg/day,
i.o.) or TMZ (10 mg/kg/day, i.o.) + VD (0.2 µg/kg/day, s.c.). Red
circles represent comparative tumor size between the TMZ- and TMZ +
VD-treated groups. (B) Graph of tumor volumetric analysis,
expressed as the mean ± standard error of the mean. ***P<0.001
vs. vehicle-treated rats; ###P<0.001 vs. TMZ-treated
rats. (C) Kaplan-Meier survival curves. Rats with intracranial C6
cells were observed for 6 weeks post-treatment; the arrows indicate
the timing of the 7-day scheduled regimen for each treatment. TMZ,
temozolomide; VD, vitamin D; POD, post-operative day; i.o.,
intraorally; s.c., subcutaneously. |
Discussion
The current study established that a combination of
VD and TMZ may have therapeutic potential in the treatment of GBM,
and provided evidence that chemosensitization to TMZ by VD occurs
through the enhancement of autophagy. Considering that GBM cells
commonly carry mutations that inactivate the apoptotic pathway
(31,32), the enhancement of autophagy by TMZ
and VD combination therapy could represent an alternative method
for the elimination of GBM cells. However, the mechanism by which
autophagy enhancement kills GBM cells, and whether the enhancement
of autophagy is the only mechanism involved in the action of TMZ
and VD against GBM, remain to be determined.
Autophagy is the principal cellular route used for
degrading long-lived proteins and cytoplasmic organelles, and the
catabolic advantage of increased autophagy may be critical in
stress conditions (33,34). Induced autophagy may, therefore,
reflect an adaptive mechanism used to prevent cell death. A marked
correlation between reduced autophagy and cancer has previously
been reported (35,36). A previous study has also indicated
that several proteins and signaling pathways that are associated
with autophagy are deregulated during malignant transformation,
resulting in reduced autophagic activity; this previous study
demonstrated that cytoplasmic levels of beclin-1 protein and mRNA
were lower in GBMs compared with lower-grade astrocytomas and
normal brain tissue (37). Another
previous study involving biochemical analysis of biopsied tumor
samples reported that high cytoplasmic levels of the protein
beclin-1 positively correlated with the survival and performance
status of patients, whereas a low expression level of beclin-1
correlated with an increase in cell proliferation (38). Furthermore, high LC3 expression has
been associated with improved survival in GBM patients with poor
performance scores (39). Based on
these previous reports and the current data, restoration of
autophagic activity to normal levels in GBM cells, which is
commonly downregulated, may be a potential strategy to treat GBM,
and could serve as a mechanism for the restriction of uncontrolled
tumor cell growth.
In addition to the hypothesis of ‘autophagy
restoration’ as a mechanism underlying the autophagy-induced
antitumor effect, there may be another explanation; during normal
autophagy, specific cytoplasmic constituents are isolated from the
rest of the cell within an autophagosome, which then fuses with a
lysosome and its cargo is degraded and recycled (40–42).
When autophagy is upregulated, the rates of autophagosome formation
exceed the rates of lysosomal degradation, a condition termed
‘autophagic stress’, is generated in the cell. If stress or
dysregulated autophagy persist, cell death may occur through energy
depletion or through alteration of the beclin-1/bcl-2 balance. The
cells may also apoptose due to the hyperactivity of autophagosomes,
engulfing vital cytoplasmic organelles, including the mitochondria
and the endoplasmic reticulum. It has been suggested that the
overall autophagic activity is increased in a cell predisposed to
death, when compared with the normal cytoplasmic and organelle
turnover occurring in healthy cells. As a consequence, the cell
‘cannibalizes’ itself from inside, a key feature of type II
programmed cell death. Although the precise pathway by which VD
increases the therapeutic efficacy of TMZ against GBM remains to be
elucidated, this combination treatment may exert its antitumor
effects by utilizing the two aforementioned autophagy
mechanisms.
VD has multiple modes of action, which indicates
that there may be other mechanisms underlying its synergism with
TMZ besides the autophagy enhancement demonstrated in the present
study; previous studies have demonstrated that VD exerts an
antitumor effect by interfering with the transduction pathways of
growth factor-activated receptors (receptor tyrosine kinases). This
subsequently changes transcription and alters genomic functions,
resulting in the inhibition of cell proliferation (43). VD has also been proposed to promote
angiogenesis (44) and to increase
the level of an endogenous protein, cystatin D, which possesses
antitumor and anti-metastatic properties (45). Furthermore, VD has been reported to
facilitate cancer cell apoptosis by upregulating proapoptotic p53,
p21 and Bax proteins, while downregulating anti-apoptotic bcl-2
protein (46). However, the current
study did not identify changes in the number of apoptotic nuclei
(Fig. 3A and B) or in cleaved
caspase-3 expression levels (Figs.
3C and 4B and C) in C6 cells
treated with VD alone. This discrepancy regarding the role of VD in
apoptosis may be due to a different VD dose regimen, C6 cell
sensitivity to VD or differing duration of the experiment.
In summary, to the best of our knowledge, the
current study is the first to demonstrate that combining VD with
TMZ exerts a synergistic effect on the antitumor activity of TMZ in
GBM in vivo and in vitro. Although other mechanisms
underlying these synergistic effects remain to be determined, an
increase in autophagy was identified as a crucial tumoricidal
mechanisms of VD chemosensitization during TMZ-based GBM
therapy.
Acknowledgements
The present study was equally supported by a grant
from the Development of Forest Science and Technology (no.
S111414L030100) and Korea Research Foundation (no.
NRF-2014R1A1A4A03005726).
References
|
1
|
Mahaley MS, Mettlin C, Natarajan N, Laws
ER Jr and Peace BB: National survey of patterns of care for
brain-tumor patients. J Neurosurg. 71:826–836. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Avgeropoulos NG and Batchelor TT: New
treatment strategies for malignant gliomas. Oncologist. 4:209–224.
1999.PubMed/NCBI
|
|
3
|
Bower M, Newlands ES, Bleehen NM, Brada M,
Begent RJ, Calvert H, Colquhoun I, Lewis P and Brampton MH:
Multicentre CRC phase II trial of temozolomide in recurrent or
progressive high-grade glioma. Cancer Chemother Pharmacol.
40:484–488. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yung WK, Prados MD, Yaya-Tur R, Rosenfeld
SS, Brada M, Friedman HS, Albright R, Olson J, Chang SM, O'Neill
AM, et al: Multicenter phase II trial of temozolomide in patients
with anaplastic astrocytoma or anaplastic oligoastrocytoma at first
relapse. J Clin Oncol. 17:2762–2771. 1999.PubMed/NCBI
|
|
5
|
Newlands ES, Blackledge GR, Slack JA,
Rustin GJ, Smith DB, Stuart NS, Quarterman CP, Hoffman R, Stevens
MF and Brampton MH: Phase I trial of temozolomide (CCRG 81045:
M&B 39831: NSC 362856). Br J Cancer. 65:287–291. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dehdashti AR, Hegi ME, Regli L, Pica A and
Stupp R: New trends in the medical management of glioblastoma
multiforme: The role of temozolomide chemotherapy. Neurosurg Focus.
20:E62006.PubMed/NCBI
|
|
7
|
Zhang J, Stevens MF and Bradshaw TD:
Temozolomide: Mechanisms of action, repair and resistance. Curr Mol
Pharmacol. 5:102–114. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Begemann M, Kashimawo SA, Lunn RM,
Delohery T, Choi YJ, Kim S, Heitjan DF, Santella RM, Schiff PB,
Bruce JN and Weinstein IB: Growth inhibition induced by Ro 31–8220
and calphostin C in human glioblastoma cell lines is associated
with apoptosis and inhibition of CDC2 kinase. Anticancer Res.
18:3139–3152. 1998.PubMed/NCBI
|
|
9
|
Gagliano N, Moscheni C, Torri C, Donetti
E, Magnani I, Costa F, Nowicky W and Gioia M: Ukrain modulates
glial fibrillary acidic protein, but not connexin 43 expression,
and induces apoptosis in human cultured glioblastoma cells.
Anticancer Drugs. 18:669–676. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kanzawa T, Bedwell J, Kondo Y, Kondo S and
Germano IM: Inhibition of DNA repair for sensitizing resistant
glioma cells to temozolomide. J Neurosurg. 99:1047–1052. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Takeuchi H, Kondo Y, Fujiwara K, Kanzawa
T, Aoki H, Mills GB and Kondo S: Synergistic augmentation of
rapamycin-induced autophagy in malignant glioma cells by
phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer
Res. 65:3336–3346. 2005.PubMed/NCBI
|
|
12
|
Takeuchi H, Kanzawa T, Kondo Y and Kondo
S: Inhibition of platelet-derived growth factor signalling induces
autophagy in malignant glioma cells. Br J Cancer. 90:1069–1075.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kanzawa T, Germano IM, Komata T, Ito H,
Kondo Y and Kondo S: Role of autophagy in temozolomide-induced
cytotoxicity for malignant glioma cells. Cell Death Differ.
11:448–457. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gutierrez MG, Master SS, Singh SB, Taylor
GA, Colombo MI and Deretic V: Autophagy is a defense mechanism
inhibiting BCG and Mycobacterium tuberculosis survival in
infected macrophages. Cell. 119:753–766. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Giovannucci E: Vitamin D and
cardiovascular disease. Curr Atheroscler Rep. 11:456–461. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xie Z and Klionsky DJ: Autophagosome
formation: Core machinery and adaptations. Nat Cell Biol.
9:1102–1109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Campbell GR and Spector SA: Hormonally
active vitamin D3 (1alpha,25-dihydroxycholecalciferol) triggers
autophagy in human macrophages that inhibits HIV-1 infection. J
Biol Chem. 286:18890–18902. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Picotto G, Liaudat AC, Bohl L and de
Talamoni Tolosa N: Molecular aspects of vitamin D anticancer
activity. Cancer Invest. 308:604–614. 2012. View Article : Google Scholar
|
|
19
|
Høyer-Hansen M and Jäättelä M:
AMP-activated protein kinase: A universal regulator of autophagy?
Autophagy. 3:381–383. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Woods A, Dickerson K, Heath R, Hong SP,
Momcilovic M, Johnstone SR, Carlson M and Carling D:
Ca2+/calmodulin-dependent protein kinase kinase-beta
acts upstream of AMP-activated protein kinase in mammalian cells.
Cell Metab. 2:21–33. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hannigan AM and Gorski SM: Macroautophagy:
The key ingredient to a healthy diet? Autophagy. 5:140–151. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zou J, Wang YX, Mu HJ, Xiang J, Wu W,
Zhang B and Xie P: Down-regulation of glutamine synthetase enhances
migration of rat astrocytes after in vitro injury. Neurochem Int.
58:404–413. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tanida I, Ueno T and Kominami E: LC3 and
autophagy. Methods Mol Biol. 445:77–88. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kobayashi N, Allen N, Clendenon NR and Ko
LW: An improved rat brain-tumor model. J Neurosurg. 53:808–815.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rubin JB, Kung AL, Klein RS, Chan JA, Sun
Y, Schmidt K, Kieran MW, Luster AD and Segal RA: A small-molecule
antagonist of CXCR4 inhibits intracranial growth of primary brain
tumors. Proc Natl Acad Sci USA. 100:13513–13518. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Knizhnik AV, Roos WP, Nikolova T, Quiros
S, Tomaszowski KH, Christmann M and Kaina B: Survival and death
strategies in glioma cells: Autophagy, senescence and apoptosis
triggered by a single type of temozolomide-induced DNA damage. PLoS
One. 8:e556652013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wilson EN, Bristol ML, Di X, Maltese WA,
Koterba K, Beckman MJ and Gewirtz DA: A switch between
cytoprotective and cytotoxic autophagy in the radiosensitization of
breast tumor cells by chloroquine and vitamin D. Horm Cancer.
2:272–85. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mizushima N, Yamamoto A, Hatano M,
Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y and Yoshimori
T: Dissection of autophagosome formation using Apg5-deficient mouse
embryonic stem cells. J Cell Biol. 152:657–668. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xu HD, Wu D, Gu JH, Ge JB, Wu JC, Han R,
Liang ZQ and Qin ZH: The pro-survival role of autophagy depends on
Bcl-2 under nutrition stress conditions. PLoS One. 8:e632322013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kapoor GS and O'Rourke DM: Mitogenic
signaling cascades in glial tumors. Neurosurgery. 52:1425–1435.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lefranc F, Brotchi J and Kiss R: Possible
future issues in the treatment of glioblastomas: Special emphasis
on cell migration and the resistance of migrating glioblastoma
cells to apoptosis. J Clin Oncol. 23:2411–2422. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mizushima N, Ohsumi Y and Yoshimori T:
Autophagosome formation in mammalian cells. Cell Struct Funct.
27:421–429. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Levine B, Mizushima N and Virgin HW:
Autophagy in immunity and inflammation. Nature. 469:323–335. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Feng Y, Ke C, Tang Q, Dong H, Zheng X, Lin
W2, Ke J, Huang J, Yeung SC and Zhang H: Metformin promotes
autophagy and apoptosis in esophageal squamous cell carcinoma by
downregulating Stat3 signaling. Cell Death Dis. 5:e10882014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu YQ, Cheng X, Guo LX, Mao C, Chen YJ,
Liu HX, Xiao QC, Jiang S, Yao ZJ and Zhou GB: Identification of an
annonaceous acetogenin mimetic, AA005, as an AMPK activator and
autophagy inducer in colon cancer cells. PLoS One. 7:e470492012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Miracco C, Cosci E, Oliveri G, Luzi P,
Pacenti L, Monciatti I, Mannucci S, De Nisi MC, Toscano M,
Malagnino V, et al: Protein and mRNA expression of autophagy gene
Beclin 1 in human brain tumours. Int J Oncol. 30:429–436.
2007.PubMed/NCBI
|
|
38
|
Huang X, Bai HM, Chen L, Li B and Lu YC:
Reduced expression of LC3B-II and Beclin 1 in glioblastoma
multiforme indicates a down-regulated autophagic capacity that
relates to the progression of astrocytic tumors. J Clin Neurosci.
17:1515–1519. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Aoki H, Kondo Y, Aldape K, Yamamoto A,
Iwado E, Yokoyama T, Hollingsworth EF, Kobayashi R, Hess K,
Shinojima N, et al: Monitoring autophagy in glioblastoma with
antibody against isoform B of human microtubule-associated protein
1 light chain 3. Autophagy. 4:467–475. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jain MV, Paczulla AM, Klonisch T, Dimgba
FN, Rao SB, Roberg K, Schweizer F, Lengerke C, Davoodpour P, et al:
Interconnections between apoptotic, autophagic and necrotic
pathways: Implications for cancer therapy development. J Cell Mol
Med. 17:12–29. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rubinsztein DC, Shpilka T and Elazar Z:
Mechanisms of autophagosome biogenesis. Curr Biol. 22:29–34. 2012.
View Article : Google Scholar
|
|
42
|
Monastyrska I and Klionsky DJ: Autophagy
in organelle homeostasis: peroxisome turnover. Mol Aspects Med.
27:483–494. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Audo I, Darjatmoko SR, Schlamp CL, Lokken
JM, Lindstrom MJ, Albert DM and Nickells RW: Vitamin D analogues
increase p53, p21, and apoptosis in a xenograft model of human
retinoblastoma. Invest Ophthalmol Vis Sci. 44:4192–4199. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bernardi RJ, Johnson CS, Modzelewski RA
and Trump DL: Antiproliferative effects of
1alpha,25-dihydroxyvitamin D(3) and vitamin D analogs on
tumor-derived endothelial cells. Endocrinology. 143:2508–2514.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Alvarez-Díaz S, Valle N, García JM, Peña
C, Freije JM, Quesada V, Astudillo A, Bonilla F, López-Otín C and
Muñoz A: Cystatin D is a candidate tumor suppressor gene induced by
vitamin D in human colon cancer cells. J Clin Invest.
119:2343–2358. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gorski SM, Chittaranjan S, Pleasance ED,
Freeman JD, Anderson CL, Varhol RJ, Coughlin SM, Zuyderduyn SD,
Jones SJ and Marra MA: A SAGE approach to discovery of genes
involved in autophagic cell death. Curr Biol. 13:358–363. 2003.
View Article : Google Scholar : PubMed/NCBI
|