Introduction
Coronary heart disease (CHD) is the most common
cause of mortality worldwide (1).
Acute coronary syndrome (ACS) is a severe phase of CHD and a
predominant cause of cardiac events (2). The principal pathophysiology of ACS is
the rupture of coronary plaques with subsequent thrombosis
(3). The pathological
characteristics of vulnerable plaques (VPs) include the presence of
a soft, lipid-rich core that is covered by a thin cap of fibrous
tissue and is infiltrated by a large number of inflammatory cells
(4). Plaque rupture occurs in
various pathological states and is typically associated with matrix
metalloproteinases (MMPs). MMPs are able to degrade collagen and
other extracellular matrix (ECM) components that compose the major
structure of plaques and previous studies have indicated an
association between the level of MMPs and plaque stability
(5–7). Co-expression of extracellular matrix
metalloproteinase inducer (EMMPRIN) and MMPs has been observed in
macrophages in vitro and in human atheroma, particularly in
the shoulder region of VP atheroma (8). The present study suggested that EMMPRIN
serves a key role in regulating MMP activity in cardiovascular
diseases.
Atorvastatin is an inhibitor of
3-hydroxy-3-methyl-glutaryl coenzyme A (HMG-CoA) reductase. Large
randomized clinical trials have determined that statins are able to
reduce the incidence of life-threatening vascular events in
patients with ACS (9–11). Over the last decade, statins have
become a standard treatment regimen for patients with
atherosclerosis. The anti-atherosclerotic effects and concomitant
lipid-lowering effects of atorvastatin have been demonstrated in
previous studies. Statins have many beneficial effects, including
lowering cholesterol (12,13), improving endothelial function
(14,15) and reducing systemic inflammatory
markers (16,17).
As a factor in atherosclerosis, EMMPRIN mediates
plaque destabilization in myocardial infarction, whereas
atorvastatin has a protective role in the development of VP.
However, the mechanism between EMMPRIN and atorvastatin in aortic
atherosclerotic plaques is not yet understood. Therefore, in the
present study, the effect of atorvastatin on EMMPRIN expression in
aortic atherosclerotic plaques was investigated.
Materials and methods
Reagents and equipment
A goat anti-EMMPRIN polyclonal antibody (sc-9757), a
rabbit polyclonal antibody (sc-7951) against cyclooxygenase-2
(COX-2), dithiothreitol, aprotinin, leupeptin, phenylmethylsulfonyl
fluoride (PMSF), Tween 20 and Nonidet P-40 were purchased from
Santa Cruz Biotechnology, Inc., (Dallas, TX, USA). The rabbit
anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) monoclonal
antibody (NB100-56875) was purchased from Novus Biologicals, LLC
(Littleton, CO, USA). The goat anti-rabbit (IRDye680LT) and donkey
anti-goat (IRDye800CW) secondary antibodies were purchased from
LI-COR Biosciences (Lincoln, NE, USA). The RNAiso Plus, SYBR
Premix Ex Taq II and the PrimeScript RT Reagent kit with
gDNA Eraser were purchased from Takara Bio, Inc., (Otsu, Japan).
The goat anti-rabbit (ZB-5301) and rabbit anti-goat (ZB-2306)
horseradish peroxidase (HRP)-conjugated secondary antibodies, a
diaminobenzidine (DAB) color reagent kit (ZLI-9017) and an ELISA
kit (EIA-1112) used for the determination of prostaglandin E2
(PGE2) levels were purchased from OriGene Technologies, Inc.,
(Beijing, China). Atorvastatin and ezetimibe, which is a selective
inhibitor of cholesterol absorption, were gifts from Pfizer, Inc.
(New York, NY, USA) and Merck & Co. (Kenilworth, NJ, USA),
respectively. Polymerase chain reaction (PCR) amplification was
performed using a thermal cycler PTC-200 (Bio-Rad Laboratories,
Inc., Hercules, CA, USA), a nucleic acid protein analyzer DU800
(Beckman Coulter, Inc., Brea, CA, USA), a microplate reader FO39300
(Bio-Rad Laboratories, Inc.), a ChemiImager 5500 gel imaging
analysis system (ProteinSimple, San Jose, CA, USA) and a Leica
DMIRB inverted fluorescence microscope (Leica Microsystems, Inc.,
Buffalo Grove, IL, USA). RPMI 1640 medium and fetal bovine serum
(FBS) were purchased from Gibco, Thermo Fisher Scientific, Inc.
(Waltham, MA, USA). Phorbol 12-myristate 13-acetate (PMA), oxidized
low-density lipoprotein (ox-LDL), NS-398 (an inhibitor of COX-2),
PGE2 and sodium pentobarbitone (1507002) were obtained from
Sigma-Aldrich (Merck Millipore, Darmstadt, Germany).
Mice and husbandry
Apolipoprotein E knockout (ApoE−/−) male
mice (n=72; age, 8 weeks; weight, 24±1 g) were purchased from the
Animal Centre of the Medical Department of Beijing University
(Beijing, China). Mice were maintained under standard conditions of
humidity (50–60%) and temperature (18–22°C), and were subjected to
a 12 h hour light-dark cycle with ad libitum access to feed
and water. Mice were inspected at least once every 24 h. Two types
of feed were provided, which included a normal rodent diet and a
high-fat content diet that contained 15% fat from lard and was
supplemented with 1.25% (w/w) cholesterol.
Experimental groups
All 72 mice were fed a normal rodent diet for one
week. Subsequently, mice were distributed randomly and evenly into
an early-start (ES) and a late-start (LS) treatment group (n=36
each).
Mice in each group were randomly divided into four
equal subgroups (n=9): Groups A (ES) and E (LS) were a normal diet
control (NDC) group; groups B (ES) and F (LS) were a high-fat diet
control (HDC) group; groups C (ES) and G (LS) were a low-dosage
treatment (LDT) group receiving a high-fat diet and a low dosage of
atorvastatin; and groups D (ES) and H (LS) were a high-dosage
treatment (HDT) group receiving a high-fat diet and a high dosage
of atorvastatin. Group C were orally administered 0.3 ml/day
atorvastatin suspension, at a dosage of 5-mg/kg beginning in week
7, whereas atorvastatin was administered to group G beginning in
week 11. Group D were orally administered 0.3-ml/day atorvastatin
suspension, at a dosage of 10 mg/kg, beginning in week 7, whereas
atorvastatin was administered to group H beginning in week 11.
Groups A, B, E, and F were administered an isodose of normal
saline. Following 8 weeks of statin intervention, mice in the ES
and LS groups were humanely sacrificed in weeks 15 and 19,
respectively.
Specimen harvesting
Mice were surgically anesthetized via
intraperitoneal injection of 1% sodium pentobarbitone (50 mg/kg).
The ascending aorta was removed from each mouse and immersed in 4%
paraformaldehyde at room temperature, for ≥24 h. Aortas were
subsequently embedded in paraffin and cut transversally into serial
sections 5-µm thick initiating at the aortic root. Remaining
sections of the aortas (from the descending to the iliac aorta)
were stored at −70°C.
Lipid detection
Serum harvested from the mice was analyzed to
determine total triglyceride (TG) cholesterol (TC) and LDL-C
levels, using an Olympus AU2700 High-Volume Chemistry-Immuno
Analyzer (Olympus Corp., Tokyo, Japan).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from the frozen
atherosclerotic carotid samples or THP-1 macrophages (American Type
Culture Collection, Manassas, VA, USA) using RNAiso Plus reagent
according to the manufacturer's protocol. Then, the total RNA was
reverse-transcribed to cDNA using the PrimeScript® RT
reagent kit with gDNA Eraser (RR047A) according to the
manufacturer's protocol. Primer Express® software v3.0.1
(Applied Biosystems; Thermo Fisher Scientific, Inc.) and sequence
information from the National Center for Biotechnology Information
database (http://www.ncbi.nlm.nih.gov) were
used to design the PCR primers for EMMPRIN, COX-2 and the
housekeeping gene GAPDH. Primer sequences were as follows: EMMPRIN,
forward 5′-GCAGAGGACACAGGCACTTAC-3′ and reverse
5′-ACAGGCTCAGGAAGGAAGATG-3′; GAPDH, forward
5′-AGGTCGGTGTGAACGGATTTG-3′ and reverse 5′-GGGGTCGTTGATGGCAACA-3′;
COX-2, forward 5′-GATTGCCCGACTCCCTTGG-3′ and reverse
5′-AACTGATGCGTGAAGTGCTG-3′. A total of 1 µg cDNA was used as a
template for Real-time PCR in 20-µl reaction volumes with SYBR
Premix Ex Taq II according to the manufacturer's
instructions on a Stratagene Mx3000P qPCR system (Agilent
Technologies, Inc., Santa Clara, CA, USA). The amplification
protocol used was as follows: An initial denaturation step for 5
min at 95°C followed by 40 cycles of denaturation for 10 sec at
95°C, annealing for 30 sec at 57°C and elongation for 30 sec at
72°C, and a final extension step at 72°C for 30 sec. Quantitative
measurements were determined using the comparative −2ΔΔCq method
(18). All samples were normalized
against the endogenous level of GAPDH. All results were repeated in
triplicate and were analyzed using MxPro-Mx3000P software (Agilent
Technologies, Inc.). The results for the relative expression levels
were expressed as the mean ± the standard deviation (SD).
Western blot analysis
For western blot analysis, mouse aortas were
homogenized, ground in liquid nitrogen and subsequently lysed with
radio-immunoprecipitation assay (RIPA) lysate and 100 µl
phenylmethylsulfonyl fluoride (PMSF) in ice for 30 min. Following
centrifugation at 20,000 × g for 30 min at 4°C, the protein
was obtained from the deposition. Cells were washed twice with 1 ml
ice-cold phosphate-buffered saline (PBS) and lysed by adding 100 µl
ice-cold RIPA and 1 mmol/l PMSF. Protein concentration was
determined using the Coomassie Brilliant Blue method with bovine
serum albumin (BSA; 5 mg/ml, Beyotime Institute of Biotechnology,
Haimen, China) as a standard. Equal amounts (30 µg/well) of protein
were separated by 10% SDS-PAGE and the protein was subsequently
transferred, using electroblotting for 30 min at 50 V, onto a
polyvinylidene difluoride membrane. The membrane was blocked in a
6% non-fat milk solution in Tris-buffered saline with 0.5% Tween 20
(Santa Cruz Biotechnology, Inc.). The membrane was probed
separately with a goat anti-EMMPRIN polyclonal antibody (1:1,000)
at 4°C overnight, rabbit anti-goat HRP-conjugated secondary
antibody (1:2,500) at 37°C for 1 h, a rabbit polyclonal antibody
against COX-2 (1:1,000) at 4°C overnight, goat anti-rabbit
HRP-conjugated secondary antibody (1:2,000) at 37°C for 1 h, rabbit
anti-GAPDH polyclonal antibody (1:2,000) at 4°C overnight, then
goat anti-rabbit HRP-conjugated secondary antibody (1:2,000) at
37°C for 1 h. EMMPRIN, COX-2 and GADPH were detected using Pierce™
ECL Western Blotting Substrate (Thermo Fisher Scientific Inc.,
32109) according to the manufacturer's protocol. Densitometric
signals were quantified using ImageQuant TL 1.1 software (General
Healthcare Bio-Sciences, Pittsburgh, PA, USA) and GAPDH was used as
a loading control.
Immunology, histology, hematoxylin and
eosin staining, and Sirius red staining
Following the rehydration and dewaxing of the serial
5-µm thick paraffin sections, they were incubated with 3% hydrogen
peroxide to inhibit endogenous peroxidase activity. The sections
were subsequently blocked with 5% BSA and were incubated for 30 min
at 4°C, prior to incubation overnight with the goat anti-EMMPRIN
polyclonal antibody (1:500) in 1% (w/v) BSA in PBS. Sections were
incubated at room temperature for 20 min with peroxidase-conjugated
AffiniPure donkey anti-goat secondary antibodies (LI-COR
Biosciences, IRDye800CW, 1:500) in 1% BSA in PBS. Hematoxylin
staining was performed on the sections to reveal cell nuclei, using
DAB as a substrate. Brown particles detected via light microscopy
were considered positive. Image-Pro Plus 5.1 color microscopic
image analysis software (Media Cybernetics, Inc., Rockville, MD,
USA) was used to measure the positive staining integrated optical
density (IOD) and provide semi-quantitative analysis. Five
DAB-stained paraffin sections were randomly selected from each
group and an average of five IOD measurements per group were
calculated. Paraffin sections were immunostained for an rabbit
anti-macrophage antibody (Abcam, Cambridge, MA, USA, ab56297,
1:100) at 4°C overnight and an anti-α-actin antibody (Santa Cruz
Biotechnology, sc-21078, 1:100) at 4°C overnight to confirm the
presence of macrophages and smooth muscle cells (SMCs) in the
vascular wall and plaques. Paraffin sections were deparaffinized,
rehydrated and subsequently treated with 0.3% hydrogen peroxide in
methanol for 30 min followed by 2% rabbit serum (sc-2338, Santa
Cruz Biotechnology, Inc.) to abolish endogenous peroxidase activity
and to block non-specific antibody binding, respectively. Sections
were subsequently incubated overnight at 4°C with the rabbit
anti-macrophage (sc-21078, 1:100) and anti-α-actin antibody
(ab56297, 1:100). Following incubation, the sections were washed
several times with PBS and incubated for 30 min at 37°C with the
goat anti-rabbit (IRDye680LT, 1:500) and donkey anti-goat
(IRDye800CW, 1:500) secondary antibodies visualized with DAB
Developer (OriGene Technologies, Inc.) and counterstained with 10%
Mayer's hematoxylin. Subsequently, sections were mounted in
Permamount and examined using light microscopy. Aortic paraffin
sections were stained with hematoxylin and eosin (H&E) and
Sirius red.
Pathology and evaluation of plaque
composition and lesion size
H&E staining was performed on the aortic
paraffin sections to observe the atherosclerotic plaque morphology.
Computer-assisted morphometry (Image-Pro Plus 5.1; Media
Cybernetics, Inc.) was used to analyze the cross-sectional area of
each plaque, the relative plaque area (the ratio of the plaque area
to the lumen area) and the fibrous cap thickness (FCT). The FCT was
measured at 10 equidistant points around the cap of each slice. For
each mouse, three sections (with an interval of 10 sections) from
the thickest part of the plaque were selected and the mean value
obtained was used for subsequent statistical analysis. Three
consecutive paraffin sections from each mouse were used to detect
macrophages and SMCs using an immunohistochemistry (IHC) assay, and
to detect collagen using Sirius red staining. Lipoid vesicles were
averaged directly from the H&E sections. The vulnerability
index (VI) of the plaque was calculated using the following formula
(19): VI = (macrophages + lipoid
vesicles) / (SMCs + collagen).
Cell culture
Human monocytic THP-1 macrophages were cultured in
suspension in RPMI 1640 containing 100 U/ml penicillin, 10% FBS and
100 µg/ml streptomycin at 37°C in an atmosphere containing 5%
CO2. A total of 1×106 THP-1 cells per well
were seeded in 6-well plates and stimulated with 0.1 µm PMA for 48
h until they adhered to the wells and exhibited macrophage-like
morphology. Plates were washed twice with 1 ml PBS following
culture. Cells were cultured in serum-free medium for an additional
24 h and incubated with the corresponding stimuli: 100-µm/ml ox-LDL
was used to induce COX-2 and EMMPRIN expression and no ox-LDL was
used as a control. For inhibition experiments, the cells were
pre-incubated with 5 µm atorvastatin, 5 µm ezetimibe or 10 µm
NS-398 for 1 h.
PGE2 assay
A PGE2 ELISA kit (EIA-1112) was used to measure PGE2
levels in the culture medium, according to the manufacturer's
protocol. Absorbance was measured at 405 nm with a microplate
reader (Thermo Fisher Scientific Inc. Waltham, MA, USA).
Statistical analysis
Image-Pro Plus 5.1 software was used to count
gray-scale and intensity values, and areas of interest. SPSS 13.0
software (SPSS, Inc., Chicago, IL, USA) was used for statistical
analyses. The data were presented as the mean ± the SD of three
independent measurements. Groups of data were compared by
performing one-way analysis of variance, followed by Tukey's
multiple comparison tests. Pearson's product-moment correlation
coefficient was used to analyze linear correlations. P<0.05 was
considered to indicate a statistically significant difference.
Results
Lipid levels
Under the high-fat feeding conditions, there were no
statistically significant differences observed in TC, LDL-C and TG
levels among the three groups of ApoE−/- mice.
High-fat diet leads to VPs in the
aortas of ApoE−/− mice
Plaque in the HDC groups (groups B and F) was
characterized by thin fibrous caps, extensive acellular collagenous
masses and large lipoid vesicles that extended to and narrowed the
lumen under the microscope. By contrast, the plaque in the NDC
groups (groups A and E) contained thick fibrous caps and small
lipoid vesicles (Fig. 1A). The
relative plaque area in the HDC groups was significantly larger
than that of the NDC groups (P<0.01; Fig. 1B). VP was defined as an FCT <65 mm
(20,21). The mean FCT in the HDC groups was
<65 mm and was significantly lower than that of the NDC groups
(P<0.05; Fig. 1C). The
macrophages and the SMCs in the plaque were stained brown during
the IHC assay. Sirius Red stained type I collagen yellow and red,
and type III collagen green (Fig.
1A). The HDC groups exhibited significantly increased
percentages of macrophages (P<0.05) and lipoid vesicles
(P<0.01), and decreased percentages of SMCs (P<0.05) and
collagen (P<0.01) in plaques compared with the NDC groups
(Fig. 1D). Therefore, the HDC groups
were classified as having VP. Mice in the HDC groups exhibited
decreased FCT (P<0.05; Fig. 1C)
and increased VI (P<0.01; Fig.
1E). A positive histological correlation was detected between
EMMPRIN expression and the VI (P<0.05; Fig. 1F).
 | Figure 1.Morphology and VI of the aortic
plaques of ApoE−/− male mice in the early-start groups
(n=9). (A) HE staining of the ascending aortas from
ApoE−/− male mice in the NDC and HDC groups
(magnification, ×100). Type I collagen is stained yellow and red,
and type III collagen is stained green (magnification, ×400; scale
bar, 100 µm). Macrophages and SMCs are stained brown. (B) Relative
plaque area was significantly increased in the HDC compared with
the NDC group. (C) Average FCT was significantly decreased in the
HDC compared with the NDC group. (D) Percentages of Ms and LVs were
increased and percentages of SMCs and C were decreased in plaques
in the HDC group compared with the NDC group. (E) VI was
significantly increased in the HDC compared with the NDC group. (F)
There was a positive correlation between histological expression of
EMMPRIN and VI. *P<0.05; **P<0.01. VI, vulnerability index;
ApoE−/−, apolipoprotein E knockout; HE, hemotoxylin and
eosin; SMC, smooth muscle cell; NDC, normal diet control; HDC,
high-fat diet control; FCT, fibrous cap thickness; M, macrophage;
LV, lipoid vesicle; C, collagen; EMMPRIN, extracellular matrix
metalloproteinase inducer; r, Pearson's product-moment correlation
coefficient. |
High-fat diet increases EMMPRIN
expression in the VPs of ApoE−/− mice
The results of the RT-qPCR (Fig. 2), the western blot analysis (Fig. 3) and the IHC assay (Fig. 4) revealed significantly increased
EMMPRIN expression in the HDC groups compared with the NDC
(P<0.01), LDT (ES, P<0.05; LS, P<0.01) and HDT (P<0.01)
groups, in ES and LS rats. The significant difference between the
HDC and NDC groups indicates that EMMPRIN expression was higher in
VPs than in stable plaques.
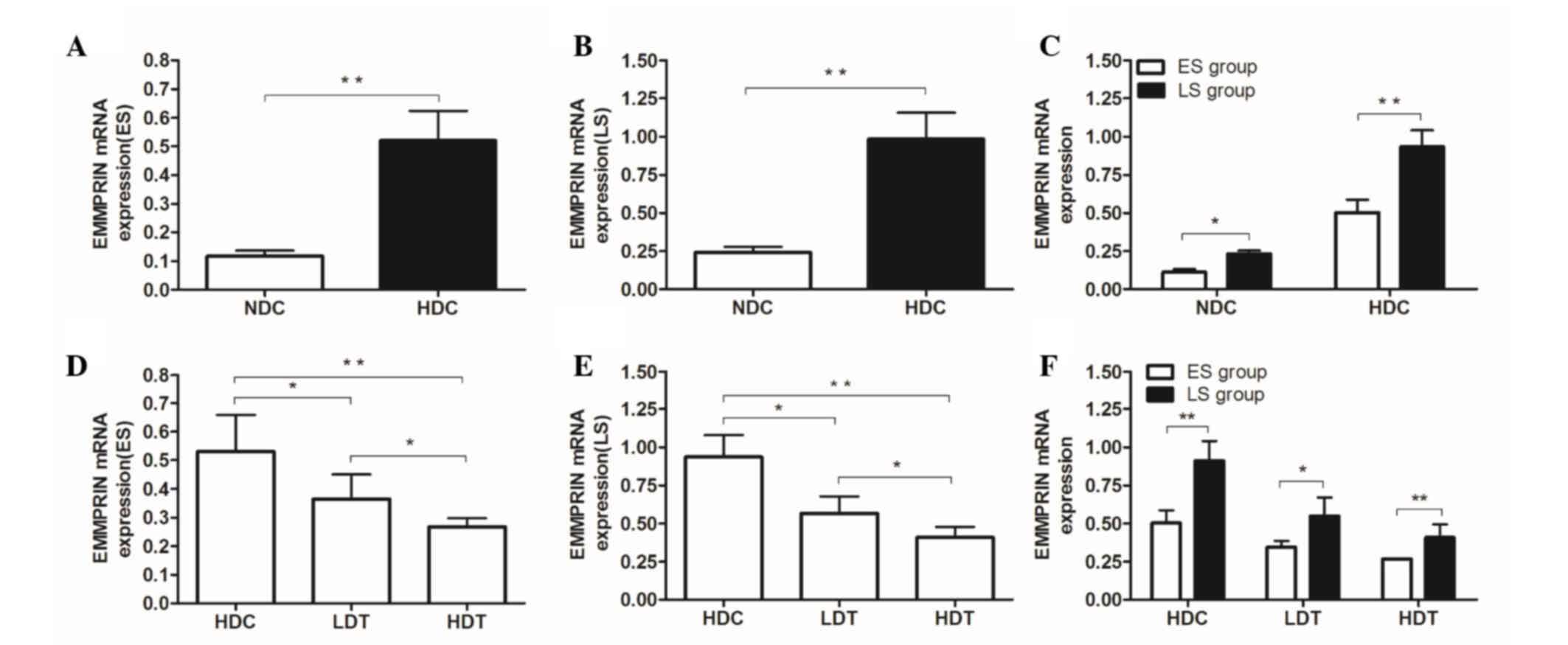 | Figure 2.mRNA expression in the aortas of
ApoE−/− male mice was determined by reverse
transcription-quantitative polymerase chain reaction. (A-C) For the
ES and LS groups, the mRNA expression of EMMPRIN in the HDC group
was significantly higher than that of the NDC group. In the (D) ES
and (E) LS groups, mRNA expression of EMMPRIN in the HDC group was
significantly higher than that of the LDT and HDT groups.
Furthermore, EMMPRIN mRNA expression in the HDT group was
significantly lower than that of the LDT group. (F) EMMPRIN mRNA
expression in the ES group was significantly lower than that of the
corresponding LS group in the HDC, LDT, and HDT subgroups.
*P<0.05; **P<0.01. ApoE−/−, apolipoprotein E
knockout; ES, early-start; LS, late-start; EMMPRIN, extracellular
matrix metalloproteinase inducer; HDC, high-fat diet control; NDC,
normal diet control; LDT, low-dose atorvastatin treatment; HDT,
high-dose atorvastatin treatment. |
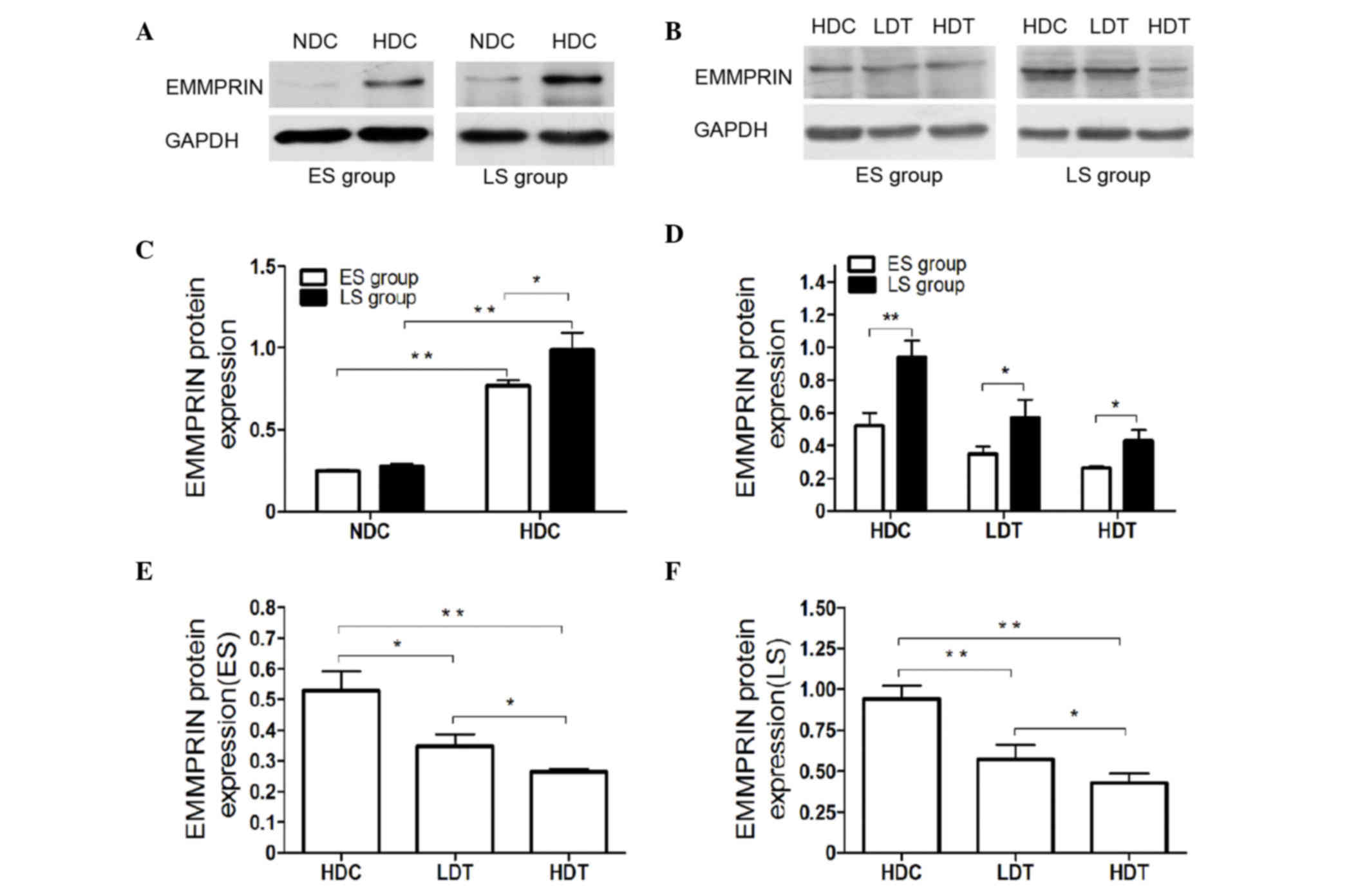 | Figure 3.(A and B) Protein expression in the
aortas of ApoE−/− male mice was determined via western
blot analyses. For the ES and LS groups, (C) the protein expression
of EMMPRIN in the HDC group was higher than that of the NDC group.
(D) EMMPRIN protein expression in the ES group was significantly
lower than in the corresponding LS group in the HDC, LDT and HDT
subgroups. EMMPRIN protein expression was significantly higher in
the HDC group than in the LDT and HDT groups, in (E) ES and (F) LS
mice. Furthermore, EMMPRIN expression was significantly higher in
the LDT than in the HDT group in ES and LS mice. *P<0.05;
**P<0.01. ApoE−/−, apolipoprotein E knockout; ES,
early-start; LS, late-start; EMMPRIN, extracellular matrix
metalloproteinase inducer; HDC, high-fat diet control; NDC, normal
diet control; LDT, low-dose atorvastatin treatment; HDT, high-dose
atorvastatin treatment; GAPDH, glyceraldehyde 3-phosphate
dehydrogenase. |
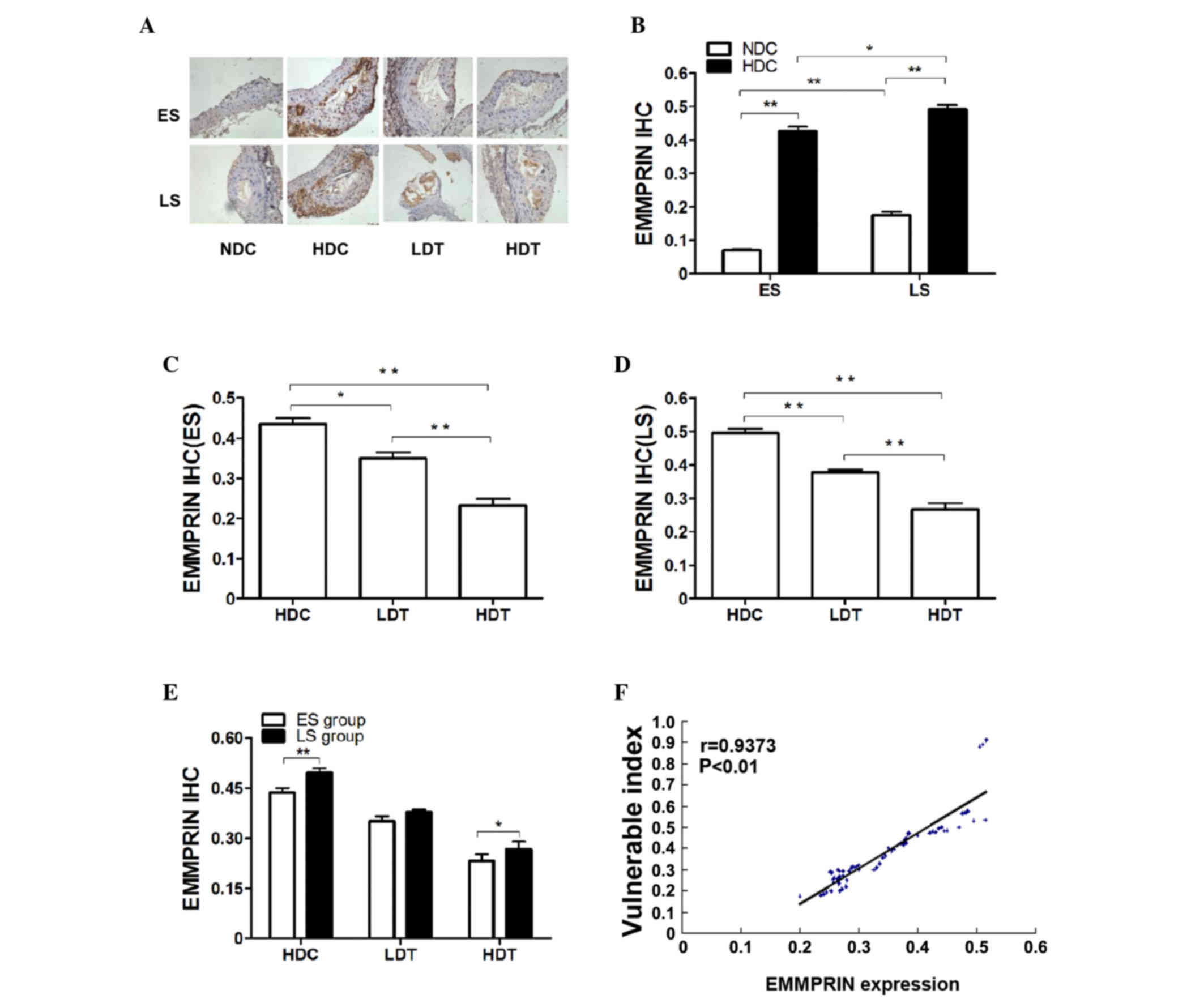 | Figure 4.IHC analysis of EMMPRIN in the aortas
of ApoE−/− male mice in the four different groups (n=9).
(A) Microscopic images (magnification, ×400) present the IHC
results. The brown color represents a positive result. (B) In the
ES and LS groups, EMMPRIN histological expression in the HDC group
was significantly higher than in the NDC group. Furthermore,
EMMPRIN histological expression was significantly higher in LS
subgroups compared with the corresponding ES subgroups. In the (C)
ES and (D) LS groups, EMMPRIN histological expression was
significantly lower in the HDT group than in the LDT group and was
significantly lower in the LDT and HDT groups compared with the HDC
group. (E) EMMPRIN histological expression in the LS group was
significantly higher in the HDC and HDT groups compared with the
corresponding ES subgroups. (F) VI was positively correlated with
the histological expression of EMMPRIN. *P<0.05; **P<0.01.
IHC, immunohistochemical; EMMPRIN, extracellular matrix
metalloproteinase inducer; ApoE−/−, apolipoprotein E
knockout; ES, early-start; LS, late-start; HDC, high-fat diet
control; NDC, normal diet control; HDT, high-dose atorvastatin
treatment; LDT, low-dose atorvastatin treatment; VI, vulnerability
index; r, Pearson's product-moment correlation coefficient. |
Atorvastatin decreases EMMPRIN
expression in the aortas and plaques of ApoE−/−
mice
Significantly decreased expression of EMMPRIN was
observed in the LDT (ES, P<0.05; LS, P<0.01) and HDT
(P<0.01) groups compared with the HDC group, according to the
mRNA (Fig. 2D-F) and protein levels
(Fig. 3B, E and F). This was
confirmed by the histological findings shown in Fig. 4 (Fig. 4A,
C and D). Histological EMMPRIN expression was positively
correlated with the VI (P<0.01; Fig.
4F).
High dose atorvastatin induced greater
downregulation of EMMPRIN expression than the low dose (P<0.05).
EMMPRIN expression was evaluated using RT-qPCR (Fig. 2F), western blotting (Fig. 3B and D) and an IHC assay (Fig. 4A and E). In the HDT and LDT groups,
EMMPRIN expression was significantly lower in the ES than in the LS
group, according to mRNA (LDT, P<0.05; HDT, P<0.01; Fig. 2F) and protein levels (P<0.05;
Fig. 3B and D). The histological
findings also indicated significantly lower EMMPRIN expression in
the ES HDT group than in the corresponding LS group (P<0.05;
Fig. 4E).
Ox-LDL induces the secretion of PGE2
and the expression of COX-2 and EMMPRIN in THP-1 macrophages
Treatment of THP-1 macrophages with 100 µg/ml ox-LDL
for 6 h induced a rapid increase in PGE2 secretion, which peaked at
12 h (Fig. 5A). RT-qPCR and western
blotting were used to examine whether COX-2 and EMMPRIN mRNA and
protein expression were upregulated in THP-1 macrophages stimulated
with ox-LDL in vitro. The mRNA and protein levels of COX-2
and EMMPRIN were increased significantly at 6 h compared with the
levels in the control group (P<0.05; Fig. 5B and C). A positive correlation was
observed between changes in mRNA and protein expression of EMMPRIN
and COX-2 (data not shown).
Atorvastatin inhibits ox-LDL-induced
expression of COX-2 and EMMPRIN in THP-1 macrophages
Following pretreatment with atorvastatin or
ezetimibe (both 5 µM) for 1 h, THP-1 macrophages were stimulated
with 100 µg/ml ox-LDL for an additional 12 h. COX-2 and EMMPRIN
protein expression levels were subsequently measured using western
blotting. Atorvastatin markedly inhibited the ox-LDL-induced
expression of COX-2 and EMMPRIN, whereas ezetimibe did not
(Fig. 5D). These results suggest
that the upregulation of EMMPRIN expression may be associated with
increased COX-2 levels in THP-1 macrophages.
Additionally, the results demonstrated that ox-LDL
was able to markedly upregulate COX-2 and EMMPRIN expression;
however, further research on the role of the COX-2/PGE2 pathway is
required. Therefore, these findings elucidate the possible effect
of COX-2/PGE2 on ox-LDL-induced EMMPRIN expression.
Following pretreatment with 5 µM atorvastatin or 10
µM NS-398 for 1 h, THP-1 macrophages were stimulated with 1 µM
ox-LDL or 0.1 µM ox-LDL and PGE2, for a further 12 h. Notably, when
atorvastatin or NS-398 was used as a co-pretreatment with PGE2, the
inhibitory effects of atorvastatin or NS-398 on ox-LDL-induced
EMMPRIN mRNA and protein expression were significantly reversed
(P<0.05; Fig. 5E and F).
Furthermore, PGE2 levels were significantly increased by ox-LDL
(P<0.05) and decreased by atorvastatin and NS-398 (Fig. 5G). These results indicate that
EMMPRIN expression in THP-1 macrophages was elevated due to an
increase in PGE2 levels, whereas atorvastatin and NS-398 each
suppressed PGE2 expression. This inhibition was reversed following
the addition of PGE2.
Discussion
In the present study, the increased EMMPRIN
expression in an ApoE−/− atherosclerotic mouse model and
the effect of atorvastatin treatment on EMMPRIN expression was
demonstrated. Atorvastatin treatment reduced artery atherosclerotic
lesion progression and EMMPRIN expression. The effect of
atorvastatin was associated with the COX-2/PGE2 signaling pathway.
These data implicate EMMPRIN as a harmful factor in the development
of VPs and indicate that atorvastatin inhibits the expression of
EMMPRIN in plaque.
EMMPRIN is a 58-kDa cell surface glycoprotein in the
immunoglobulin superfamily (22).
Previous experimental research has demonstrated that EMMPRIN is
localized in macrophage-rich regions of human atherosclerotic
plaques (8) and clinical researchers
have previously demonstrated that EMMPRIN is upregulated in
monocytes in acute myocardial infarction (AMI), whereas it is not
in patients with stable angina (23). Therefore, EMMPRIN may serve a role in
the destabilization of atheroma by inducing MMP-mediated ECM
degradation (24). The present study
demonstrated the increased infiltration of macrophages into VPs, in
which EMMPRIN was highly expressed. Macrophages secrete MMPs, which
promote the progression of VPs by degrading the ECM. It has been
suggested that nuclear factor κB (NF-κB) is a critical promoter of
MMPs (25,26). Therefore, downregulating EMMPRIN
expression in macrophages may reduce the potential damage caused by
MMPs via the NF-κB pathway and inhibit the development of VPs.
ApoE−/− mice contain the entire spectrum
of lesions that typically occur during atherogenesis and this mouse
model was the first to develop lesions similar to those observed in
humans (27). Previous studies have
demonstrated that feeding ApoE−/− mice either chow or a
high fat, Western-type diet may lead to plaque rupture (4,28–30). In
one such study, the brachiocephalic arteries of 62% of mice
exhibited acutely ruptured plaques after 8 weeks of feeding either
normal chow or a high-fat diet (29). Fragmentation and the loss of SMCs and
elastin in the fibrous caps were observed in relatively small and
lipid-rich plaques that overlay large complex lesions,
characterizing the rupture (4).
In the present study, lesions were immunostained to
identify macrophages, collagen, SMCs and the percentage of the
lesional area to determine the lesion composition in the plaques.
Apoptosis of vascular SMCs and macrophages may promote
pro-coagulation, plaque growth and rupture, which are the major
consequences of atherosclerosis in humans (31). Results from previous studies have
suggested that preventing ECM degradation or increasing ECM
components, including elastin and collagen, may decrease the risk
of plaque progression, thereby improving outcomes in
atherosclerotic disease (32–34).
Therefore, the present study evaluated the stability of the plaques
(the VI). The results indicated that EMMPRIN was increased in VPs
with a high ratio of macrophages to lipoid vesicles and in VPs with
a low ratio of SMCs to collagen, which suggests a latent link
between EMMPRIN and these plaque compositions. Previous studies
have indicated that EMMPRIN is upregulated during monocyte
differentiation into macrophages and that foam cells induce the
proliferation and migration of SMCs (8,35,36).
This suggests that EMMPRIN may induce VPs through these
pathways.
Atorvastatin is an inhibitor of HMG-CoA reductase
and is widely used to treat patients with atherosclerosis.
Atorvastatin was selected for the current study due to its
anti-atherosclerotic effect. Following confirmation of the
correlation between EMMPRIN and VP, atorvastatin was used as an
intervention in ApoE−/− mice that were fed a high-fat
diet. The results suggested that atorvastatin was able to
downregulate EMMPRIN expression in the aortas of ApoE−/−
mice, according to mRNA and protein levels and histological
findings. Additionally, artery atherosclerotic lesion progression
was inhibited. In the present study, ApoE−/− mice were
23 weeks old when the experiment in the ES group was completed. The
treatment subgroups had been treated with atorvastatin for 8 weeks,
whereas treatment of LS group mice had been initiated later and
lasted for 4 weeks. The results demonstrated that plaque VI and
EMMPRIN expression were significantly greater in the LS group. This
suggests that differences in the initiation time and duration of
the statin intervention may have affected plaque vulnerability and
that the difference observed following early statin treatment may
be due to the early reduction in the expression of EMMPRIN during
SMC migration and macrophage aggregation.
A number of studies have indicated that EMMPRIN
levels may be increased by various stimuli, including free
radicals, cytokines and ox-LDL, that have key regulatory roles in
MMP activity (37,38). MMPs make an essential contribution to
the pathophysiology of atherosclerosis (23,39,40). A
link has been identified between the presence of membrane type
1-MMP, MMP-2 and MMP-9 within vascular walls and unstable plaque
phenotypes that are prone to rupture (41–43). In
cardiac disease, patients with AMI exhibit increased EMMPRIN, MMP-2
and MMP-9 levels, whereas patients with stable angina do not
(44). In unstable plaques, levels
of MMP-9 activity are increased, whereas in stable plaques, levels
of MMP-2 activity levels are increased (45). MMP expression levels are associated
with different glycosylated EMMPRIN forms (23). Additionally, different EMMPRIN forms
have been observed among different plaque phenotypes (46). The present data suggested that
atorvastatin may have prevented the process of atherosclerosis and
plaque rupture by reducing EMMPRIN expression in plaques.
Atorvastatin had no significant effect on the plasma lipid and
cholesterol concentrations in the ApoE−/− mice, which
Bea et al (47) and Johnson
et al (29) have previously
documented for simvastatin and pravastatin, respectively.
Atorvastatin may have a pleiotropic effect, similar to other
statins (48).
In previous studies conducted by the present authors
(49,50), it was demonstrated that angiotensin
II upregulated EMMPRIN expression in macrophages via the COX-2
pathway. Following the finding that atorvastatin induced COX-2
expression in macrophages and monocytes, the mechanism used in the
atorvastatin-induced inhibition of EMMPRIN expression in
ApoE−/− mice was investigated. In an in vitro
study, it was demonstrated that the expression of the COX-2 gene
and protein induced by ox-LDL were also inhibited by atorvastatin
and that PGE2 restored the EMMPRIN inhibition induced by reductase
HMG-CoA inhibitors or COX-2 inhibitors. These observations
indicated that atorvastatin was able to inhibit ox-LDL-mediated
upregulation of EMMPRIN expression via the COX-2/PGE2 signaling
pathway.
In conclusion, the present study evaluated EMMPRIN
expression in aortic plaques in ApoE−/− mice according
to dietary fat content. It was demonstrated that EMMPRIN expression
was upregulated and was positively correlated with the development
of VPs. Atorvastatin treatment was able to reduce atherosclerotic
plaque vulnerability by downregulating EMMPRIN expression. This
inhibitory effect of atorvastatin on EMMPRIN may occur via the
COX-2/PGE2 signaling pathway. Further studies are necessary to
confirm that this is the case.
References
|
1
|
Aronson D and Edelman ER: Coronary artery
disease and diabetes mellitus. Cardiol Clin. 32:439–455. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pagidipati NJ and Peterson ED: Acute
coronary syndromes in women and men. Nat Rev Cardiol. 13:471–480.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lusis AJ: Atherosclerosis. Nature.
407:233–241. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rosenfeld ME, Polinsky P, Virmani R,
Kauser K, Rubanyi G and Schwartz SM: Advanced atherosclerotic
lesions in the innominate artery of the ApoE knockout mouse.
Arterioscler Thromb Vasc Biol. 20:2587–2592. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schonbeck U and Libby P: CD40 signaling
and plaque instability. Circ Res. 89:1092–1103. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rajagopalan S, Meng XP, Ramasamy S,
Harrison DG and Galis ZS: Reactive oxygen species produced by
macrophage-derived foam cells regulate the activity of vascular
matrix metalloproteinases in vitro. Implications for
atherosclerotic plaque stability. J Clin Invest. 98:2572–2579.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fabunmi RP, Moore KJ, Libby P and Freeman
MW: Stromelysin-1 (MMP-3) expression driven by a
macrophage-specific promoter results in reduced viability in
transgenic mice. Atherosclerosis. 148:375–386. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Major TC, Liang L, Lu X, Rosebury W and
Bocan TM: Extracellular matrix metalloproteinase inducer (EMMPRIN)
is induced upon monocyte differentiation and is expressed in human
atheroma. Arterioscler Thromb Vasc Biol. 22:1200–1207. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Patti G, Pasceri V, Colonna G, Miglionico
M, Fischetti D, Sardella G, Montinaro A and Di Sciascio G:
Atorvastatin pretreatment improves outcomes in patients with acute
coronary syndromes undergoing early percutaneous coronary
intervention: Results of the ARMYDA-ACS randomized trial. J Am Coll
Cardiol. 49:1272–1278. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Soeda T, Uemura S, Okayama S, Kawakami R,
Sugawara Y, Nakagawa H, Matsumoto T, Sung JH, Nishida T, Senoo A,
et al: Intensive lipid-lowering therapy with rosuvastatin
stabilizes lipid-rich coronary plaques. -Evaluation using
dual-source computed tomography. Circ J. 75:2621–2627. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Murphy SA, Cannon CP, Wiviott SD, de Lemos
JA, Blazing MA, McCabe CH, Califf RM and Braunwald E: Effect of
intensive lipid-lowering therapy on mortality after acute coronary
syndrome (a patient-level analysis of the Aggrastat to Zocor and
Pravastatin or Atorvastatin Evaluation and Infection
Therapy-Thrombolysis in Myocardial Infarction 22 trials). Am J
Cardiol. 100:1047–1051. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Farmer JA and Gotto AM Jr: Currently
available hypolipidaemic drugs and future therapeutic developments.
Baillieres Clin Endocrinol Metab. 9:825–847. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Illingworth DR and Bacon S: Hypolipidemic
effects of HMG-CoA reductase inhibitors in patients with
hypercholesterolemia. Am J Cardiol. 60:33G–42G. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
O'Driscoll G, Green D and Taylor RR:
Simvastatin, an HMG-coenzyme A reductase inhibitor, improves
endothelial function within 1 month. Circulation. 95:1126–1131.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Stroes ES, Koomans HA, de Bruin TW and
Rabelink TJ: Vascular function in the forearm of
hypercholesterolaemic patients off and on lipid-lowering
medication. Lancet. 346:467–471. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Meredith IT, Plunkett JC, Worthley SG,
Hope SA and Cameron JD: Systemic inflammatory markers in acute
coronary syndrome: Association with cardiovascular risk factors and
effect of early lipid lowering. Coron Artery Dis. 16:415–422. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ridker PM: Inflammation, infection, and
cardiovascular risk: How good is the clinical evidence?
Circulation. 97:1671–1674. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Williams H, Johnson JL, Carson KG and
Jackson CL: Characteristics of intact and ruptured atherosclerotic
plaques in brachiocephalic arteries of apolipoprotein E knockout
mice. Arterioscler Thromb Vasc Biol. 22:788–792. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Burke AP, Farb A, Malcom GT, Liang YH,
Smialek J and Virmani R: Coronary risk factors and plaque
morphology in men with coronary disease who died suddenly. N Engl J
Med. 336:1276–1282. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Virmani R, Burke A and Farb A: Coronary
risk factors and plaque morphology in men with coronary disease who
died suddenly. Eur Heart J. 19:678–680. 1998.PubMed/NCBI
|
|
22
|
Biswas C, Zhang Y, DeCastro R, Guo H,
Nakamura T, Kataoka H and Nabeshima K: The human tumor cell-derived
collagenase stimulatory factor (renamed EMMPRIN) is a member of the
immunoglobulin superfamily. Cancer Res. 55:434–439. 1995.PubMed/NCBI
|
|
23
|
Schmidt R, Bültmann A, Ungerer M,
Joghetaei N, Bülbül O, Thieme S, Chavakis T, Toole BP, Gawaz M,
Schömig A and May AE: Extracellular matrix metalloproteinase
inducer regulates matrix metalloproteinase activity in
cardiovascular cells: Implications in acute myocardial infarction.
Circulation. 113:834–841. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yoon YW, Kwon HM, Hwang KC, Choi EY, Hong
BK, Kim D, Kim HS, Cho SH, Song KS and Sangiorgi G: Upstream
regulation of matrix metalloproteinase by EMMPRIN; Extracellular
matrix metalloproteinase inducer in advanced atherosclerotic
plaque. Atherosclerosis. 180:37–44. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ghattas A, Griffiths HR, Devitt A, Lip GY
and Shantsila E: Monocytes in coronary artery disease and
atherosclerosis: Where are we now? J Am Coll Cardiol. 62:1541–1551.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen F, Eriksson P, Hansson GK, Herzfeld
I, Klein M, Hansson LO and Valen G: Expression of matrix
metalloproteinase 9 and its regulators in the unstable coronary
atherosclerotic plaque. Int J Mol Med. 15:57–65. 2005.PubMed/NCBI
|
|
27
|
Nakashima Y, Plump AS, Raines EW, Breslow
JL and Ross R: ApoE-deficient mice develop lesions of all phases of
atherosclerosis throughout the arterial tree. Arterioscler Thromb.
14:133–140. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jawien J, Nastalek P and Korbut R: Mouse
models of experimental atherosclerosis. J Physiol Pharmacol.
55:503–517. 2004.PubMed/NCBI
|
|
29
|
Johnson J, Carson K, Williams H, Karanam
S, Newby A, Angelini G, George S and Jackson C: Plaque rupture
after short periods of fat feeding in the apolipoprotein E-knockout
mouse: Model characterization and effects of pravastatin treatment.
Circulation. 111:1422–1430. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rosenfeld ME, Carson KG, Johnson JL,
Williams H, Jackson CL and Schwartz SM: Animal models of
spontaneous plaque rupture: The holy grail of experimental
atherosclerosis research. Curr Atheroscler Rep. 4:238–242. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gao P, Guo RW, Chen JF, Chen Y, Wang H, Yu
Y and Huang L: A meprin inhibitor suppresses atherosclerotic plaque
formation in ApoE−/− mice. Atherosclerosis. 207:84–92.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cheng XW, Kuzuya M, Sasaki T, Arakawa K,
Kanda S, Sumi D, Koike T, Maeda K, Tamaya-Mori N, Shi GP, et al:
Increased expression of elastolytic cysteine proteases, cathepsins
S and K, in the neointima of balloon-injured rat carotid arteries.
Am J Pathol. 164:243–251. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Grainger DJ, Witchell CM and Metcalfe JC:
Tamoxifen elevates transforming growth factor-beta and suppresses
diet-induced formation of lipid lesions in mouse aorta. Nat Med.
1:1067–1073. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sukhova GK, Zhang Y, Pan JH, Wada Y,
Yamamoto T, Naito M, Kodama T, Tsimikas S, Witztum JL, Lu ML, et
al: Deficiency of cathepsin S reduces atherosclerosis in LDL
receptor-deficient mice. J Clin Invest. 111:897–906. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang J, Ge H, Wang C, Guo TB, He Q, Shao
Q and Fan Y: Inhibitory effect of PPAR on the expression of EMMPRIN
in macrophages and foam cells. Int J Cardiol. 117:373–380. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Seizer P, Schonberger T, Schött M, Lang
MR, Langer HF, Bigalke B, Krämer BF, Borst O, Daub K, Heidenreich
O, et al: EMMPRIN and its ligand cyclophilin A regulate MT1-MMP,
MMP-9 and M-CSF during foam cell formation. Atherosclerosis.
209:51–57. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gabison EE, Hoang-Xuan T, Mauviel A and
Menashi S: EMMPRIN/CD147, an MMP modulator in cancer, development
and tissue repair. Biochimie. 87:361–368. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Haug C, Lenz C, Díaz F and Bachem MG:
Oxidized low-density lipoproteins stimulate extracellular matrix
metalloproteinase Inducer (EMMPRIN) release by coronary smooth
muscle cells. Arterioscler Thromb Vasc Biol. 24:1823–1829. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dutta P, Courties G, Wei Y, Leuschner F,
Gorbatov R, Robbins CS, Iwamoto Y, Thompson B, Carlson AL, Heidt T,
et al: Myocardial infarction accelerates atherosclerosis. Nature.
487:325–329. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kampoli AM, Tousoulis D, Papageorgiou N,
Antoniades C, Androulakis E, Tsiamis E, Latsios G and Stefanadis C:
Matrix metalloproteinases in acute coronary syndromes: Current
perspectives. Curr Top Med Chem. 12:1192–1205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Satoh K, Nigro P, Matoba T, O'Dell MR, Cui
Z, Shi X, Mohan A, Yan C, Abe J, Illig KA and Berk BC: Cyclophilin
A enhances vascular oxidative stress and the development of
angiotensin II-induced aortic aneurysms. Nat Med. 15:649–656. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sukhanov S, Higashi Y, Shai SY, Vaughn C,
Mohler J, Li Y, Song YH, Titterington J and Delafontaine P: IGF-1
reduces inflammatory responses, suppresses oxidative stress, and
decreases atherosclerosis progression in ApoE-deficient mice.
Arterioscler Thromb Vasc Biol. 27:2684–2690. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Newby AC: Metalloproteinases and
vulnerable atherosclerotic plaques. Trends Cardiovasc Med.
17:253–258. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nie R, Xie S, Du B, Liu X, Deng B and Wang
J: Extracellular matrix metalloproteinase inducer (EMMPRIN) is
increased in human left ventricle after acute myocardial
infarction. Arch Med Res. 40:605–611. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Fiotti N, Altamura N, Orlando C, Simi L,
Reimers B, Pascotto P, Zingone B, Pascotto A, Serio M, Guarnieri G
and Giansante C: Metalloproteinases-2, −9 and TIMP-1 expression in
stable and unstable coronary plaques undergoing PCI. Int J Cardiol.
127:350–357. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yang LX, Yang ZH, Guo RW, Ye JS and Liu H:
Angiotensin II induces extracellular matrix metalloproteinase
inducer expression via an AT1R dependent pathway in aortic
atherosclerotic plaque in apolipoprotein E knockout mice. J Renin
Angiotensin Aldosterone Syst. 13:67–75. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bea F, Blessing E, Bennett B, Levitz M,
Wallace EP and Rosenfeld ME: Simvastatin promotes atherosclerotic
plaque stability in apoE-deficient mice independently of lipid
lowering. Arterioscler Thromb Vasc Biol. 22:1832–1837. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ray KK and Cannon CP: Atorvastatin and
cardiovascular protection: A review and comparison of recent
clinical trials. Expert Opin Pharmacother. 6:915–927. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Shao Q, Shen LH, Hu LH, Pu J, Jing Q and
He B: Atorvastatin suppresses inflammatory response induced by
oxLDL through inhibition of ERK phosphorylation, IκBα degradation,
and COX-2 expression in murine macrophages. J Cell Biochem.
113:611–618. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Deng P, Zhao SP, Dai HY, Guan XS and Huang
HG: Atorvastatin reduces the expression of COX-2 mRNA in peripheral
blood monocytes from patients with acute myocardial infarction and
modulates the early inflammatory response. Clin Chem. 52:300–303.
2006. View Article : Google Scholar : PubMed/NCBI
|















