Introduction
Osteoporosis is a skeletal disease characterized by
the loss of bone mass and degeneration of bone microstructure,
which results in an increased risk of fracture (1,2). Various
physiological or pathological factors, such as menopause, age,
sustained glucocorticoid therapy and endocrine disorders,
contribute to osteoporosis (2–4). Over
half of women aged >50 years old will suffer from osteoporosis
or osteoporotic fracture during their lifetime (5). Previous evidence has demonstrated that
food intake and glucose metabolism are directly linked to bone
remodeling (6), and they act via
regulating the systemic release of the osteoblast-specific protein
osteocalcin (7). This link is also
suggested by the close association between obesity, osteoporosis
and diabetes (8,9). However, the pathogenesis of
osteoporosis requires clarification.
Oxidative stress (OS) has been recognized to serve a
role in the pathogenesis of various diseases, including
cerebrovascular disorders (10),
diabetes mellitus (11) and
cardiovascular disease (12,13). Studies have indicated that the
formation and reabsorption of dynamic bone tissue are also affected
by OS and/or antioxidant enzyme deficiency (14–17). OS
is a biochemical disequilibrium promoted by excessive production of
free radicals and reactive oxygen species (ROS), including hydrogen
peroxide (H2O2) and the superoxide anion
(O2−), which are produced from an endogenous
oxidative metabolism, including the mitochondria, peroxisomes and
inflammatory cell activation (18),
in addition to exogenous sources. Excessive ROS are a major cause
of cellular damage and death in a variety of pathological
conditions (19,20), acting by regulating the processes
involved in the mitochondrial dysfunction and the promotion of
apoptosis. An increased generation of ROS induces cytochrome
c release from mitochondria (21) and promotes apoptosis (22). However, the role of ROS-induced
mitochondrial dysfunction and apoptosis in osteoporosis remains
unclear.
Multiple natural products and drugs with antioxidant
properties have been investigated for their therapeutic use and
prevention of OS injury. In particular, proanthocyanidins from
grape seeds inhibit H2O2-induced apoptosis in
MC3T3-E1 cells by ameliorating mitochondrial dysfunction (23). The present study used allicin, a
natural ingredient that is derived from garlic (24). Allicin was reported to exert multiple
biological antimicrobial, immune-modulatory and anti-cancer effects
(25). Furthermore, allicin also
exerts antioxidant effects at the physiological level (25), particularly against OS in
cardiovascular diseases that are closely associated with oxidative
stress (26,27). Recently, it was indicated to regulate
the pro-inflammatory cytokines in postmenopausal osteoporotic women
(28), implying a possible and
bright prospect for an osteoporosis treatment.
The present study investigated the protective
effects of allicin against H2O2-induced
mitochondrial dysfunction and apoptosis in the osteoblast-like
MC3T3-E1 cells, which are commonly used to study osteogenic
development.
Materials and methods
Reagents, cell culture and
treatment
Allicin (98% purity; Shaanxi Ciyuan Biotech Co.,
Ltd., Xi'an, China) was dissolved in deionized water. The same
batch of allicin was used for all of the experiments. Minimum
essential medium (MEM)-α and fetal bovine serum (FBS) were
purchased from Gibco (Thermo Fisher Scientific, Inc., Waltham, MA,
USA). H2O2 was purchased from Sigma-Aldrich
(St. Louis, MO, USA). Murine osteoblastic MC3T3-E1 cells were
purchased from American Type Culture Collection (Manassas, VA, USA)
and cultured at 37°C under 5% CO2 in MEM-α supplemented
with 10% FBS (or 2% FBS for cell maintenance) and 1%
penicillin/streptomycin (CSPC Pharmaceutical Group Limited,
Shijiazhuang, China). The cells were treated with 200 µM
H2O2 for 6 h in order to induce OS, and
allicin treatment was performed with a concentration of 10, 30 or
100 µg/ml for 24 h.
Cell viability and apoptosis
assay
The viabilities of the MC3T3-E1 cells were evaluated
by MTT assay, subsequent to the various treatments. Briefly,
MC3T3-E1 cells were seeded in 96-well plates, cultured to ~85%
confluence and were then treated with 200 µM
H2O2 for 6 h with or without allicin
treatment at 10, 30 or 100 µg/ml for 24 h at 37°C. Next, MTT
solution was added to each well and the plates were incubated for
an additional 2 h at 37°C. Subsequent to the removal of the
solutions from the well, dimethyl sulfoxide (Thermo Fisher
Scientific, Inc.) was added to dissolve formazan products. Finally,
the absorbance of each well was recorded on a microplate
spectrophotometer (PowerWave XS2; BioTek Instruments, Inc.,
Winooski, VT, USA) at 570 nm.
Apoptosis of MC3T3-E1 cells subsequent to treatment
was examined with an Annexin V-FITC Apoptosis Detection kit
(Sigma-Aldrich). In brief, 5×105 cells were collected
subsequent to treatments and were stained with annexin V-FITC and
propidium iodide at 25°C for 5–15 min (placed in the dark).
Apoptotic cells were then detected using a flow cytometer (BRYTE
HS; Bio-Rad Laboratories, Inc., Hercules, CA, USA) and were
quantified by calculating the percentage of apoptotic cells
relative to the total number of cells using FlowJo 7.6 (Tree Star,
Inc., Ashland, OR, USA).
Protein sample isolation and western
blot analysis
Following treatment with 200 µM
H2O2 for 6 h with or without 10, 30 or 100
µg/ml allicin for 24 h, MC3T3-E1 cells were collected and lyzed
with an ice-cold cell lysis reagent (FNN0091; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions, and
were then supplemented with protease inhibitors (04693116001; Roche
Diagnostics, GmbH, Mannheim, Germany). The protein concentration of
each sample was measured using the Pierce bicinchoninic acid assay
reagent (Thermo Fisher Scientific, Inc., Rockford, IL, USA).
Samples were separated using 8–12% SDS-PAGE gel, with a sample
volume of 25 µg each, on a protein electrophoresis apparatus
(Mini-PROTEAN Tetra Cell; Bio-Rad Laboratories, Inc.) at 80 V for
25 min and 120 V for 75 min. Next, the separated proteins were
transferred to a polyvinylidene difluoride membranes (EMD
Millipore, Bedford, MA, USA) and were blocked with 2% bovine serum
albumin (BioVision, Mountain View, CA, USA) at 4°C overnight.
Membranes were inoculated with rabbit polyclonal antibody against
caspase 3 (cat. no. 9662; 1:400; Cell Signaling Technology, Inc.,
Danvers, MA, USA) and poly adenosine diphosphate-ribose polymerase
(PARP; cat. no. 5625, 1:800; Cell Signaling Technology, Inc.),
cytochrome c (ab90529, 1:600; Abcam, Cambridge, UK), Akt and
caspase 9 (C7729, 1:300; Sigma-Aldrich), cAMP response
element-binding protein (CREB) with (sc-101663; 1:600) or without
S133 phosphorylation (sc-25785; 1:800), extracellular
signal-regulated kinases (ERKs) with (sc-101761; 1:600) or without
T204 phosphorylation (sc-292838; 1:800; Santa Cruz Biotechnology,
Inc.) or to tubulin (13918-T16-100; 1:1,000; Sino Biological, Inc.,
Beijing, China) at 4°C overnight. A goat anti-rabbit IgG conjugated
to horseradish peroxidase secondary antibody was used (A27036;
1:600; Thermo Fisher Scientific, Inc.) and a Novex ECL
Chemiluminescent Substrate Reagent Kit (WP20005; Thermo Fisher
Scientific, Inc.) was used to detect the specific binding of each
target protein to its antibody. The results were expressed as
relative protein levels to tubulin.
Determination of complex IV activity
and mitochondrial membrane potential
Complex IV activity was measured with a Complex IV
Rodent Enzyme Activity Microplate Assay kit (Abcam) according to
the manufacturer's instructions. Briefly, MC3T3-E1 cell samples
underwent detergent extraction with 1:10 volume detergent followed
by 12,000 × g centrifugation for 20 min to extract the
supernatants. Supernatant samples were quantified to an optimal
concentration of 5 mg/ml, and were serially diluted using 1X
phosphate-buffered saline. Samples were then loaded onto plates
(100 µl per well), with a positive control sample and buffer
control included as null reference. This step was performed to
enable the binding of complex IV to the bound monoclonal antibody,
included in the kit, which immobilized the enzyme in the wells.
Following a 3-h incubation at 25°C, each well was rinsed twice with
solution 1 (100 µl per well), then assay solution was added (100 µl
per well). Finally, the reaction was measured for optical density
at 550 nm absorbance at 1–5 min intervals for 2 h at 30°C using a
spectrophotometer (PowerWave XS2). The results were expressed as a
percentage of the complex IV activity of the control MC3T3-E1
cells.
A tetramethylrhodamine, ethyl ester (TMRE)
Mitochondrial Membrane Potential Assay kit (Abcam) was used
according to the manufacturer's instructions. TMRE is a
cell-permeant, positively-charged, red-orange dye that readily
accumulates in active mitochondria due to their relative negative
charge. Depolarized or inactive mitochondria have a decreased
membrane potential and fail to sequester TMRE. A total of
1×105 MC3T3-E1 cells per treatment group were incubated
with the mitochondrial membrane potential (MMP)-sensitive
fluorescent TMRE for 20 min at 37°C, 5% CO2 (1,000 nM
FCCP was added to the positive control cells 10 min prior to TMRE
addition). Following incubation, the cells were trypsinized (0.25%;
Ameresco, Inc., Framingham, MA, USA), centrifuged and cell pellets
were resuspended in 0.4 ml of Dulbecco's phosphate-buffered saline
(Sigma-Aldrich) with 0.2% bovine serum albumin (BioVision) and
analyzed for TMRE fluorescence by flow cytometry using FlowJo 7.6,
with an excitation/emission fluorescence for TMRE of 549/575
nm.
Measurement of ROS and mitochondrial
superoxide
The intracellular level of ROS was quantified by the
oxidation of the ROS-sensitive fluorophore
dichloro-dihydro-fluorescein diacetate (Thermo Fisher Scientific,
Inc., Waltham, MA, USA) according to the manufacturer's
instructions. In brief, confluent MC3T3-E1 cells in 6-well plates
(5×105 cells/well) were loaded with a 5 µM probe
(5-chloromethyl-2,7-dichlorodihydrofluorescein diacetate) in Hanks'
balanced salt solution (HBSS; Thermo Fisher Scientific, Inc.) and
were incubated at 37°C and 5% CO2 for 30 min. The cells
were rinsed with HBSS subsequent to the removal of the probe and
the 2,7-dichlorofluorescein fluorescence was measured using a
luminescence spectrometer (2390–0000; PerkinElmer, Inc., Waltham,
MA, USA) with an excitation source at 488 nm and an emission at 530
nm. The mitochondrial superoxide levels were detected using a
Molecular Probes MitoSOX Red Mitochondrial Superoxide Indicator
(Thermo Fisher Scientific, Inc.), which is a fluorogenic dye
(excitation/emission = 510/580 nm) for highly selective detection
of superoxide in the mitochondria of cells. Subsequent to the
different treatments, cells were incubated with 5 µM MitoSOX Red at
37°C for 20 min and then the MitoSOX Red fluorescence was detected
following removal and washing of the cells.
Measurement of phosphoinositide
3-kinase (PI3K) and Akt activities, and phosphorylated CREB
PI3K activity in lysates of MC3T3-E1 cells following
the various treatments was evaluated using a PI3-Kinase Activity
ELISA kit (Echelon Biosciences, Inc., Salt Lake City, UT, USA). The
assay was a competitive ELISA in which the signal is inversely
proportional to the amount of phosphatidylinositol (3,4,5) trisphosphate (PIP3) produced.
Subsequent to the completion of the PI3K reactions, the reaction
products were first mixed and incubated with a PIP3
antibody and then added to a PIP3-coated microplate for
competitive binding. A peroxidase-linked secondary antibody and
colorimetric detection were used to detect the PIP3
detector binding to the plate. The colorimetric signal is inversely
proportional to the amount of PIP3 produced by PI3K.
Statistical analysis
All the experiments were conducted in triplicate and
the results are expressed as the mean ± standard error of at least
three independent experiments. The statistical significance was
determined by analysis of variance and subsequently applying
Student's t-test. P<0.05 was considered to indicate a
statistically significant difference.
Results
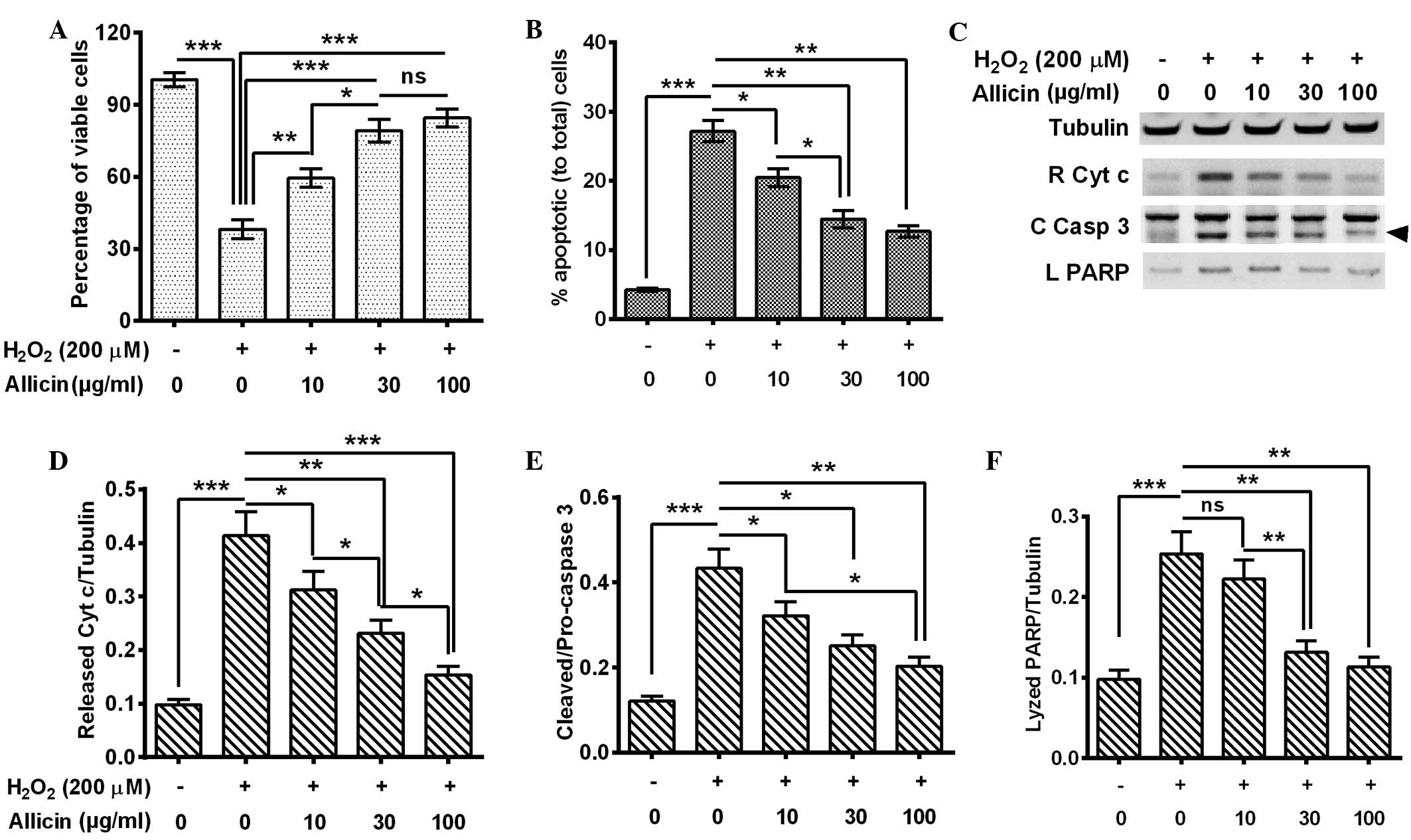
Allicin inhibits the
H2O2-promoted apoptosis in MC3T3-E1
osteoblast cells
In order to investigate the anti-OS effects of
allicin in MC3T3-E1 cells, they were exposed to
H2O2 to mimic ROS elevation, and the effects
of the exposure on cell viability and apoptosis were evaluated. The
MC3T3-E1 cells were treated with 200 µM H2O2
for 6 h and the cell viability was examined by MTT assay. The
results indicated that the cell viability was significantly reduced
by H2O2 treatment for 6 h (P=0.00082;
Fig. 1A). Next, the apoptosis levels
were examined by flow cytometry of the MC3T3-E1 cells following 200
µM H2O2 treatment for 6 h, and consistent
with the cell viability results, Fig.
1B indicated that there was a significantly higher percentage
of apoptotic cells in the 200 µM H2O2-treated
group (P=0.00034). The MC3T3-E1 cells were treated with 200 µM
H2O2 and with 0, 10, 30 or 100 µg/ml allicin
and then the cell viability and apoptosis levels were measured.
Compared with the H2O2 treatment alone, the
combined treatment of H2O2 with 10 (P=0.033),
30 or 100 µg/ml (P=0.00090) allicin significantly ameliorated the
cell viability. This effect was increased in the 30 µg/ml group
compared with the 10 µg/ml group (P=0.042; Fig. 1A). Furthermore, a combined treatment
of H2O2 with allicin demonstrated that
H2O2-induced apoptosis was significantly
inhibited by the treatment of 10 (P=0.040), 30 P=0.0076) or 100
µg/ml (P=0.0065) allicin and the inhibition was significantly
increased in the 30 µg/ml group compared with the 10 µg/ml group
(P=0.022) (Fig. 1B).
In order to confirm the effect of allicin on
H2O2-induced apoptosis in MC3T3-E1 cells,
cytochrome c release and caspase 3 activation (catalyzed
from pro-caspase 3 into 17- and 12-kDa subunits) were measured in
the H2O2-treated cells by western blot assay.
It was demonstrated that the 200 µM H2O2
treatment for 6 h promoted increased levels of cytochrome c
release P=0.00041), activated caspase-3 (P=0.00037) and lyzed PARP
(P=0.00083) by activated caspase-3 (Fig.
1C–F). However, allicin treatment with 10, 30 and 100 µg/ml
partially reversed the effect of H2O2
treatment and led to reduced cytochrome c release (P=0.041,
P=0.0085 and P=0.00047, respectively; Fig. 1D), caspase 3 activation (P=0.040,
P=0.028 and P=0.0066, respectively; Fig.
1E) and PARP lysis (P>0.05, P=0.0080 and P=0.0043,
respectively; Fig. 1F). These
effects were also observed to increase in a dose-dependent
manner.
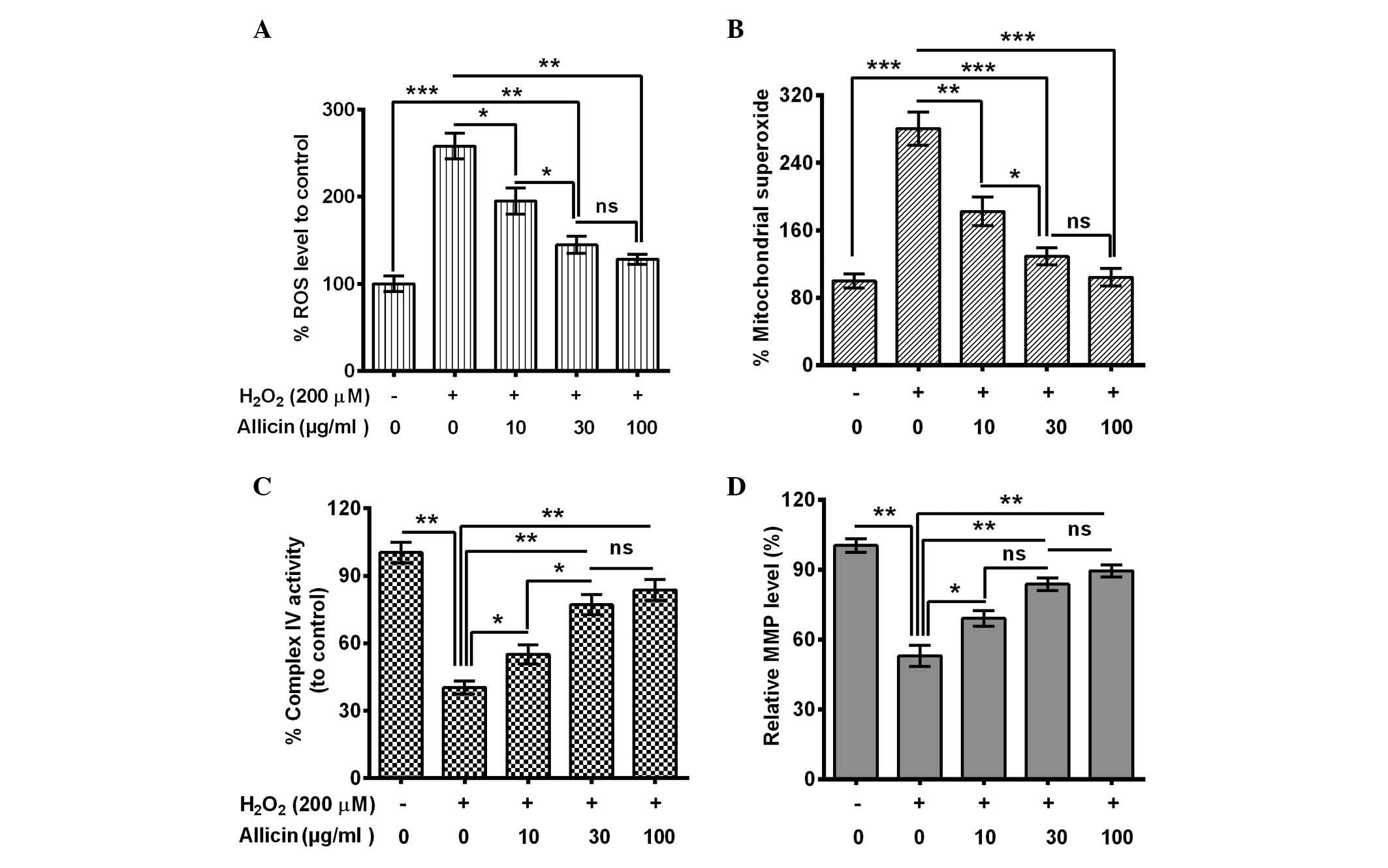
Allicin ameliorates mitochondrial
dysfunction in H2O2-treated MC3T3-E1
cells
The influence of allicin on the
H2O2-induced osteoblast mitochondrial
dysfunction was also evaluated. ROS generation in MC3T3-E1 cells
with or without the treatment of 200 µM H2O2
was assessed. A significantly elevated ROS release from
H2O2-treated cells was indicated (P=0.00093;
Fig. 2A). However, this increase was
blocked by 10 (P=0.038), 30 (P=0.0086) or 100 µg/ml (P=0.0075)
allicin, and the 30 µg/ml group was significantly reduced compared
with the 10 µg/ml group (P=0.048). Secondly, mitochondrial
superoxide was measured using MitoSOX Red, a live-cell-permeable
and mitochondrial-localizing superoxide indicator that further
assessed the effect of allicin on mitochondrial superoxide
production. It was demonstrated that H2O2
treatment enhanced mitochondrial MitoSOX Red fluorescence
(P=0.00071; Fig. 2B) whereas
treatment with allicin with a concentration of 10 (P=0.0094), 30
(P=0.00087) and 100 µg/ml (P=0.00092) substantially inhibited the
stimulatory effect of H2O2 to the
mitochondrial superoxide production (Fig. 2B).
In addition, the activity of the mitochondrial
respiratory chain complex IV and mitochondrial membrane potential
was examined. The complex IV activity decreased dramatically in the
H2O2-treated cells (P=0.0032; Fig. 2C). However, allicin significantly
ameliorated this effect (10 µg/ml, P=0.044; 30 µg/ml, P=0.0063; or
100 µg/ml, P=0.0053; Fig. 2C) and
the activity was significantly higher in the 30 µg/ml group
compared with the 10 µg/ml group (P=0.028; Fig. 2C). MMP, as a marker of mitochondrial
oxidative phosphorylation activity, is a well-established indicator
of mitochondrial function. As TMRE is a cell-permeable,
positively-charged, red-orange dye that readily accumulates in
active mitochondria due to their relative negative charge, the
mitochondrial function was assessed by examining the TMRE
accumulation in mitochondria. TMRE accumulation was significantly
reduced by the H2O2 treatment (P=0.0037), and
it was reversed by 10, 30 or 100 µg/ml allicin (Fig. 2D). In conclusion, the results of the
present study confirmed that allicin may protect from mitochondrial
damage in MC3T3-E1 cells following OS.
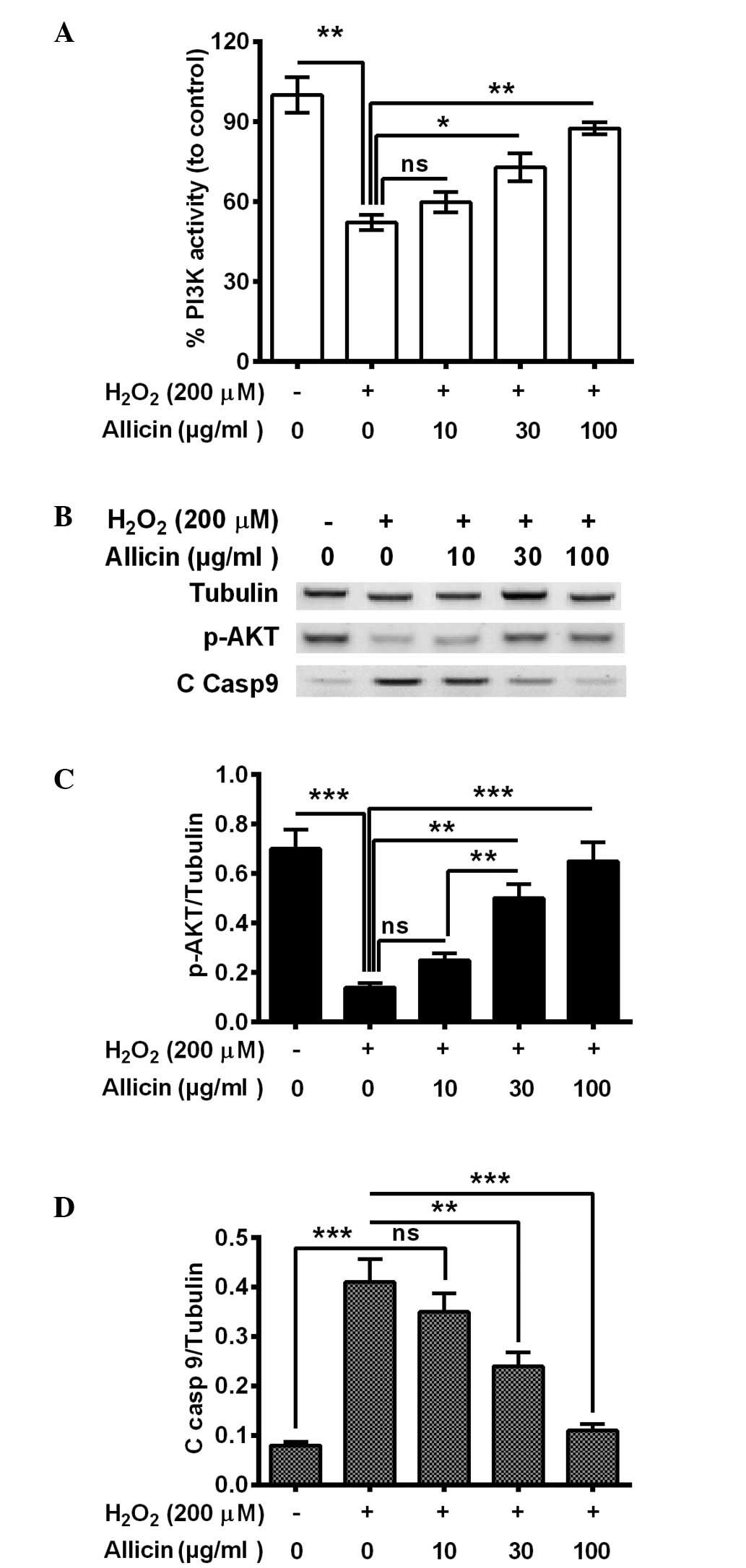
Allicin ameliorates the reduction of
PI3K/AKT activity in osteoblasts by H2O2
The PI3K/AKT pathway is important in cell survival,
particularly against OS (29,30). To
further deduce the mechanism of the cytoprotective effect of
allicin, the present study investigated whether PI3K/AKT activation
was implicated in the cytoprotective effect of allicin in
osteoblastic MC3T3-E1 cells. It was demonstrated that PI3K activity
was significantly decreased in the MC3T3-E1 cells subsequent to
H2O2 treatment (P=0.0036; Fig. 3A). The treatment with 30 (P=0.024) or
100 µg/ml (P=0.0035) allicin markedly ameliorated this effect
(Fig. 3A). Furthermore, western
blotting indicated that phosphorylated (p)-AKT was also
significantly downregulated (P=0.00037), whereas the cleaved
caspase 9 (C Casp 9) was markedly promoted (P=0.00040) by
H2O2 treatment (Fig. 3B–D). Notably, 30 and 100 µg/ml
allicin significantly increased the level of p-AKT (P=0.0057 and
P=0.00048, respectively; Fig. 3C)
and reduced levels of C casp 9 (P=0.0063 and 0.00056, respectively;
Fig. 3C) compared with the
H2O2-only group. In summary, allicin was
suggested to ameliorate the H2O2-induced
PI3K/AKT pro-survival signal inhibition in osteoblasts by
H2O2.
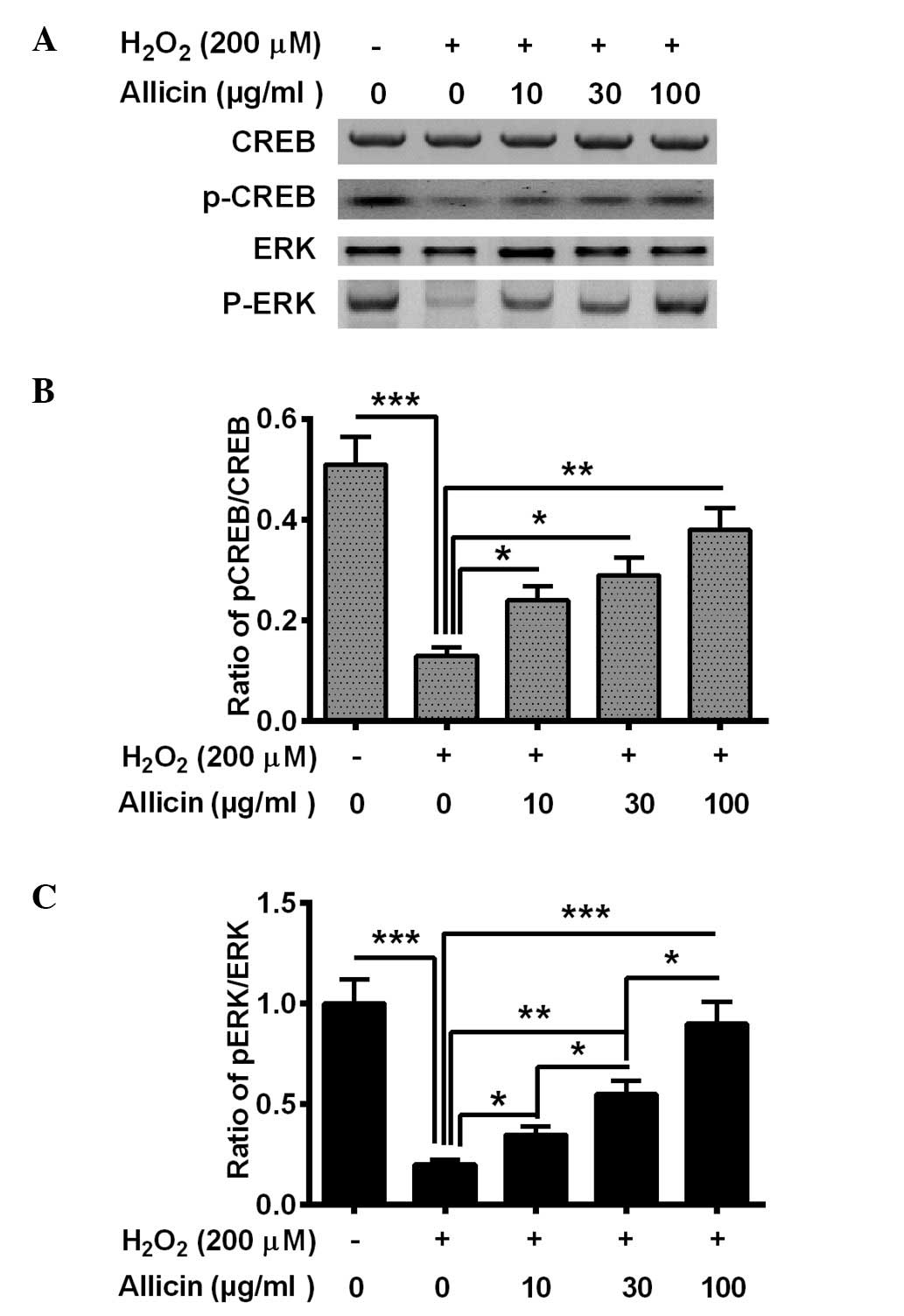
Allicin ameliorates the reduction of
CREB/ERK signaling in osteoblasts by
H2O2
CREB/ERK signaling was also demonstrated to be
involved in OS (31). In the present
study, the activation of CREB/ERK signaling was also demonstrated,
and p-CREB and p-ERK levels were altered in the MC3T3-E1 cells
following the treatment with H2O2 and
allicin. The H2O2 treatment significantly
reduced the level of p-CREB (P=0.00071) and p-ERK (P=0.00033) in
the MC3T3-E1 cells (Fig. 4). Allicin
treatment with 10, 30 or 100 µg/ml markedly increased p-CREB
(P=0.028, P=0.015 and P=0.0022, respectively; Fig. 4B) and p-ERK (P=0.036, P=0.0035 and
P=0.00045, respectively; Fig. 4C)
levels in MC3T3-E1 cells compared with the
H2O2-only group. In summary, it was suggested
that allicin ameliorates the H2O2-induced
CREB/ERK pro-survival signaling inhibition in osteoblasts via
H2O2.
Discussion
Mitochondria are the key targets for ROS, which have
been suggested to regulate the processes involved in mitochondrial
dysfunction and the promotion of apoptosis. Increased generation of
ROS induces cytochrome c release from mitochondria (21). In a previous study, the mimicking of
ROS elevation by exposure to H2O2 resulted in
the release of cytochrome c, which promoted apoptosis
(22). ROS has also been
demonstrated to cause structural and functional damage to
mitochondria (32). The elevated ROS
production causes mitochondrial damage, such as collapse of the
mitochondrial membrane potential and complex IV inactivation, which
opens up the mitochondrial permeability transition pores and leads
to the release of proapoptotic proteins into the cytoplasm
(33,34).
In the present study, allicin was indicated to
ameliorate the viability of cells by decreasing and inhibiting
apoptosis in MC3T3-E1 cells. In the present study, apoptosis was
induced by manipulating ROS elevation through exposure of MC3T3-E1
cells to H2O2. The inhibitory effect of
allicin was confirmed by the inhibition of the
H2O2-induced cytochrome c release and
the activation of caspase-3 after treatment. Additionally,
apoptosis inhibition was hypothesized to be associated with the
counteraction to H2O2-induced mitochondrial
dysfunction. Allicin treatment inhibited the
H2O2-induced ROS generation and mitochondrial
superoxide activation, whereas it ameliorated the
H2O2-induced reduction of complex IV activity
and mitochondrial membrane potential. Furthermore, the
counteraction of H2O2-induced mitochondrial
dysfunction was also verified by the inhibition to
H2O2-induced nitrotyrosine production and the
amelioration of the H2O2-induced thioredoxin
reductase activity decreasing. In addition, allicin improved the
pro-survival signal inhibition in osteoblasts via
H2O2. However, it remains unclear how allicin
reduces the H2O2-induced ROS generation. It
is considered that the protective effect of allicin on H9c2 cells
may inhibit intracellular ROS production (35). Therefore, the present study
speculated that the inhibition of ROS production may be a key
mechanism underlying the reduction of H2O2
toxicity by allicin. PI3K and CREB are established to function as
pro-survival signals (36–40). In the present study, PI3K activity
and CREB phosphorylation were reduced in MC3T3-E1 cells after
H2O2 treatment, whereas allicin treatment
prevented the reduction of PI3K activity and CREB
phosphorylation.
OS has been indicated to be involved in the
pathogenesis of osteoporosis. It is conceivable that
mitochondrial-mediated ROS generation under OS leads to damage to
ROS-produced cells or peripheral osteoblasts. As major sources of
ROS production, mitochondria also are major targets for ROS. The
present study demonstrated that mimicking OS by
H2O2 exposure significantly exerted
mitochondrial dysfunction in osteoblastic MC3T3-E1 cells. The
anti-OS effect exerted by allicin has been previously identified in
ischemic/reperfused rats by inhibiting the activation of c-Jun and
c-Jun N-terminal kinase, and thus inhibiting apoptosis further
(41). However, the present study
indicated that allicin inhibited ROS generation and ameliorated
ROS-induced mitochondrial disfunction. In addition, the allicin
treatment also ameliorated the repressed PI3K/AKT and CREB/ERK
signaling by H2O2 that may also be associated
with the anti-OS effect of allicin. Previously, PI3K and CREB were
demonstrated to be involved in osteoblast-like cell proliferation
and differentiation (42,43). In the present study, allicin
treatment on MC3T3-E1 cells led to increased PI3K activity,
phosphorylation of AKT, CREB and ERK. PI3K is a lipid kinase and
generates PIP3, which directly or indirectly affects
CREB phosphorylation (44). CREB is
a ubiquitous transcription factor in the higher eukaryotes that,
once phosphorylated, promotes the synthesis of mitochondrially
encoded subunits of oxidative phosphorylation complexes (45). By contrast, since the PI3K/AKT and
CREB/ERK signaling pathways were suppressed by
H2O2, the attenuation of the repressed PI3K
and CREB by allicin may be caused by reduced
H2O2 toxicity. In addition, the attenuation
of the repressed PI3K and CREB may ameliorate the mitochondrial
function via regulating the expression of B-cell lymphoma-2 and
Bcl-extra large (46), which are
located in the outer membrane of the mitochondria and regulate
mitochondrial function (47).
To conclude, allicin protects osteoblasts from
H2O2-induced OS and apoptosis in osteoblastic
MC3T3-E1 cells by improving mitochondrial function and by
activation of the PI3K/AKT and CREB/ERK signaling pathways. The
present study implies a promising role for allicin in OS-associated
osteoporosis.
References
|
1
|
Graat-Verboom L, Wouters EF, Smeenk FW,
van den Borne BE, Lunde R and Spruit MA: Current status of research
on osteoporosis in COPD: A systematic review. Eur Respir J.
34:209–218. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhao Y, Liu Y and Zheng Y: Osteoporosis
and related factors in older females with skeletal pain or
numbness: A retrospective study in East China. J Int Med Res.
41:859–866. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Abe E, Sun L, Mechanick J, Iqbal J, Yamoah
K, Baliram R, Arabi A, Moonga BS, Davies TF and Zaidi M: Bone loss
in thyroid disease: Role of low TSH and high thyroid hormone. Ann
NY Acad Sci. 1116:383–391. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Compston JE: Risk factors for
osteoporosis. Clin Endocrinol (Oxf). 36:223–224. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chrischilles EA, Butler CD, Davis CS and
Wallace RB: A model of lifetime osteoporosis impact. Arch Intern
Med. 151:2026–2032. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shah M, Kola B, Bataveljic A, Arnett TR,
Viollet B, Saxon L, Korbonits M and Chenu C: AMP-activated protein
kinase (AMPK) activation regulates in vitro bone formation and bone
mass. Bone. 47:309–319. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD,
Confavreux C, Dacquin R, Mee PJ, McKee MD, Jung DY, et al:
Endocrine regulation of energy metabolism by the skeleton. Cell.
130:456–469. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pei L and Tontonoz P: Fat's loss is bone's
gain. J Clin Invest. 113:805–806. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rosen CJ and Bouxsein ML: Mechanisms of
disease: Is osteoporosis the obesity of bone? Nat Clin Pract
Rheumatol. 2:35–43. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Slemmer JE, Shacka JJ, Sweeney MI and
Weber JT: Antioxidants and free radical scavengers for the
treatment of stroke, traumatic brain injury and aging. Curr Med
Chem. 15:404–414. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lopes JP, Oliveira SM and Soares FJ:
Oxidative stress and its effects on insulin resistance and
pancreatic beta-cells dysfunction: Relationship with type 2
diabetes mellitus complications. Acta Med Port. 21:293–302.
2008.(In Portuguese). PubMed/NCBI
|
|
12
|
Kingwell BA: Nitric oxide-mediated
metabolic regulation during exercise: Effects of training in health
and cardiovascular disease. Faseb J. 14:1685–1696. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Maharjan BR, Jha JC, Adhikari D Akila,
Risal S, Alurkar VM and Singh PP: Oxidative stress, antioxidant
status and lipid profile in ischemic heart disease patients from
western region of Nepal. Nepal Med Coll J. 10:20–24.
2008.PubMed/NCBI
|
|
14
|
Sendur OF, Turan Y, Tastaban E and Serter
M: Antioxidant status in patients with osteoporosis: A controlled
study. Joint Bone Spine. 76:514–518. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sánchez-Rodríguez MA, Ruiz-Ramos M,
Correa-Muñoz E and Mendoza-Núñez VM: Oxidative stress as a risk
factor for osteoporosis in elderly Mexicans as characterized by
antioxidant enzymes. BMC Musculoskelet Disord. 8:1242007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ozgocmen S, Kaya H, Fadillioglu E, Aydogan
R and Yilmaz Z: Role of antioxidant systems, lipid peroxidation,
and nitric oxide in postmenopausal osteoporosis. Mol Cell Biochem.
295:45–52. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Basu S, Michaëlsson K, Olofsson H,
Johansson S and Melhus H: Association between oxidative stress and
bone mineral density. Biochem Biophys Res Commun. 288:275–279.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Steinberg GR and Kemp BE: AMPK in Health
and Disease. Physiol Rev. 89:1025–1078. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Morgan MJ and Liu ZG: Reactive oxygen
species in TNFalpha-induced signaling and cell death. Mol Cells.
30:1–12. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Freitas I, Griffini P, Bertone V, Bertone
R, Fenoglio C, Milliery R and Vairetti M: In situ detection of
reactive oxygen species and nitric oxide production in normal and
pathological tissues: Improvement by differential interference
contrast. Exp Gerontol. 37:591–602. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tan S, Sagara Y, Liu Y, Maher P and
Schubert D: The regulation of reactive oxygen species production
during programmed cell death. J Cell Biol. 141:1423–1432. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kirkland RA, Windelborn JA, Kasprzak JM
and Franklin JL: A Bax-induced pro-oxidant state is critical for
cytochrome c release during programmed neuronal death. J Neurosci.
22:6480–6490. 2002.PubMed/NCBI
|
|
23
|
Zhang Z, Zheng L, Zhao Z, Shi J, Wang X
and Huang J: Grape seed proanthocyanidins inhibit
H2O2-induced osteoblastic MC3T3-E1 cell
apoptosis via ameliorating H2O2-induced mitochondrial dysfunction.
J Toxicol Sci. 39:803–813. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chan SW: Panax ginseng, Rhodiola
rosea and Schisandra chinensis. Int J Food Sci Nutr.
63(Suppl 1): S75–S81. 2012. View Article : Google Scholar
|
|
25
|
Borlinghaus J, Albrecht F, Gruhlke MC,
Nwachukwu ID and Slusarenko AJ: Allicin: Chemistry and biological
properties. Molecules. 19:12591–12618. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kita T, Kume N, Minami M, Hayashida K,
Murayama T, Sano H, Moriwaki H, Kataoka H, Nishi E, Horiuchi H, et
al: Role of oxidized LDL in atherosclerosis. Ann NY Acad Sci.
947:199–206. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Arzanlou M, Bohlooli S, Jannati E and
Mirzanejad-Asl H: Allicin from garlic neutralizes the hemolytic
activity of intra- and extra-cellular pneumolysin O in vitro.
Toxicon. 57:540–545. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mozaffari-Khosravi H, Hesabgar HA, Owlia
MB, Hadinedoushan H, Barzegar K and Fllahzadeh MH: The effect of
garlic tablet on pro-inflammatory cytokines in postmenopausal
osteoporotic women: A randomized controlled clinical trial. J Diet
Suppl. 9:262–271. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Padiya R, Chowdhury D, Borkar R, Srinivas
R, Bhadra Pal M and Banerjee SK: Garlic attenuates cardiac
oxidative stress via activation of PI3K/AKT/Nrf2-Keap1 pathway in
fructose-fed diabetic rat. PLoS One. 9:e942282014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Umoh NA, Walker RK, Al-Rubaiee M, Jeffress
MA and Haddad GE: Acute alcohol modulates cardiac function as
PI3K/Akt regulates oxidative stress. Alcohol Clin Exp Res.
38:1847–1864. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang L and Jope RS: Oxidative stress
differentially modulates phosphorylation of ERK, p38 and CREB
induced by NGF or EGF in PC12 cells. Neurobiol Aging. 20:271–278.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chomyn A and Attardi G: MtDNA mutations in
aging and apoptosis. Biochem Biophys Res Commun. 304:519–529. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Balaban RS, Nemoto S and Finkel T:
Mitochondria, oxidants and aging. Cell. 120:483–495. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Choi EM: Magnolol protects osteoblastic
MC3T3-E1 cells against antimycin A-induced cytotoxicity through
activation of mitochondrial function. Inflammation. 35:1204–1212.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chan JY, Tsui HT, Chung IY, Chan RY, Kwan
YW and Chan SW: Allicin protects rat cardiomyoblasts (H9c2 cells)
from hydrogen peroxide-induced oxidative injury through inhibiting
the generation of intracellular reactive oxygen species. Int J Food
Sci Nutr. 65:868–873. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Saavedra A, García-Martínez JM, Xifró X,
Giralt A, Torres-Peraza JF, Canals JM, Díaz-Hernández M, Lucas JJ,
Alberch J and Pérez-Navarro E: PH domain leucine-rich repeat
protein phosphatase 1 contributes to maintain the activation of the
PI3K/Akt pro-survival pathway in Huntington's disease striatum.
Cell Death Differ. 17:324–335. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bhattacharya D, Singh MK and Chaudhuri S,
Acharya S, Basu AK and Chaudhuri S: T11TS impedes glioma
angiogenesis by inhibiting VEGF signaling and pro-survival
PI3K/Akt/eNOS pathway with concomitant upregulation of PTEN in
brain endothelial cells. J Neurooncol. 113:13–25. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hsu HH, Cheng LH, Ho TJ, Kuo WW, Lin YM,
Chen MC, Lee NH, Tsai FJ, Tsai KH and Huang CY: Apicidin-resistant
HA22T hepatocellular carcinoma cells massively promote pro-survival
capability via IGF-IR/PI3K/Akt signaling pathway activation. Tumour
Biol. 35:303–313. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Das S, Tosaki A, Bagchi D, Maulik N and
Das DK: Potentiation of a survival signal in the ischemic heart by
resveratrol through p38 mitogen-activated protein kinase/mitogen-
and stress-activated protein kinase 1/cAMP response element-binding
protein signaling. J Pharmacol Exp Ther. 317:980–988. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dworkin S, Malaterre J, Hollande F, Darcy
PK, Ramsay RG and Mantamadiotis T: cAMP response element binding
protein is required for mouse neural progenitor cell survival and
expansion. Stem Cells. 27:1347–1357. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sato M, Bagchi D, Tosaki A and Das DK:
Grape seed proanthocyanidin reduces cardiomyocyte apoptosis by
inhibiting ischemia/reperfusion-induced activation of JNK-1 and
C-JUN. Free Radic Biol Med. 31:729–737. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Carpio L, Gladu J, Goltzman D and Rabbani
SA: Induction of osteoblast differentiation indexes by PTHrP in
MG-63 cells involves multiple signaling pathways. Am J Physiol
Endocrinol Metab. 281:E489–E499. 2001.PubMed/NCBI
|
|
43
|
Matsuo N, Tanaka S, Gordon MK, Koch M,
Yoshioka H and Ramirez F: CREB-AP1 protein complexes regulate
transcription of the collagen XXIV gene (Col24a1) in osteoblasts. J
Biol Chem. 281:5445–5452. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
45
|
De Rasmo D, Signorile A, Roca E and Papa
S: cAMP response element-binding protein (CREB) is imported into
mitochondria and promotes protein synthesis. FEBS J. 276:4325–4333.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Boucher MJ, Morisset J, Vachon PH, Reed
JC, Lainé J and Rivard N: MEK/ERK signaling pathway regulates the
expression of Bcl-2, Bcl-X(L) and Mcl-1 and promotes survival of
human pancreatic cancer cells. J Cell Biochem. 79:355–369. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chan SL and Yu VC: Proteins of the bcl-2
family in apoptosis signalling: From mechanistic insights to
therapeutic opportunities. Clin Exp Pharmacol Physiol. 31:119–128.
2004. View Article : Google Scholar : PubMed/NCBI
|