Introduction
Cervical cancer ranks as the second most common
cause of mortality in women with malignant tumors. Each year,
~450,000 women are newly diagnosed with cervical cancer worldwide,
with 80% of cases occurring in developing countries, and with
mortality observed in ~280,000 of these cases (1). In China, ~130,000 new cases of cervical
cancer are annually diagnosed, accounting for 28% of the total
cases diagnosed worldwide (2–4).
Currently, cervical cancer is the only malignant tumor that can be
definitively diagnosed and possibly prevented. Human papillomavirus
(HPV) serves a key role in cancer development processes, and the
persistent infection of HPV is considered to be the most important
cause of cervical cancer development. In total, ~80% of women will
be infected with HPV during their lifetime; however, in the
majority of cases the body is able to clear the virus within 24
months without treatment (5).
Therefore, only a small number of women are infected persistently,
which can result in HPV infection progressing into precancerous
lesions and eventually invasive cervical cancer after 10–15 years
(6). To date, >180 subtypes of
HPV have been identified, including HPV 16 and HPV 18, which are
two high risk HPV subtypes and the main causes of cervical cancer
due to persistent infection of host cells. E6 and E7 are two genes
expressed in these high risk HPVs, and are the main oncogenes of
cervical cancer (7).
To date, HPV treatment focuses on the symptoms of
the infection. If the HPV infection causes abnormal cell changes,
there are four main treatment options: Cryotherapy, surgical
removal, laser therapy and loop electrosurgical excision procedure.
Primary prevention with HPV vaccination remains the most effective
strategy; however, vaccines may not offer protection against all
cancer-associated HPV types. Furthermore, there is still no FDA
approved anti-HPV drug listed so far but the search for potential
drug candidates against HPV strains is increasing. The current
anti-HPV drugs are predominantly oral hormonal medicines, including
acyclovir, ganciclovir, interferon and interleukin. Nevertheless, a
number of traditional Chinese medicines (including Chaihu,
Youdujing, Paiteling and Xinfuning) have been used for the
prevention and treatment of HPV.
In terms of treatment, cidofovir that is formulated
as a gel can be safe and effective for the treatment of epithelial
hyperplasia in those with human immunodeficiency virus infection
(8). Furthermore, chemotherapy is
considered as the standard treatment for patients with advanced or
recurrent cervical cancer, and cisplatin appears to treat the
disease effectively.
The present study aimed to compare the inhibitory
effects of cidofovir (CDV) on the proliferation of HPV-18 positive
HeLa cells with the effect of cisplatin (DDP), in order to evaluate
the potential application value of CDV in the prevention and
treatment of cervical HPV infection.
Materials and methods
Reagents
Dulbecco's modified Eagle's medium (DMEM) and
penicillin-streptomycin solution used in cell cultures were
purchased from Hyclone (GE Healthcare Life Sciences, Logan, UT,
USA). Newborn calf serum was purchased from Gibco (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). Nuclear and Cytoplasmic
Protein Extraction kit (163–2089) was purchased from Bio-Rad
Laboratories Inc., (Hercules, CA, USA). Mouse anti-human monoclonal
E6 antibody (ab51931) was purchased from Abcam (Cambridge, UK). In
addition, anti-mouse FITC secondary antibody (200-032-037) was
purchased from Jackson Immunoresearch Inc., (West Grove, PA, USA),
while the p53 anti-mouse (BA0521) and anti-mouse β-actin (BA2305)
antibodies were purchased from Boster Biological Technology, Ltd.,
(Wuhan, China). CDV, Goat anti-mouse IgG-HRP (sc-2005) and
anti-rabbit IgG-HRP (sc-2004) were purchased from Santa Cruz
Biotechnology Inc., (Shanghai, China). Alexa Fluor 594-conjugated
donkey anti-rabbit IgG secondary antibody (R37119) for
immunofluorescence staining was purcahsed from Thermo Fisher
Scientific, Inc. DDP and dimethyl sulfoxide (DMSO) reagents were
purchased from Sigma-Aldrich (St. Louis, MO, USA), all of which
were dissolved in 0.01 M phosphate-buffered saline (PBS) to make a
50 µM stock solution, and diluted with DMEM to the required
concentration prior to use.
HeLa cell culture
HeLa cells (gifted from Professor Liu Cong; West
China Second University Hospital) were cultured in complete DMEM
supplemented with 10% fetal bovine serum (both Gibco; Thermo Fisher
Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml streptomycin.
Cells were harvested by trypsinization and an assessment was made
of their density using a hemocytometer to a density of
1.0×105 cells/ml and 5.0×104 cells were added
to each well of a 96-well tissue culture-treated plate (Costar;
Sigma-Adrich). The cells were inoculated and then incubated at 37°C
with 5% CO2 for 14 days. The DMEM culturing medium was
preheated at 37°C in a water bath for a minimum of 30–45 mins to
ensure they were at the right temperature, and adherent cells were
washed with new pre-warmed media and aspirated to remove any traces
of the old media. The media was replaced daily. In the present
study, the following groups were used: Control group, which
included HeLa cells without any drug treatment; CDV-treated group,
in which HeLa cells were treated with an appropriate concentration
of CDV (2.5, 5, 10 and 20 µM); and DDP-treated group, in which HeLa
cells were treated with an appropriate concentration of DDP (2.5,
5, 10 and 20 µM).
Detection of HeLa cell viability by
MTT assay
HeLa cells in the logarithmic growth phase were
selected, digested with 0.25% trypsin (Gibco; Thermo Fisher
Scientific, Inc.), made into cell suspensions of 5×104
cells/ml, and transferred to 96-well plates with 100 µl cell
suspension in each well. After the cells adhered to the plates, any
solution was discarded. Cells were treated with different
concentrations of CDV (2.5, 5, 10 and 20 µM) and DDP (2.5, 5, 10
and 20 µM) in DMEM for 24, 48 or 72 h. A blank control well was
set, and each group was cultured and treated in triplicate. At
every time point, 10 µl 5 mg/ml MTT solution from an MTT cell
proliferation assay kit (11465007001; Sigma-Aldrich) was added into
each well and the cells were cultured for a further 4 h. Next, 150
µl DMSO was added to terminate the culturing. The plate was then
shaken for 10–15 min at a low speed. Subsequently, the optical
density (OD) value was identified by enzyme-linked immunosorbent
assay using the MTT cell proliferation assay kit (11465007001;
Sigma-Aldrich) and a microplate reader (Thermo Fisher Scientific,
Inc.) at a wavelength of 490 nm. In addition, the viability of HeLa
cells was calculated by the following formula: Cell viability (%) =
(OD490nm – ODblank)CDV /
(OD490nm – ODblank)DDP × 100%.
Formation of HeLa cell colonies
HeLa cells in the logarithmic growth phase were
selected, digested with 0.25% trypsin and made into single-cell
suspensions with DMEM. Next, the cells were transferred into 6-well
plates with 1 ml cell suspension per well, which contained 300
cells per well, and were incubated overnight at 37°C with 5%
CO2. Any previous solutions were discarded, and 2.5 ml
fresh complete DMEM with corresponding concentrations of drugs were
added into each well. The cells were then incubated at 37°C with 5%
CO2 for approximately 14 days. A blank control well was
set and each group was cultured and treated in triplicate. When
visible colonies formed, the culturing was terminated and 2 ml pure
methanol was added for 15 min for cell fixation. Subsequently, any
stationary solution was discarded, Giemsa stain (A0909-0010;
Applichem GmbH, Darmstadt, Germany) was added for 10–15 min, and
then the plates were slowly washed with water and dried in air.
Cell colonies containing >50 cells were counted under the
microscope (low magnification), or by Image-Pro Plus 6.0 software
(Media Cybernetics, Inc., Rockville, MD, USA). The effects of CDV
and DDP on proliferation and apoptosis in HeLa cells were
determined based on the survival rate, as follows: Colony formation
rate = (number of colonies / number of cells inoculated) × 100%;
Survival rate = (colony formation rate in the drug group / colony
formation rate in the control group) × 100%. Morphology of the HeLa
cells was observed under an inverted microscope (CKX41-A32PH;
Olympus Corp., Tokyo, Japan).
Apoptosis determined by Giemsa
staining
Sterile coverslips (174950, NUNC, Roskilde, Denmark)
were placed into 24-well plates, which were inoculated with
5×105 cells per well and incubated overnight at 37°C
with 5% CO2. Next, fresh complete DMEM containing
appropriate concentrations of drugs was added into each well. A
blank control well was set and each group was cultured and treated
in triplicate. Cells were incubated at 37°C with 5% CO2
for 48 h, and the culture medium was discarded. The cells were
gently washed twice with PBS for 2 min each time, and then 500 µl
methanol was added into each well for 3 min. Any remaining solution
was discarded, cells were washed twice with PBS, and Giemsa stain
was added to completely cover the cells on the slides for 30 min.
Following staining, the cells were washed with pure water until the
solution was colorless. Next, 1–2 drops of neutral gum (Bioworld
Technology, Inc., St. Louis Park, MN, USA) were added onto a clean
glass slide. A small corner of the coverslip was gently clamped
using ophthalmic forceps, and the coverslip containing the cells
was placed with the cells facing down. When neutral gum fully
expanded along the coverslip, any remaining neutral gum was
absorbed with absorbent paper. At the same time, the slides were
laid flat to avoid the formation of air bubbles and were stored at
room temperature until further use. The morphology of
Giemsa-stained apoptotic cells was observed under the microscope
and images were captured.
Cell cycle progression and apoptosis
determined by flow cytometry
Cells were inoculated into 6-well plates at a
density of 1×105 cells per well and incubated overnight
at 37°C with 5% CO2. Next, they were treated with 15 µM
CDV or 15 µM DDP for 48 h, digested with 0.25% trypsin solution
(without EDTA), collected by centrifugation at a speed of 200 ×
g for 2 min at 4°C, and washed twice with cold PBS.
Subsequently, 70% pre-cooled ethanol (diluted with PBS) was added
to suspend the cells, and they were stored at 4°C overnight, or at
−20°C for a longer storage period. Cells were harvested by
centrifugation at 200 × g for 10 min at 4°C, washed twice
with pre-cooled PBS, centrifuged again and collected by discarding
the supernatant. Pre-cooled PBS was added to suspend the cells and
obtain a concentration of 1×106 cells/ml. Subsequently,
the RNase A enzyme was added to a final concentration of 1 mg/ml.
The suspension were mixed and placed in a water bath at 37°C for 30
min, followed by addition of propidium iodide (GT21008;
Sigma-Aldrich) stain to a final concentration of 50 µg/ml.
Following gentle mixing, the cells were stored at 4°C to avoid
light exposure and tested by flow cytometry. The red fluorescence
at 490 nm was recorded using a microplate reader (Thermo Fisher
Scientific Inc.), and the results were analyzed by CELLQUEST MODFIT
LT computer systems (BD Biosciences, Franklin Lakes, NJ, USA)
(9).
Expression levels of E6 and p53
proteins determined by western blot analysis
The effects of CDV and DDP on the expression levels
of E6 and p53 proteins were detected by western blot analysis.
Cytoplasm and nucleus extracts were prepared using a Nuclear and
Cytoplasmic Protein Extraction kit according to the manufacturer's
instructions. Total protein was extracted from HeLa cells, and a
15% separation gel and 5% stacking gel were used to separate the
proteins. A total of 10 µl of sample were loaded onto each well,
the samples were run on the separation gel for 15 min and the
stacking gel for 80 min. Next, the gel was transferred to a
membrane, which was then blocked for more than one hour. The
following primary mouse antibodies were added to the membrane and
incubated overnight at room temperature: Anti-human E6 (1:1,000),
anti-p53 (1:1,000) and anti-β-actin (1:500). Following washing, the
goat anti-mouse and anti-rabbit FITC secondary antibodies (both
1:5,000) were added and incubated for 1 h at room temperature.
β-actin was used as a loading control. After washing with
Tris-buffered saline with Tween 20 three times, the membrane was
developed and an image was captured with Vilber Lourmat (Bio-Rad
Laboratories Inc.).
Cell immunofluorescence staining
Sterile slides were inserted into 24-well plates.
HeLa cells were plated into each well at a concentration of
5×104/ml and incubated at 37°C with 5% CO2
for 48 h. The culture media were discarded, and cells were washed
twice with PBS for 1 min each time. Next, 4% cool paraformaldehyde
was added for 10 min at room temperature. The cells were then
washed three times with PBS for 1 min each time, and blocked with
milk for 30–60 min at room temperature. Subsequently, the blocking
solution was discarded, and mouse anti-human E6 and mouse anti-p53
antibody diluted in blocking solution were added. The plates were
placed in an immunohistochemistry dampness box and then in an
incubator at 37°C for 30 min, or 4°C overnight. Cells were washed
2–3 times with PBS for 1 min each time and secondary fluorescent
antibodies diluted by milk were added, placed in the wet boxes,
followed by incubation at 37°C for 30 min. The secondary antibody
used was Alexa Fluor 594-conjugated donkey anti-rabbit IgG (1:400).
Subsequently, the cells were washed 2–3 times with PBS for 1 min
each time. Mounting solution containing
4′,6-diamidino-2-phenylindole was dropped onto the glass slides. A
tiny corner of the miniature coverslip was gently clamped by
ophthalmic forceps and placed on the glass slide with the cells
growing on the coverslip slide. Images of the cells were captured
under fluorescence microscopes and analyzed by Image-Pro Plus 6.0
software.
Statistical analyses
Data were statistically processed and analyzed by
SPSS version 17.0 (SPSS, Inc., Chicago, IL, USA). Measurement data
are presented at a format of mean ± standard deviation. Data were
analyzed by one-way analysis of variance to compare the differences
between groups, and P<0.05 was considered to demonstrate
statistically significant differences.
Results
Effect of CDV on HeLa cell viability,
determined by MTT assay
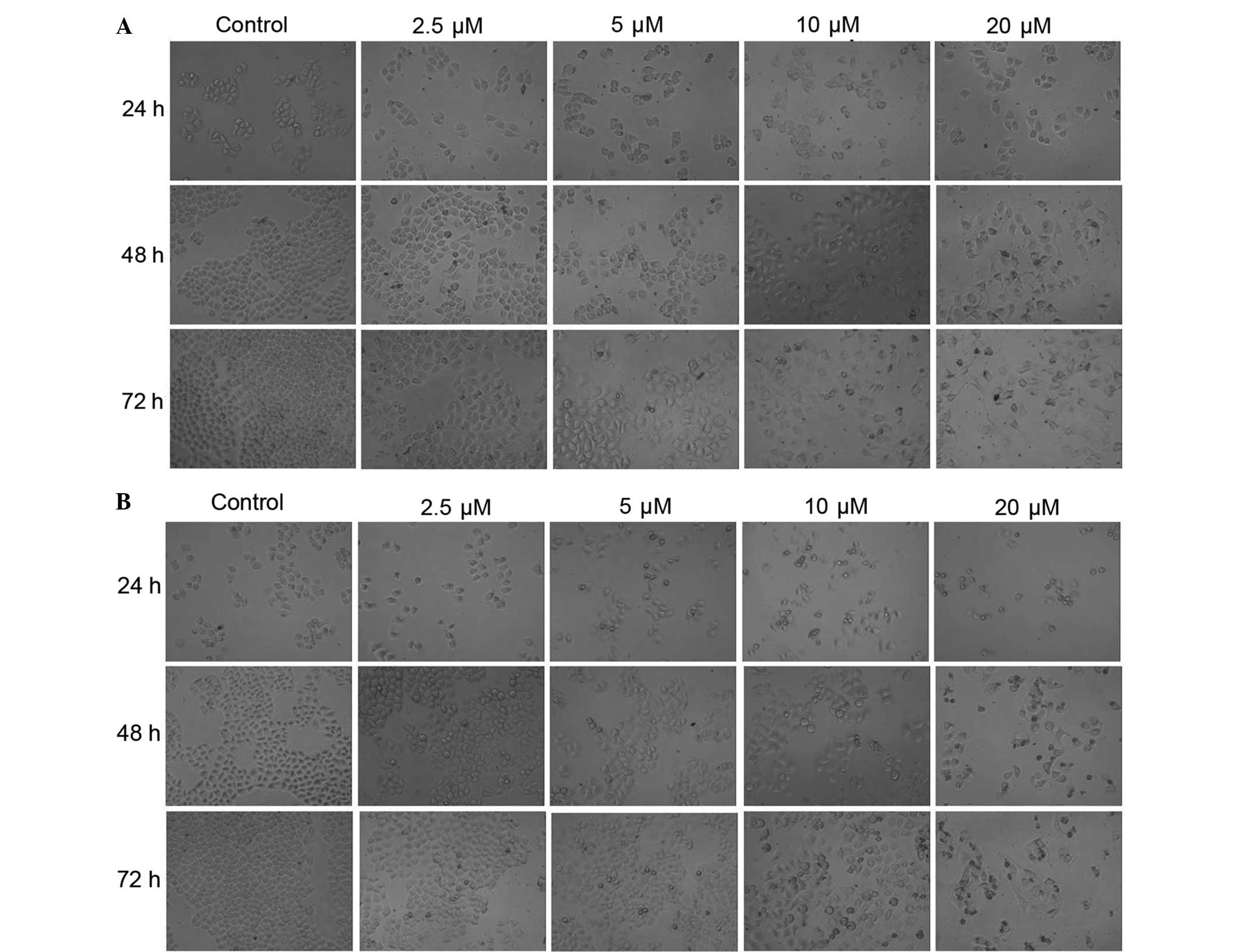
As observed under an inverted microscope, the
morphology of HeLa cells appeared to change significantly after
treatment with CDV (Fig. 1A) and DDP
(Fig. 1B). The treated cells with
CDV and DDP had irregular shapes, smaller sizes, dissociation of
cell membrane, karyopyknosis and dark stained nuclei. HeLa cells in
the control group were stained as light blue or light purple by
Giemsa reagent, whereas apoptotic cells in the CDV and DDP groups
were stained dark blue or dark purple. In the control group, cells
were characterized by loose cytoplasm that were stained light
purple, large nucleoli, and separated nucleoli and cytoplasm.
However, the characteristics of cells in the CDV and DDP treatment
group suggested that the size of the numerous cells was reduced,
some cells were long and spindle-shaped, whereas agglutination and
karyopyknosis in the nucleus chromatin were stained with a dark
color, similar to the typical apoptotic cells. The results showed
that treatment with CDV or DDP was able to induce apoptosis and
morphological changes in HeLa cells. At the same time points,
treatment with CDV and DDP had a similar inhibitory effect on the
proliferation of HeLa cells in a concentration-dependent manner,
with an increased number of apoptotic cells observed. Additionally,
at the same drug concentrations, the two treatment groups presented
increased numbers of apoptotic cells in a time-dependent manner.
All these aforementioned observations demonstrated that CDV and DDP
treatments can inhibit the proliferations of HeLa cells depending
on the incubation time and drug concentration.
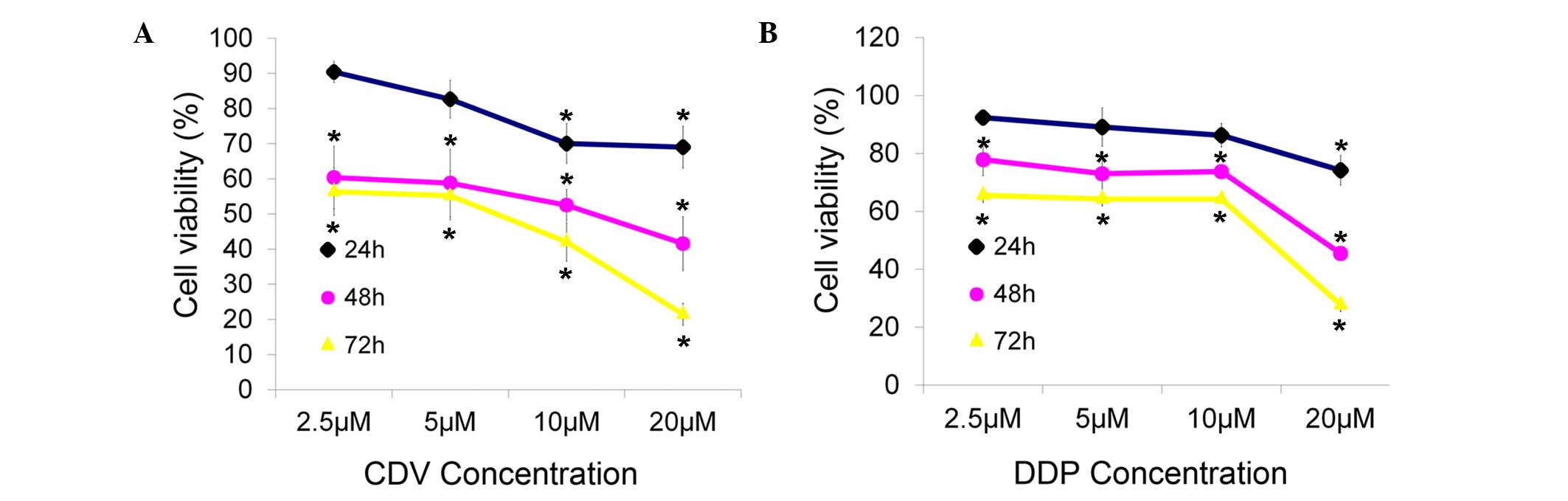
The inhibitory effects of CDV and DDP on the
proliferation of HeLa cells changed markedly in time- and
concentration-dependent manners, when compared with the
proliferation observed in the control group (Fig. 2). Cell viability was reduced at
higher concentrations and incubation times. Through this MTT cell
viability detection and data analysis, it was concluded that the
concentration of CDV and DDP that was able to inhibit the viability
of HeLa cells by 50% after 48 h of treatment was 15 µM for both
drugs. Thus, the treatments of 15 µM CDV and 15 µM DDP were adopted
for 48 h in subsequent experiments in the present study.
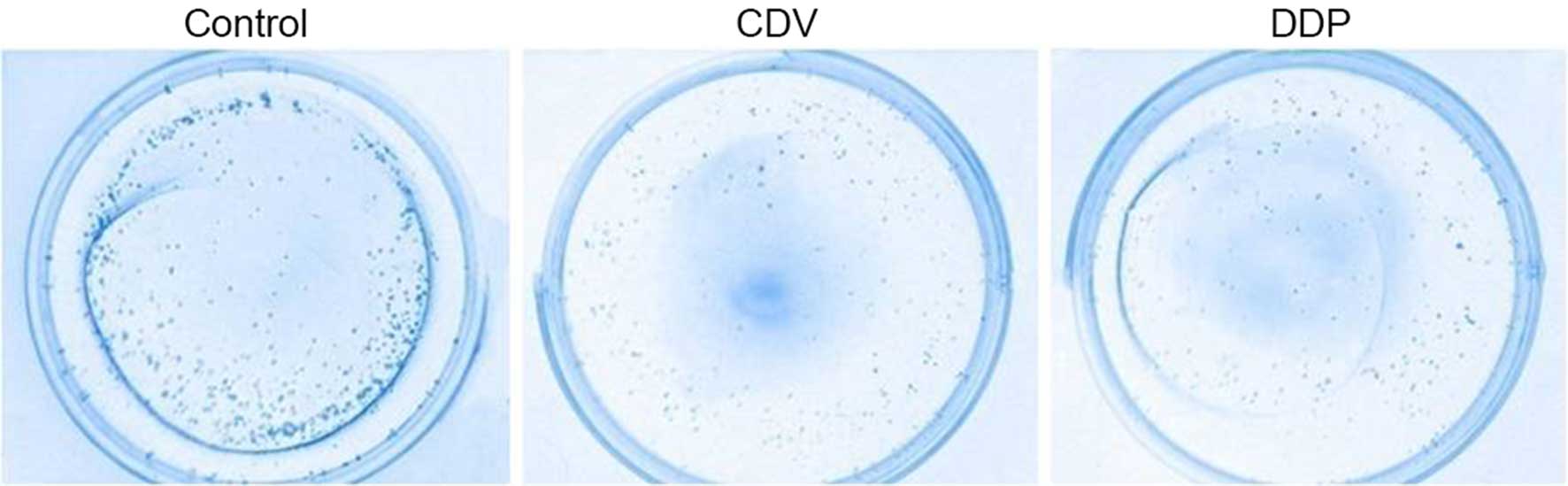
Formation of HeLa cell colonies
As shown in Fig. 3,
the results of Giemsa staining of HeLa cell colonies demonstrated
that the CDV and DDP groups had a reduced number of colonies when
compared with the control group. In addition, the cell survival
rate of the CDV group (75.35±1.14%) was significantly higher
compared with that in the DDP group (63.71±0.82%), demonstrating
that cell proliferation was inhibited to a greater extent in the
DDP group.
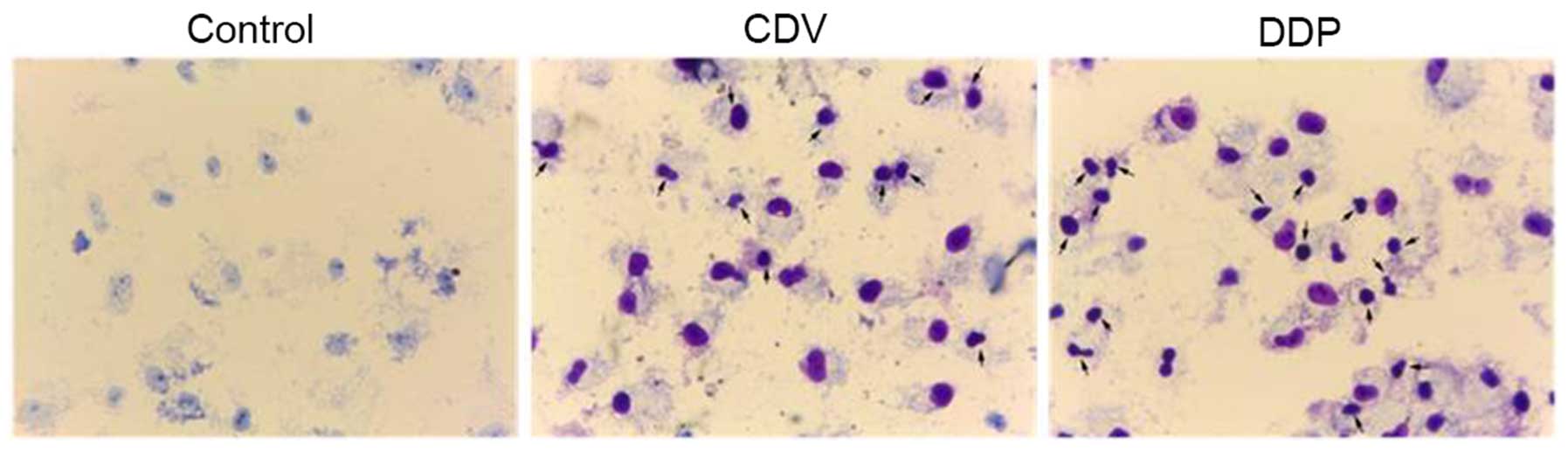
Apoptotic cell detection by Giemsa
staining
As shown in Fig. 4,
HeLa cells in the control group were stained as light blue or light
purple by Giemsa reagent, while apoptotic cells in the CDV and DDP
group were stained as dark blue or dark purple. In the control
group, cells were characterized by loose cytoplasms that were
stained light purple, large nucleoli, separated nucleoli and
cytoplasms, and nucleoli that were visible with a large number of
visible nucleoli. However, the characteristics of cells were
observed by an inverted microscope (Figs. 4 and 5). The morphology of HeLa cells changed
significantly. They exhibited irregular shapes, were smaller in
size, had dissociation of the cell membrane, karyopyknosis and
dark-stained nuclei. At the same time, the inhibitory function of
CDV and DDP on the proliferation of HeLa cells increased as the
concentration of the drugs increased. In addition, the proportion
of HeLa cells increased as the concentration of drugs and the
proportion of apoptotic cells increased, and at the same drug
concentration the proportion of apoptotic cells increased with
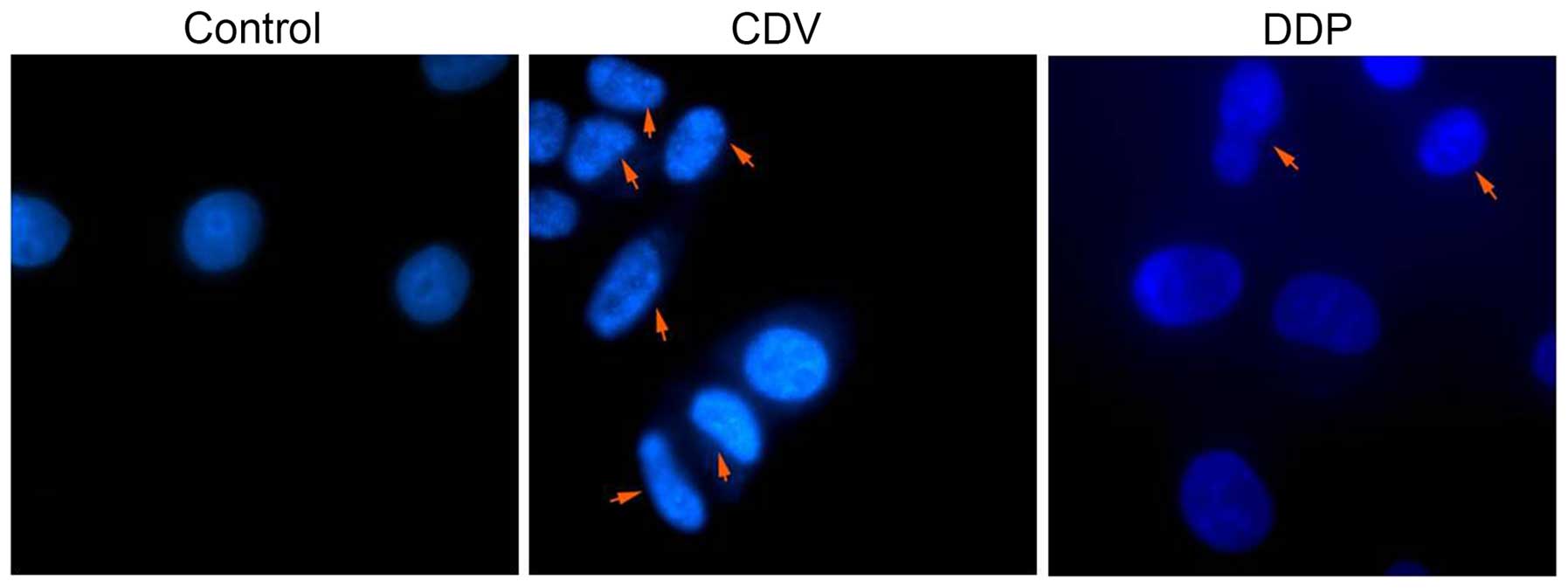
time. As shown in Fig. 5, with the
arrows indicating apoptotic cells and apoptotic bodies. The size of
apoptotic cells was reduced, the cytoplasm was condensed and the
membrane was intact with a foaming phenomenon detected. At the late
phase of apoptosis, nuclei were lysed into pieces, producing
apoptotic bodies. Fig. 5 suggested
that treatment with CDV or DDP was able to induce apoptosis and
morphological changes in HeLa cells, and the effect of CDV was
stronger. Therefore, these findings demonstrated that treatment
with CDV or DDP was able to induce apoptosis and morphological
changes in HeLa cells.
Cell cycle progression and apoptosis
detection by flow cytometry
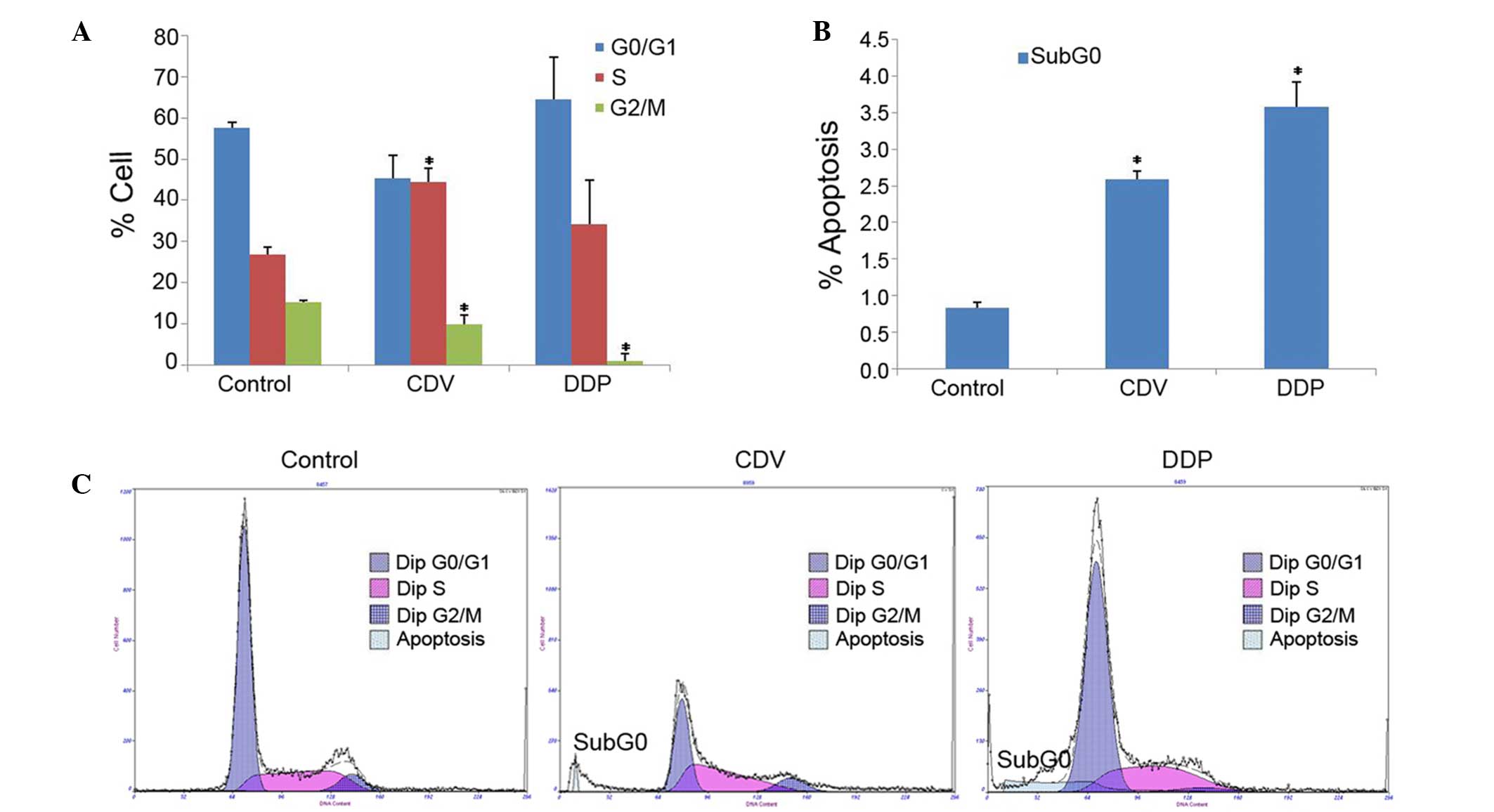
The results of cell cycle detection showed that CDV
and DDP treatments had an impact on the progression of the HeLa
cell cycle (Fig. 6A). Compared with
the control group, the proportion of cells in S-phase was
significantly increased in the CDV group (P<0.05), demonstrating
that CDV was able to block the HeLa cell cycle in the S-phase. In
addition, the proportion of HeLa cells in G0/G1 phase and S-phase
in the DDP group increased slightly, demonstrating that DDP
possibly resulted in the arrest of certain HeLa cells in the G0/G1
phase and of others in the S-phase. These results indicated that
the two drugs inhibited the progression of the HeLa cell cycle.
Furthermore, the results of the cell apoptosis test
demonstrated that an apoptosis peak was evidently observed in the
CDV and DDP groups (Sub-G0 peak), whereas this was not observed in
the control group (Fig. 6B and C).
This indicated that CDV and DDP induced the apoptosis of HeLa
cells. The apoptosis rates of HeLa cells in CDV (2.593±0.103%;
P<0.05) and DDP groups (3.573±0.348%; P<0.05) were
significantly higher when compared with the rate in the control
group.
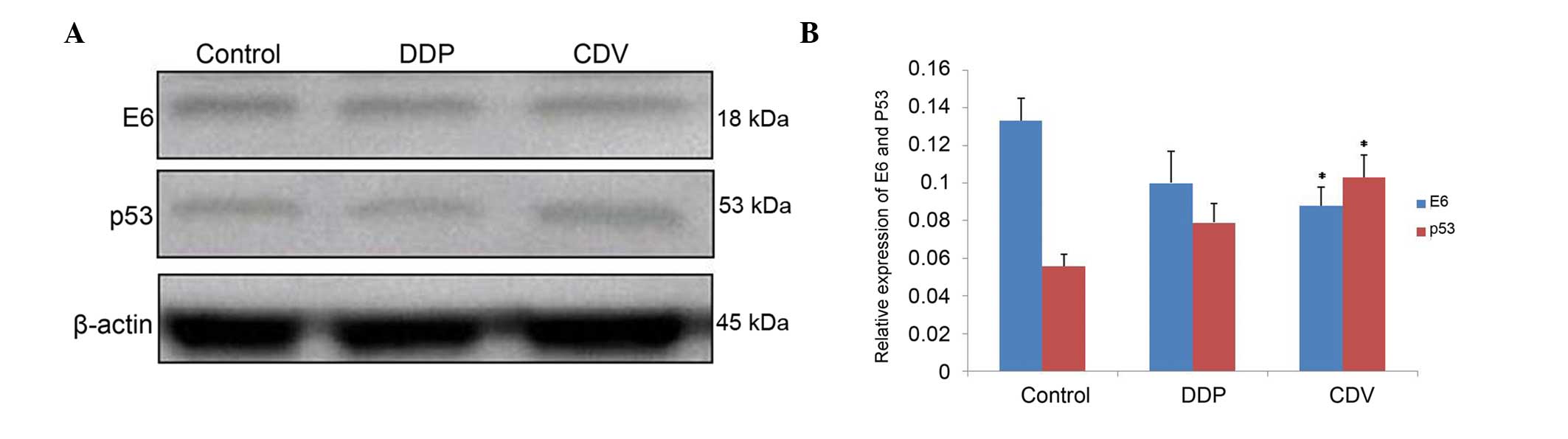
Effects of CDV and DDP on the
expression levels of E6 and p53 protein
The results (Fig. 7;
Table I) showed that, compared with
the control group, the expression of E6 protein decreased
subsequent to treatment with the two drugs. E6 expression in the
CDV group decreased more significantly (0.088±0.010; P<0.01),
compared with that in the DDP group (0.100±0.017). By contrast,
compared with the control group, the expression of p53 protein
increased subsequent to treatment with these drugs, with the
expression increasing more significantly in the CDV group
(0.103±0.012; P<0.01). Overall, the decreases in E6 protein and
the increases in p53 protein were not statistically
significant.
 | Table I.Expression levels of E6 and p53
proteins treated with DDP and CDV. |
Table I.
Expression levels of E6 and p53
proteins treated with DDP and CDV.
| Groups | E6 | p53 |
|---|
| Control | 0.133±0.012 | 0.056±0.006 |
| DDP | 0.100±0.017 | 0.079±0.010 |
| CDV |
0.088±0.010a |
0.103±0.012a |
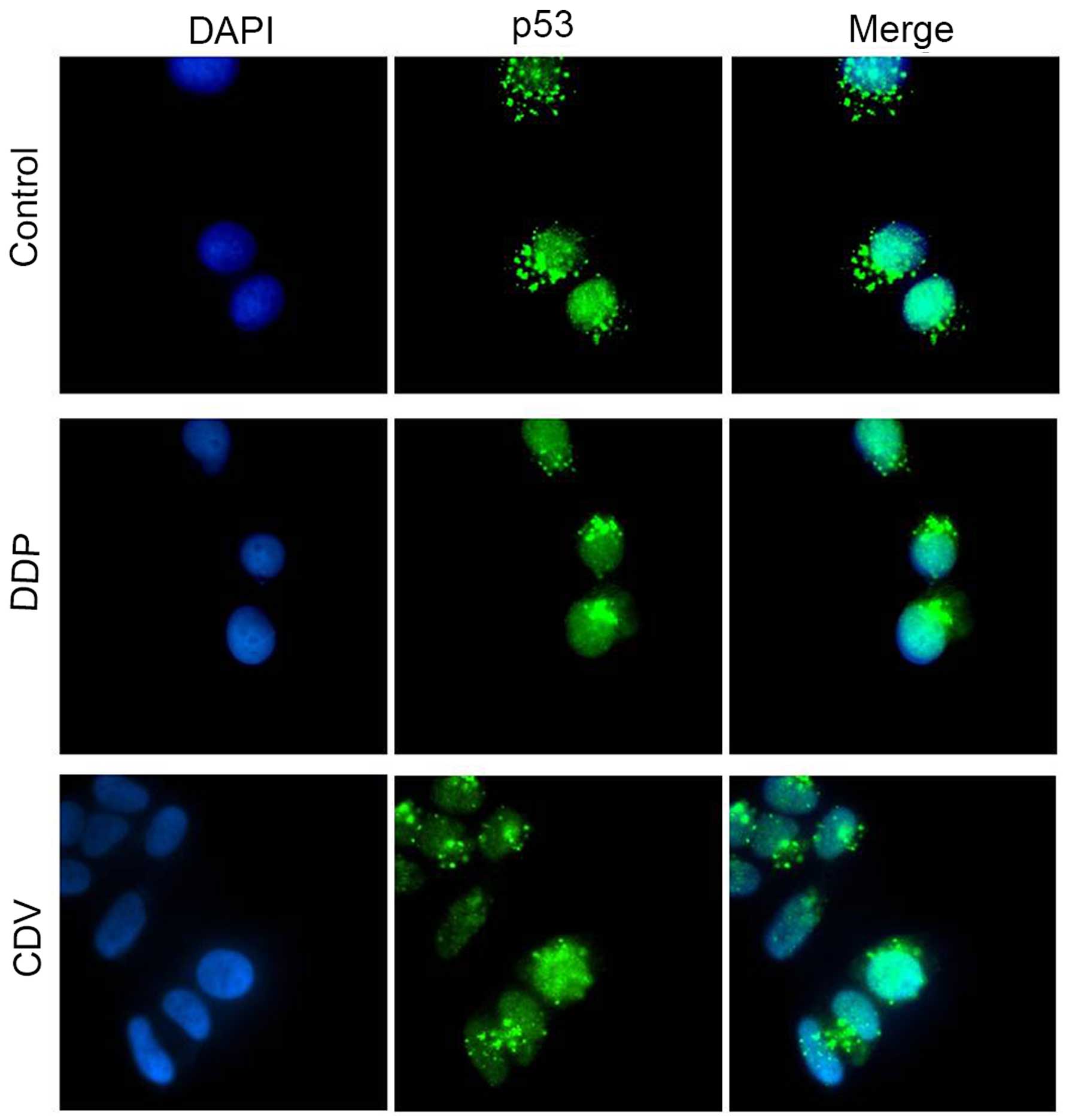
The results of immunofluorescence staining (Fig. 8) revealed that p53 mainly gathered in
the nuclei of HeLa cells in the control group, while only a small
part was distributed in the cytoplasm. However, compared with the
control group, p53 protein accumulated mainly in the nuclei of
cells treated with CDV or DDP, which was possibly caused by the
decrease in E6 protein expression. This is assumed because analysis
of other complex structures of the p53 core showed that the
E6-binding surface of p53 was available for interaction not only in
‘free’ p53 tetramers but also in p53 molecules bound to DNA or
other cellular proteins (10). These
results were consistent with the findings of western blot analysis.
In conclusion, cidofovir and cisplatin inhibited proliferation and
induced apoptosis of HeLa cells. Furthermore, cidofovir may also be
used for cervical cancer therapy.
Discussion
Cervical HPV vaccines can be divided into two types,
including preventive and therapeutic vaccines. Preventive vaccines
mainly include anti-HPV 16 and 18 bivalent vaccines, as well as
anti-HPV 6, 11, 16 and 18 tetravalent vaccines, which can provide
protection for nearly 70% of patients (11).
DDP is widely used in anti-cancer treatment, since
it is able to induce DNA damage, activate p53 and induce programmed
cell death, thus making it one of the most common drugs in the
treatment of cervical cancer (12).
CDV was originally approved by the Food and Drug Administration for
the treatment of acquired immune deficiency syndrome patients with
retinitis caused by cytomegalovirus (13). In recent years, the clinical
indications of CDV have been expanded. However, there are few
studies focusing on using CDV as antiviral therapy (14).
In high-risk HPV infection, E6 targets p53 protein,
binds to p53 through the E6-associated protein (E6-AP), induces
permanent cell proliferation, and impairs the regulatory role of
p53 in cell apoptosis and cell cycle arrest. Ultimately, all these
effects of E6 led to cell immortalization and the occurrence of
cervical cancer (15,16). Studies have demonstrated that p53
protein serves an important role in cell cycle regulation and
apoptosis, as well as in the progression and development of tumors
(17). p53 is located in the nuclei,
and can maintain cell genome stability by inhibiting cell
proliferation, regulating the cell cycle and gene transcription,
and activating programmed cell death (18).
The present study identified that CDV and DDP
inhibited the growth and proliferation of HeLa cells. This
inhibitory effect was depended on the drug concentration and
incubation time, and was enhanced by higher concentration and
longer treatment times. Under the same experimental conditions, DDP
had a relatively higher inhibitory effect on the proliferation of
HeLa cells compared with CDV. According to the results of flow
cytometry, CDV and DDP had an impact on the HeLa cell cycle
progression and induction of cell apoptosis. CDV caused HeLa cell
arrest in S-phase, and DDP resulted in the arrest of certain HeLa
cells in S-phase and others in G0/G1 phase. This indicates that the
two drugs had inhibitory effects on the cell cycle progression of
HeLa cells and can induce HeLa cell death; thus, CDV and DDP may
restore the activity of p53 protein in certain ways.
Comparative analysis of the above findings suggests
that CDV and DDP may reduce the expression levels of E6 protein or
activate the p53 protein pathway in HeLa cells. The expression
activity of p53 recovers under the effects of DDP, since DDP may
reduce the E6 activity in HeLa cells directly or indirectly by
binding with the sulfhydryl on the surface of E6. In addition, DDP
and p53 protein competitively bind with E6 protein, which can cause
the release of p53 and then recovery of p53 expression, and
ultimately inhibit cell proliferation and induce cell apoptosis
(19). In the present study, western
blot analysis revealed that, following treatment by CDV or DDP, the
E6 protein levels in HeLa cells were reduced, resulting in an
increase in p53 protein levels. This suggests that CDV and DDP are
able to inhibit the expression of E6 protein in HeLa cells, and
resume the activity of p53 protein. The study of Donne et al
(20) showed that the upregulated
expression of E6 mRNA was observed using semi-quantitative or
quantitative polymerase chain reaction, which may be associated
with the specific concentration of CDV, the experimental time or
the types of cells.
The expression of E6 protein has been shown to be
associated with the location and translocation of p53 protein in
the nuclei of HPV-positive cells (21). In cervical cancer cells, p53 tumor
suppressor protein normally exists as wild-type. In HPV-infected
cervical cancer cells, E6 combines with p53 protein into a stable
E6-p53 complex by E6-AP. In HeLa cells without the presence of
mouse double minute 2 homolog, HPV 18 E6 and E6-AP, which contain a
nuclear export sequence, are capable of transporting the p53
protein to the cytoplasm (22). E6
can degrade p53 protein by the proteasome system in the nuclei and
cytoplasm. These previous findings are consistent with the current
study results showing that p53 protein can be observed in the
nucleus and cytoplasm in the control group. Currently, it is
considered that the degradation of p53 by HPV E6 is, at least
partly, based on the nuclear export of the protein. The common
mechanism involves E6-AP degrading and regulating p53 by shuttling
between the nuclei and cytoplasm (23). In the current study, subsequent to
CDV and DDP treatment, p53 protein in HeLa cells was mainly located
in the nuclei rather than the cytoplasm. According to the results
of western blot analysis, it can be inferred that CDV and DDP
reduced the expression of E6 protein, activating the wild-type p53
protein expression.
In conclusion, treatment with CDV or DDP can inhibit
the proliferation of HeLa cells in a concentration- and
time-dependent manner. CDV and DDP can affect the expression levels
of E6 and p53 proteins in HeLa cells, and consequently regulate
cell cycle arrest and cell apoptosis. According to the findings of
the present study, CDV can inhibit HeLa cell proliferation and
induce cell apoptosis similar to DDP, which indicates that CDV may
be a possible agent for cervical cancer treatment.
Acknowledgements
This study was supported by the Initial Project for
Post-Graduates of Hubei University of Medicine (grant no
2015QDJZR08) and the Scientific Research and Technological
Development Project of Shiyan Science and Technology Bureau (grant
no. 16K70).
Glossary
Abbreviations
Abbreviations:
|
HPV
|
human papillomavirus
|
|
CDV
|
cidofovir
|
|
DDP
|
cisplatin
|
|
PBS
|
phosphate buffer
|
|
E6-AP
|
E6-associated protein
|
References
|
1
|
Drain PK, Holmes KK, Hughes JP and Koutsky
LA: Determinants of cervical cancer rates in developing countries.
Int J Cancer. 100:199–205. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Faridi R, Zahra A, Khan K and Idrees M:
Oncogenic potential of Human Papillomavirus (HPV) and its relation
with cervical cancer. Virol J. 8:2692011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ajay AK, Meena AS and Bhat MK: Human
papillomavirus 18 E6 inhibits phosphorylation of p53 expressed in
HeLa cells. Cell Biosci. 2:22012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shi JF, Canfell K, Lew JB and Qiao YL: The
burden of cervical cancer in China: Synthesis of the evidence. Int
J Cancer. 130:641–652. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cox JT: Epidemiology and natural history
of HPV. J Fam Pract Suppl. 3–9. 2006.
|
|
6
|
Tommasino M: The human papillomavirus
family and its role in carcinogenesis. Semin Cancer Biol. 26:13–21.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rosales R and Rosales C: Immune therapy
for human papillomaviruses-related cancers. World J Clin Oncol.
5:1002–1019. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Collette DC and Zechel MA: Novel:
treatment of atypical human papillomavirus-associated epithelial
hyperplasia with cidofovir. J Oral Maxillofac Surg. 69:2383–2386.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang YH, Peng HY, Xia GH, Wang MY and Han
Y: Anticancer effect of two diterpenoid compounds isolated from
Annona glabra Linn. Acta Pharmacol Sin. 25:937–942. 2004.PubMed/NCBI
|
|
10
|
Travé G and Zanier K: HPV-mediated
inactivation of tumor suppressor p53. Cell Cycle. 15:2231–2232.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kjær SK, Frederiksen K, Munk C and Iftner
T: Long-term absolute risk of cervical intraepithelial neoplasia
grade 3 or worse following human papillomavirus infection: Role of
persistence. J Natl Cancer Inst. 102:1478–1488. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Koivusalo R, Krausz E, Ruotsalainen P,
Helenius H and Hietanen S: Chemoradiation of cervical cancer cells:
Targeting human papillomavirus E6 and p53 leads to either augmented
or attenuated apoptosis depending on the platinum carrier ligand.
Cancer Res. 62:7364–7371. 2002.PubMed/NCBI
|
|
13
|
De Clercq E and Holý A: Acyclic nucleoside
phosphonates: A key class of antiviral drugs. Nat Rev Drug Discov.
4:928–940. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Topalis D, Nogueira TC, De Schutter T, El
Amri C, Krecmerova M, Naesens L, Balzarini J, Andrei G and Snoeck
R: Resistance to the nucleotide analogue cidofovir in HPV(+) cells:
a multifactorial process involving UMP/CMP kinase 1. Oncotarget.
7:10386–10401. 2016.PubMed/NCBI
|
|
15
|
Scheffner M, Werness BA, Huibregtse JM,
Levine AJ and Howley PM: The E6 oncoprotein encoded by human
papillomavirus types 16 and 18 promotes the degradation of p53.
Cell. 63:1129–1136. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Werness BA, Levine AJ and Howley PM:
Association of human papillomavirus types 16 and 18 E6 proteins
with p53. Science. 248:76–79. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jin Y, Wei Y, Xiong L, Yang Y and Wu JR:
Differential regulation of survivin by p53 contributes to cell
cycle dependent. Cell Research. 15:361–370. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bargonetti J and Manfredi JJ: Multiple
roles of the tumor suppressor p53. Curr Opin Oncol. 14:86–91. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huang H, Huang SY, Chen TT, Chen JC, Chiou
CL and Huang TM: Cisplatin restores p53 function and enhances the
radiosensitivity in HPV16 E6 containing SiHa cells. J Cell Biochem.
91:756–765. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Donne AJ, Hampson L, He XT, Rothera MP,
Homer JJ and Hampson IN: Cidofovir induces an increase in levels of
low-risk and high-risk HPV E6. Head Neck. 31:893–901. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Abdulkarim B, Sabri S, Deutsch E,
Chagraoui H, Maggiorella L, Thierry J, Eschwege F, Vainchenker W,
Chouaïb S and Bourhis J: Antiviral agent Cidofovir restores p53
function and enhances the radiosensitivity in HPV-associated
cancers. Oncogene. 21:2334–2346. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Stewart D, Ghosh A and Matlashewski G:
Involvement of nuclear export in human papillomavirus type 18
E6-mediated ubiquitination and degradation of p53. J Virol.
79:8773–8783. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hillemanns P, Jentschke M, Evans TG,
Soergel P and Hass R: Detection of E6-AP as a potential therapeutic
target in cervical specimen of HPV-infected women. Arch Gynecol
Obstet. 289:1281–1286. 2014. View Article : Google Scholar : PubMed/NCBI
|